Introduction
Eighty percent of patients with ovarian cancer are
diagnosed with pathological stage III/IV, suffering incurable
progression within 3 years (1,2).
Anti-ER treatments are well-tolerated and appealing in this
setting, but are frequently met with anti-estrogen resistance
(3). Estrogen (E2) is implicated in
the ovarian carcinogenesis. E2 drives tumor proliferation via
estrogen receptor α (ERα) and anti-estrogen can markedly inhibit
ovarian cancer mitogenesis (3–5). As
compared with a relatively lower level of ERβ in ovarian cancers,
ERα in ovarian cancers is expressed at a level similar to that in
breast cancers, 60–70% (6,7). Despite a relatively higher level of ER
expression in ovarian cancers, numerous small clinical trials of
ER-blockers were disappointing (3–6).
Tamoxifen yielded a low response rate for recurrent ovarian cancers
(mean, 13%). In parallel, the pure ER-antagonist, fulvestrant, had
the overall response of ~10% in advanced and heavily pretreated
recurrent ovarian cancer (3–5). In
sum, therapeutic regimens that block pro-tumorigenic estrogen
effects in ovarian cancer have not been studied in well-designed
trials.
Moreover, 12–34% of ovarian cancer that do initially
respond invariably and frequently develop anti-estrogen resistance
(8,9). Estrogen efficiently promotes
transition from quiescence to G1 in ER-sensitive cells (10). Cyclin-bound cyclin-dependent kinase
(Cdk) and Cdk inhibitors mutually orchester cell cyclin
progression. In breast cancer cells, ER blockade is able to induce
G1 phase arrest by upregulating p27 expression (11,12).
Notably, p27 is frequently downregulated in ovarian cancers
(13,14). This may result from constitutive
activation of Src, which facilitates degradation of p27 by
Src-induced phosphorylation. Crosstalk between estrogen-bound ER
and Src drives mitogenic pathways that are frequently activated in
ovarian cancer (15). On the other
hand, the lack of effective treatments for recurrent ovarian cancer
has stimulated development of targeted ovarian cancer therapies.
Mitogenic pathways, including Src, Ras/Raf/MEK and PI3K/AKT, are
frequently activated in ovarian cancer (16). Src is overexpressed and activated in
the majority of ovarian cancers and regulates cell anti- and
pro-apoptosis (17,18). It has been demonstrated that the
intercross action between liganded ER and Src contributes to
mitogenesis and ER-activated gene expression (3,17,19). A
potent inhibitor of Abl and Src family kinases, saracatinib
(AZD0530), is able to inhibit cell invasion in vitro and
xenograft growth in vivo, and is well-tolerated in phase I
trial (3). Nevertheless, the
anti-estrogen effect has not been evaluated in ovarian cancer.
In the present study, we investigated whether
constitutive Src activation contributed to resistance to
ER-blockade in ovarian cancer. We found that saracatinib reversed
fulvestrant resistance in vitro and in vivo, cell
cycle arrest, autophagy and apoptosis. The data provide novel
insights for crosstalk between ER and Src pathways in ovarian
cancer.
Materials and methods
Patients and tissue samples
A total of 40 ovarian cancers with
immunohistochemically positive ER expression were selected and
collected following radical surgery at the Department of Pathology
of the First Affiliated Hospital of Xi’an Jiaotong University from
1999 to 2000. The cases of ovarian cancer displayed ER-positive
expression determined by two individual pathologists according to
immunohistochemistry (IHC) assay. All patients in this study were
classified as International Federation of Gynecology and Obstetrics
(FIGO) stage III (FIGO). Subsequently, postoperative therapeutic
strategy was determined by a multi-disciplinary team (MDT),
including an oncosurgeon, an oncologist and a radiologist.
Accordingly, ER antagonist, fulvestrant, was recommended to treat
ER-positive ovarian cancer as a single agent, 500 mg
intramuscularly on day 1, 250 mg intramuscularly on day 15, and 250
mg intramuscularly on day 29 and every 28 days thereafter until
either in-tolerance or disease progression.
The patients with ovarian cancer were routinely
scheduled for life-long follow-up. The enrolled patients were
followed up at the outpatient clinic at intervals of three months
during the first two years and every six months for three more
years. Metastatic events were diagnosed at the outpatient clinic.
Whenever a distant metastasis was suspected, radiologic, endoscopic
or histologic confirmation was compulsory. The calculation of
disease-free survival (DFS) started at the date of surgery and
ended at the date of one of the following events: recurrence,
distant metastasis or oncological death. As reported thus far, no
participants were lost during follow-up.
Immunohistochemistry
After routine deparaffinization and hydration,
tissue sections were treated with 3% hydrogen peroxide and heated
in EDTA (pH 8.0) for antigen retrieval. Following serum
deprivation, ERα and p-Src (phospho-Y416-Src) antigen-antibody
reactions took place at 4°C overnight. The streptavidin/peroxidase
kit (Invitrogen) was used to detect antigen-antibody reactions. The
purified rabbit/rat monoclonal antibodies against human ERα
(ab-81086) and Src (ab-106271) (both from Abcam) were used at 2
μg/ml and goat anti-rabbit/rat biotin-conjugated IgG was secondary
antibody. Immunohistochemical signals were scored by two
independent observers. The scores were calculated as the number of
stained cells divided by the total number of cancer cells counted.
Four high-power fields (x400) per slide were calculated and
outcomes were averaged. Unequivocal staining of cytoplasm in
>50% of cancer cells was considered as positive.
Cell culture and transfection
Ovarian cancer cell lines, SKOV-3 and an inherent
anti-estrogen-resistant variant, SKOV-3R, established after
prolonged passage, were cultured in DMEM supplemented with 10% FBS.
Cells (293T) were a gift from Professor Chen Huang from the Medical
College of Xi’an Jiaotong University. Cell lines in the present
study were authenticated using American Type Culture Collection
(ATCC) guidelines. Asynchronous culture was treated with
10−6 mol/l saracatinib, 10−6 mol/l
fulvestrant or both for 48 h. SKOV-3R with 0.1% cFBS for 48 h were
then treated with ERα and ERβ agonists/antagonists. Then, SKOV-3R
cells were transduced with lentivirus carrying pGIPZ-luc-flag
encoding an Src-specific shRNA (5′-CAGATTGTCAACAACACAG-3′) or
control shRNA with the selection of puromycin (8 μg/ml) for stably
transfected cells. Lentivirus-shRNA was purchased from GenePharma,
Shanghai, China.
Agents and drugs
Saracatinib (10−6 mol/l) and fulvestrant
(10−6 mol/l) were dissolved in dimethyl sulfoxide (DMSO)
(AstraZeneca). The pure ERα agonist
4,40,400-[4-propyl-(1H)-pyrazole-1,3,5-triyl]trisphenol (PPT), the
selective ERβ agonist 2,3-bis(4-hydroxyphenyl)-propionitrile (DPN),
the dual ERα agonist/ERβ antagonist
(R,R)-5,11-diethyl-5,6,11,12-tetrahydro-2,8-chrysenediol (THC), and
the ERβ antagonist
4-[2-phenyl-5,7-bis(trifluoromethyl)pyrazolo[1,5-a]pyrimidin-3-yl]-phenol
(PHTPP) were obtained from GenePharma.
Immunofluorescence microscopy
Cells were serum/E2-deprived for 24 h, then treated
with 10−8 mol/l E2 for the indicated times.
Immunofluorescence (IF) was processed via anti-ERα (ab-12223) or
anti-p-Src (ab-4816) (both from Abcam). Cells were counterstained
by 40,6-diamidino-2-phenylindole (DAPI) and analyzed by IF
microscopy. Dual ERα and p-Src staining were linked to Texas Red
for p-Src and GFP for ERα.
Subcellular fractionation
Cellular subcellular fractionations were
serum/E2-deprived for 24 h and then treated with E2
(10−8 mol/l) for 4 h. Fractionation was performed as
previously described (20).
Cell cycle analysis
Cells were bromodeoxyuridine (BrdUrd)-labeled,
counterstained with propidium iodide, and cell cycle distribution
was assayed as previously described (21).
Annexin V staining
Drug-treated cells were analyzed by FITC Annexin V
apoptosis detection kit I (BD Biosciences) as per the
manufacturer’s instructions.
Detection of autophagic vesicles
Drug effects on autophagic vesicles were evaluated
by Cyto-ID Autophagy Detection kit (Enzo) as per the manufacturer’s
instructions.
Immunoblotting, immunoprecipitation and
kinase assay
Western blot analysis was conducted and quantitated
by densitometry. ERα, MAPK, p-MAPK antibodies were purchased from
Santa Cruz; p27, Akt, p-Akt, Src and p-Src antibodies from Abcam.
ER-Src complexes were precipitated after 24 h of serum/E2
deprivation. At intervals after E2 repletion, Src and ERα were
detected by western blot analysis.
Xenografts
Luciferase-positive SKOV-3R cells re-suspended with
Matrigel were implanted at the axillary top of female BALB/C nude
mice. Xenograft growth was compared in nude mice (10 per group)
with supplemental estradiol at 0.72 mg/90 days (Invitrogen).
E2-supplemented animals (10 per group) either received no
treatment, fulvestrant 3.5 mg per week SQ, fulvestrant 5 mg per
week SQ plus saracatinib 25 mg/kg daily (via oral gavage) or both
starting either 11 or 26 days after tumor implantation. Viable
tumor burden (luciferase activity) was assessed weekly. Mice were
imaged by IVIS-100 bioluminescence imager (IVIS). Animals were
weighed twice per week. Entire experimental data was detailed and
documented.
Statistical analysis
All assays of cell cycle distribution, IF and
IP/western blot analysis were at least triplicated. Xenograft
studies were repeated twice. Data were characterized as means ±
SEM. One- or two-way ANOVA was conducted to assess differences
between treatment groups. The significance was a statistical
indication of synergism, suggesting that the combined effect of two
agents was manifested in a non-additive manner. Following a
significant ANOVA result (P<0.05) rejecting the null hypothesis,
Tukey honestly significant difference (HSD) test was used for all
pairwise mean comparisons. These multiple comparison procedures
ensured actual error rates no greater than prespecified 5%.
Analysis was conducted in SPSS 19.0.
Results
Expression of p-Src in ER-positive
ovarian cancers
Forty ER-positive ovarian cancer patients with FIGO
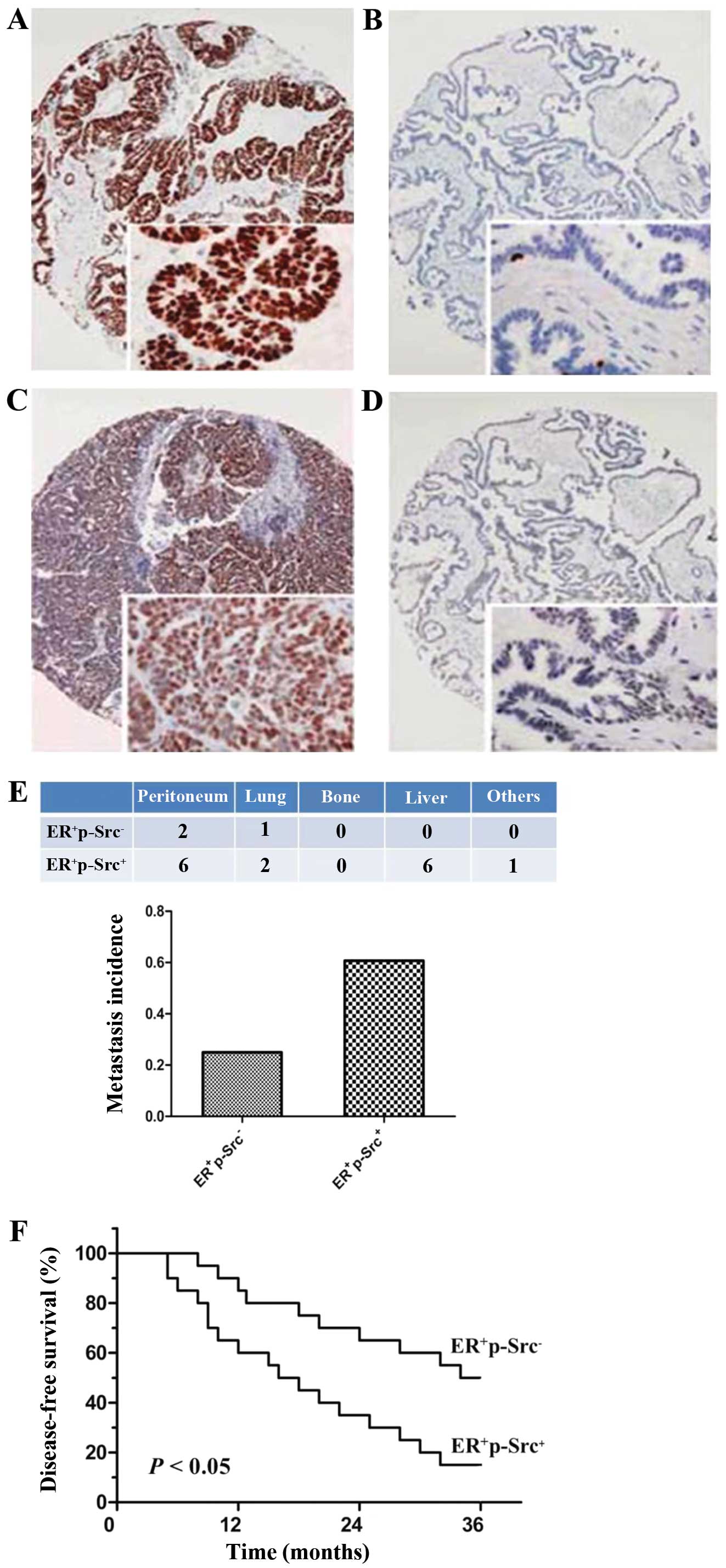
stage III were eligible in the present study (Fig. 1A). The participating patients with
ER-positive ovarian cancer received post-operative treatment of ER
antagonist, fulvestrant. We observed that activated Src, p-Src, was
positively expressed in 20/40 ER-positive ovarian cancer (50%;
Fig. 1C). Notably, p-Src was not
significantly associated with any clinicopathological
characteristics with a favorable balance between two groups
demonstrated in Table I. It was
unclear whether ER antagonist was absolutely beneficial for disease
progression of patients with ovarian cancer. Hence, we investigated
the effects of ER antagonist on disease progression in patients
with ovarian cancer. As compared with ovarian cancer patients with
ER+p-Src+, ovarian cancer patients with
ER+p-Src− presented a markedly higher
metastatic incidence and a significant advantage for DFS at 3 years
after ER antagonist treatment (P<0.01; Fig. 1E and F), indicating the possibility
that Src activation exerted the positive effects on resistance to
ER antagonist associated with the propensity for metastasis or
disease progression in patients with ER+ ovarian
cancer.
 | Table IAssociation of p-Src expression with
clinicopathological characteristics of patients with ER-positive
ovarian cancer (n=40). |
Table I
Association of p-Src expression with
clinicopathological characteristics of patients with ER-positive
ovarian cancer (n=40).
| Clinicopathological
characteristics | p-Src expression n
(%) | P-value |
|---|
|
|---|
| Positive | Negative |
|---|
| Age (years) | | | >0.05 |
| ≤60 | 8 (40.0) | 8 (40.0) | |
| >60 | 12 (60.0) | 12 (60.0) | |
| Median age
(years) | 67 | 66 | |
| FIGO grade | | | >0.05 |
| Well | 3 (15.0) | 2 (10.0) | |
| Moderate | 7 (35.0) | 7 (35.0) | |
| Poor | 10 (50.0) | 11 (55.0) | |
| Lymph node
metastasis | | | >0.05 |
| Positive | 5 (25.0) | 5 (25.0) | |
| Negative | 15 (75.0) | 15 (75.0) | |
| FIGO stage | | | >0.05 |
| IIIA | 4 (20) | 5 (25.0) | |
| IIIB | 11 (55.0) | 10 (50.0) | |
| IIIC | 5 (25.0) | 5 (25.0) | |
ERα-ER binding triggers Src
activation
SKOV-3R, a variant of ascites ovarian cancer SKOV-3,
displayed the resistance against estrogen during prolonged culture.
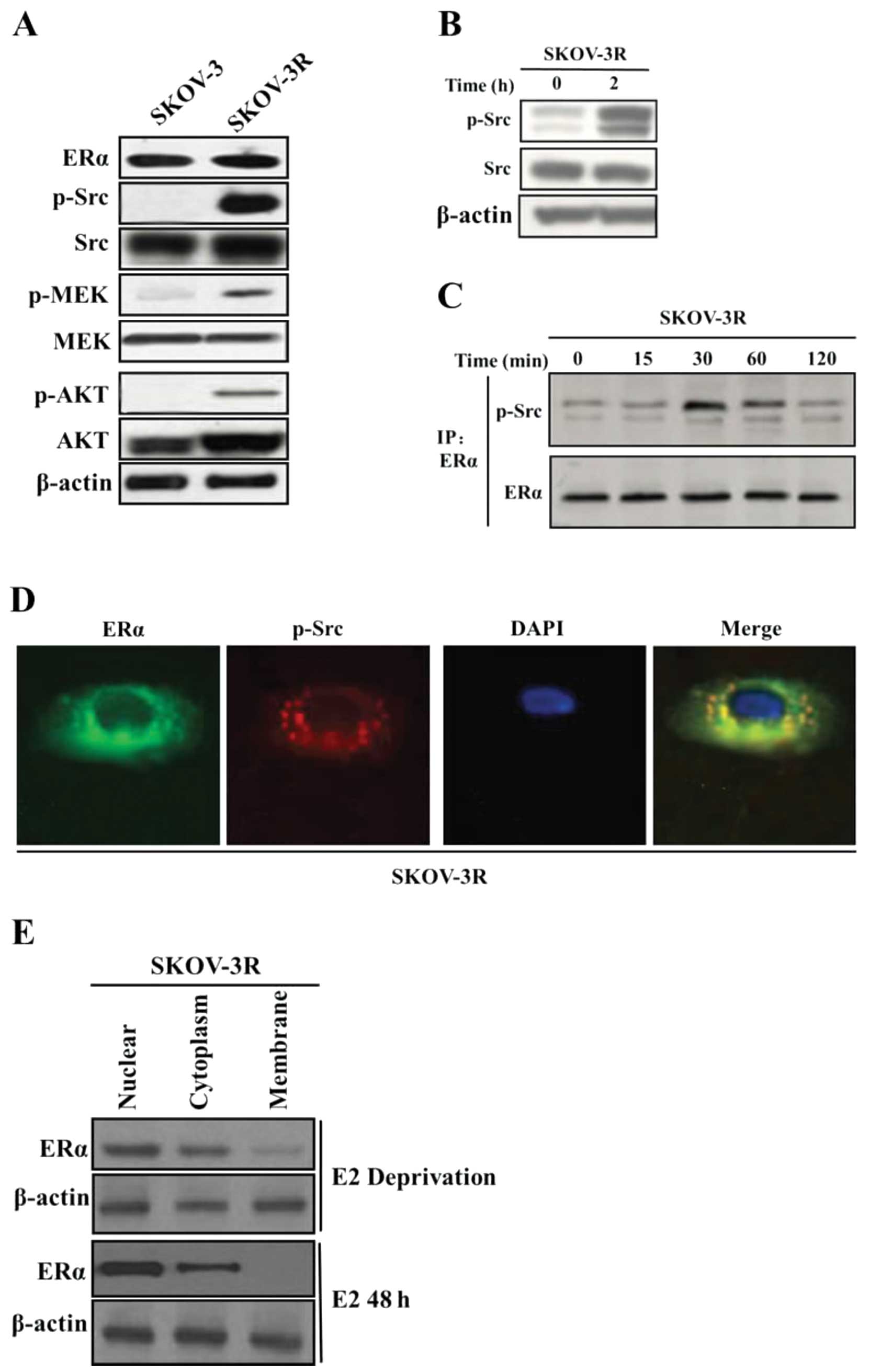
Both SKOV-3R and SKOV-3 expressed ER (Fig. 2A), and SKOV-3R had clearly activated
Src, MEK and AKT at higher levels than those in SKOV-3 (Fig. 2A). To explore the involvement of Src
in E2-stimulated ovarian cancer proliferation, SKOV-3 was E2- and
serum-deprived for 48 h and in turn treated with E2
(10−8 mol/l) for the times indicated. It was observed
that estrogen activated Src in SKOV-3R (Fig. 2B). ER-estrogen binding activated Src
in ERα immunoprecipitation at 30 min after E2 triggering (Fig. 2C), supported by the observation that
ER and Src co-localized in the perinuclear zone by
immunofluoresence at 15 min (Fig.
2D). Thus, it was likely that E2 stimulated ER-Src interaction
and was subject to Src activation, which may contribute to its
mitogenic effects. Furthermore, subcellular fractionation analysis
showed that ERα was strongly cytoplasmic in the E2-deprived cells,
but rapidly transferred into the nucleus 48 h after E2 addition
(Fig. 2E). As compared with breast
cancer where ER was predominantly nuclear distribution (11,12),
the ovarian cancer cell line expressed cytoplasmic ER in the
absence of E2, which translocated to the nucleus with E2
addition.
E2 enhances ovarian cancer growth via the
receptor of ERα
Subsequently, estrogen deprivation with or without
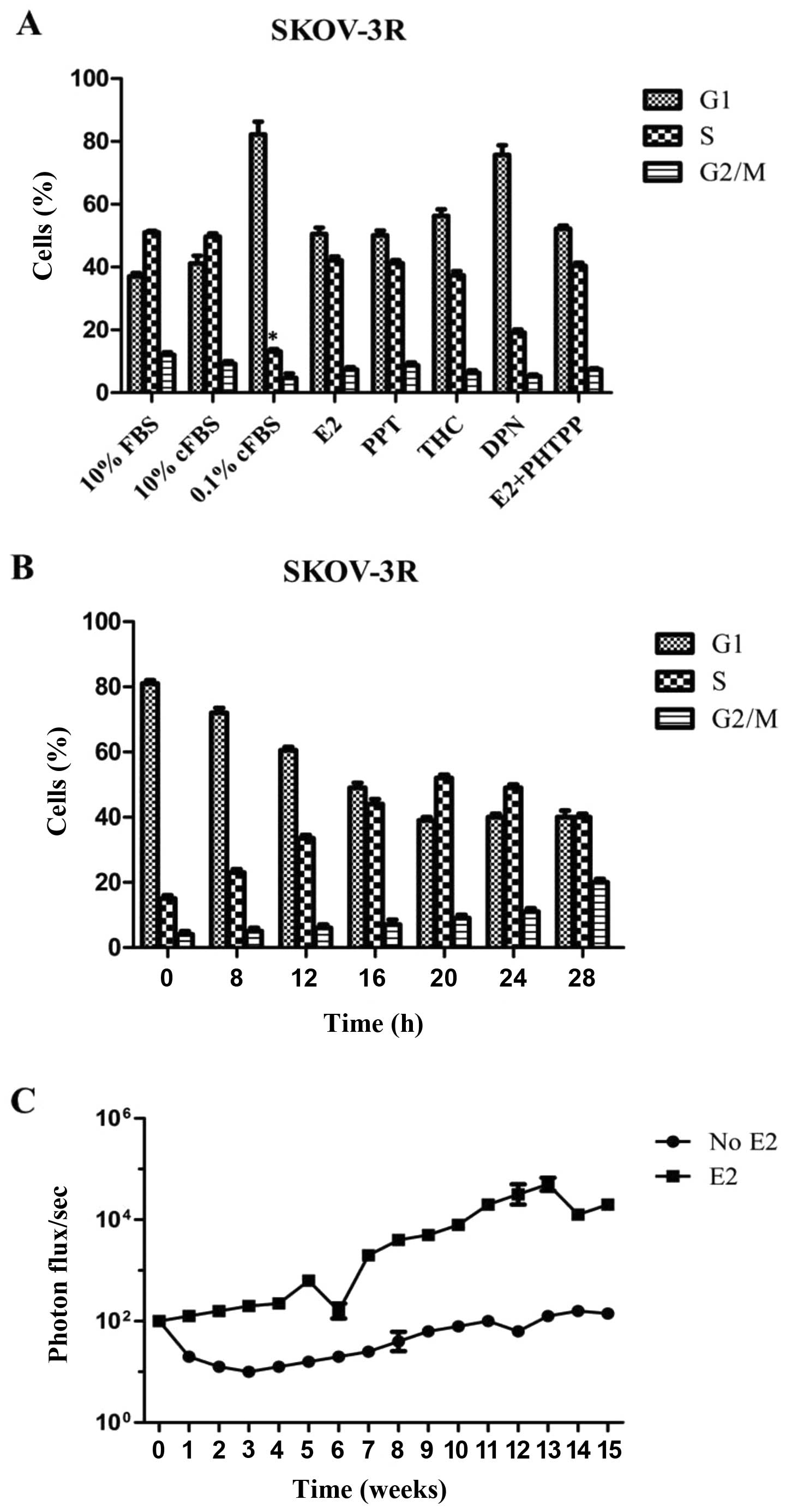
growth factor depletion was examined. SKOV-3R growth was not
inhibited after 48 h of E2 depletion alone (media, 10% cFBS), but
estrogen and serum depleted media (0.1% cFBS) for 48 h diminished
the percentage of S-phase cells from 50 to 15%
(*P<0.01 vs. 10% FBS or 10% cFBS; Fig. 3A). It was of note that the addition
of E2 alone restored 0.1% cFBS-induced growth arrest (Fig. 3A). It indicated that estrogen alone
was able to maintain SKOV-3R cell proliferation in the absence of
growth factors. To examine the ability of E2 to drive SKOV-3R cell
proliferation, SKOV-3R cells were preliminarily arrested in 0.1%
cFBS for 48 h. Then, the addition of E2 alone stimulated cell cycle
re-entry with % S-phase peaking at 20 h (Fig. 3B). Owing to binding of either E2 or
fulvestrant to ERs, including ERα and ERβ, we tested which receptor
induced SKOV-3R cell growth arrest of estrogen. SKOV-3R cells were
transferred to 0.1% cFBS for 48 h, then treated with various
hormone receptor agonists or antagonists for 48 h before cell cycle
analysis. The pure ERα agonist, PPT and the ERα agonist/ERβ
antagonist, THC, rescued E2 deprivation in SKOV-3R cells.
Furthermore, the ERβ agonist, DPN, did not induce tumor cell growth
while the ERβ antagonist PHTPP with E2 addition did not abrogate
E2-inducing cell growth (Fig. 3A).
The mean % S-phase did not significantly differ between control and
those treated with E2, PPT, THC or PHTPP, nor did they differ
between DPN-treated and E2-deprived in SKOV-3R cells. Thus, the
proliferative function of E2 in ovarian cancer was determined by
ERα independent of ERβ. Then, E2 function was further assayed in
nude mice models in vivo (Fig.
3C). After SKOV-3R cell pellets were implanted with or without
E2 manipulation, estrogen-supplemented xenographs showed
considerable growth with markedly higher photon flux/sec at 16
weeks as compared with those without E2 treatment (Fig. 3C).
Saracatinib reverses resistance to
fulvestrant via Src inhibition
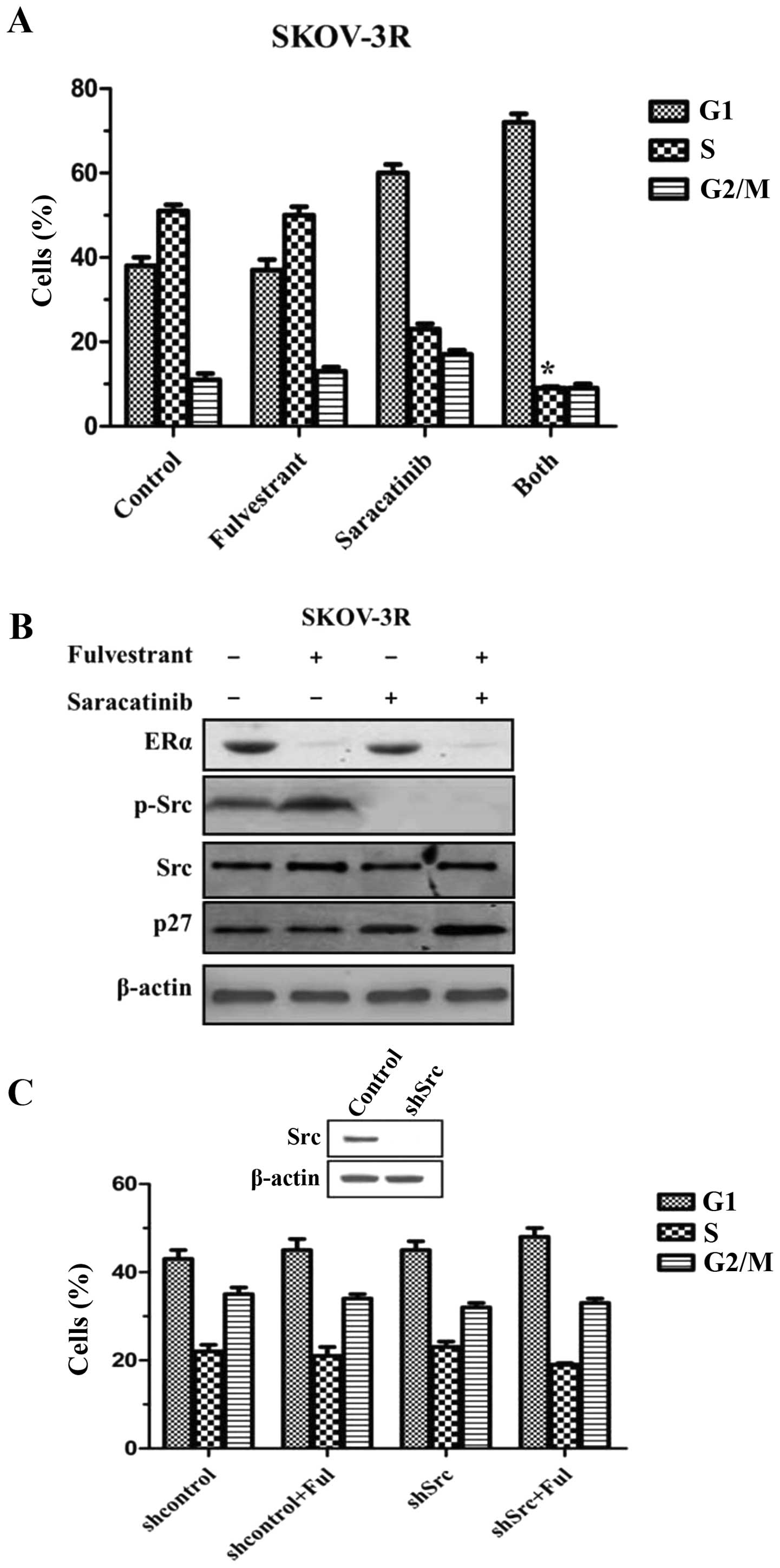
On the basis of observation that estrogen rapidly
activated Src, it was hypothesized that combined Src and ER
blockade would arrest tumor growth more efficiently than either
alone. SKOV-3R was treated with an inhibitor specific for Src,
saracatinib, fulvestrant or both. There was no cell cycle arrest
after 48 h of fulvestrant (Fig.
4A). A modest decrease of % S-phase cells was detected by using
saracatinib alone (Fig. 4A).
Combined Src and ER blockade significantly reduced % S-phase cells
as compared with Src-specific inhibitor alone
(*P<0.05 vs. saracatinib alone group; Fig. 4A). In SKOV-3R cells, the reduction
in % S-phase cells following dual therapy was much more than a sum
of decreases of either saracatinib or fulvestrant alone. It was
reported that estrogen efficiently activated Src to promote p27
proteolysis via the phosphorylation of activated Src (22). Therefore, we investigated the
expressions of activated Src and p27 in SKOV-3R cells. It was
demonstrated that activated Src, phospho-Y416 Src (p-Src), was
reduced with no change in total Src in saracatinib-treated SKOV-3R
(Fig. 4B). Both drugs together
significantly increased p27 expression as compared with either drug
alone (Fig. 4B), indicating that
there was a significant synergistic interaction between fulvestrant
and saracatinib for cell growth arrest via Src-regulated p27
expression.
To test the contribution of Src to saracatinib
effects, SKOV-3R was stably transduced with shRNA to knockout Src
(shSrc), the contribution of fulvestrant was in turn evaluated.
Although saracatinib weakened SKOV-3R cell growth (Fig. 4C), the lack of Src had little effect
on cell cycle arrest as compared with control cells (Fig. 4C). Thus, it was likely that
saracatinib effects were partially ascribed to the inhibition of
other Src family members. Nevertheless, shSrc-transfected cells
with the addition of fulvestrant reduced SKOV-3R cell growth by a
one-third cut of % S-phase cells, suggesting Src knockout
facilitated the fulvestrant treatment to some extent. Since Src
downregulation did not produce an equal effect to saracatinib
alone, it was supposed that the antiproliferative effects of
saracatinib were partially due to its inhibition of other Src
family members.
Src inhibition in combination with ER
blockade causes cell death
Anticancer efficacy results from dual directions,
including antiproliferation or cell death. We assayed autophagy and
apoptosis by flow cytometry following 48 h treatment of
saracatinib, fulvestrant or both. To examine autophagy, SKOV-3R
cells were labeled with Cyto-ID-Green autophagy dye, that was
examined by flow cytometry. Accordingly, fulvestrant or saracatinib
alone did not increase autophagic vesicle formation, whereas
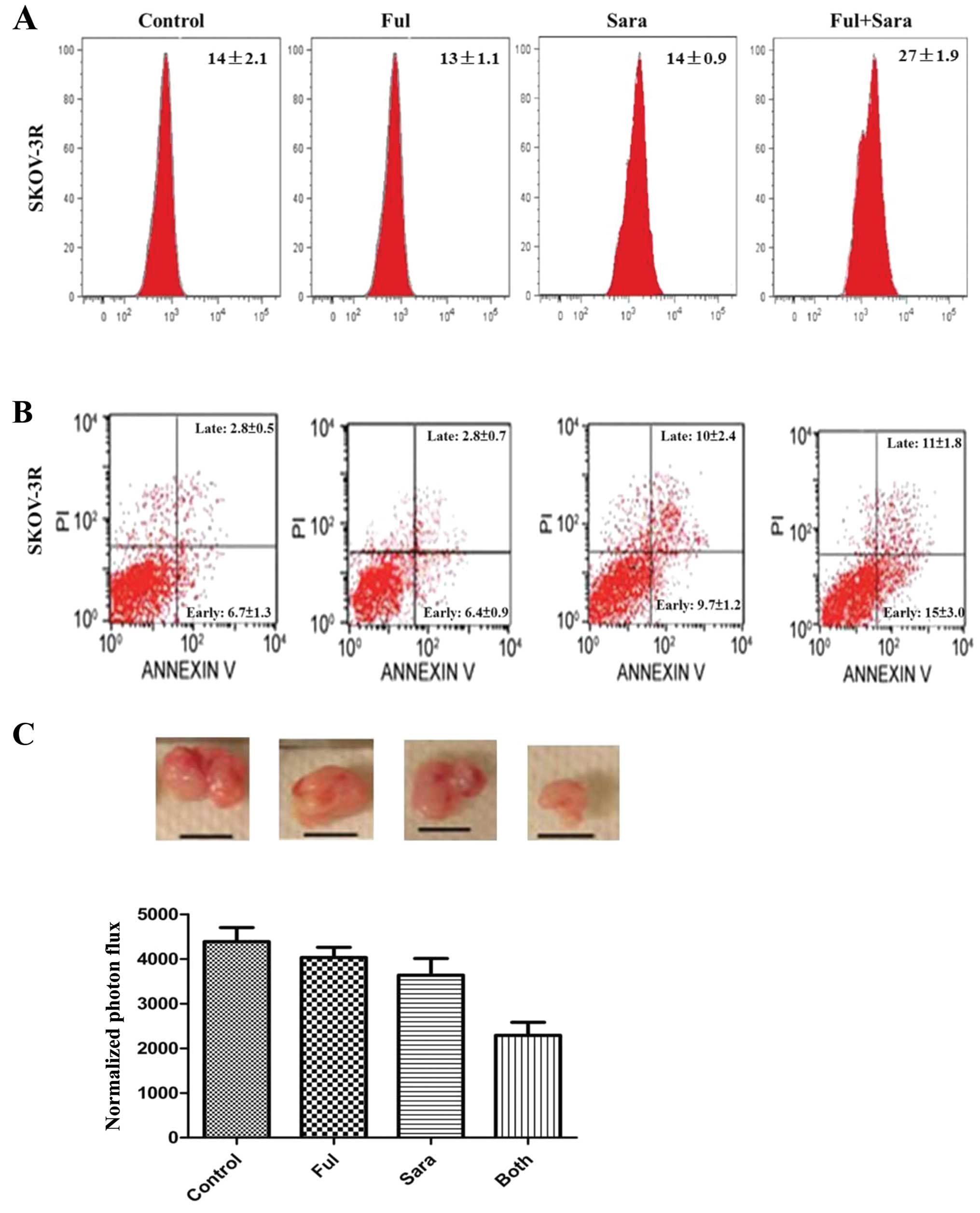
autophagic vesicles increased significantly in SKOV-3R (Fig. 5A; P<0.01). In parallel,
saracatinib-associated SKOV-3R growth inhibition resulted from
increased events of early and late apoptosis. (Fig. 5B; P<0.05, control vs.
saracatinib), and combined therapy had a significantly additional
effect relative to saracatinib alone (Fig. 5B; P<0.05, both vs. saracatinib).
We showed the combined drug effects in nude mice model in
vivo. Implanted tumors were harvested at 11 weeks after
implantation. Regarding the combination effects, combination of
fulvestrant with saracatinib significantly decreased tumor growth
as compared with either drug alone (Fig. 5C).
Discussion
More than 55% of ovarian cancers express ER on the
cell surface (23,24). Although ER expression in ovarian
cancer is similar to breast cancer, the efforts to target ER merely
present a short-term benefit in ~28% of ovarian cancer cases
(4,8,9). In
patients with breast cancer, the survival benefit of ER blockade
has been confirmed in the ER-positive breast cancers (25,26).
Therefore, the potential efficacy of anti-estrogen in ovarian
cancer may be underestimated. Numerous small trials of ER-blockers
have been disappointing (3,6). Only 8% overall response and 35%
disease stabilization were detected in advanced heavily pretreated
ovarian cancer cases (4). Hence,
understanding the mechanisms of anti-estrogen resistance may
provide new insight for the treatment of ovarian cancer. In
estrogen-sensitive cancers, estrogen is able to stimulate Src
activation to phosphorylate p27 promoting its degradation and
increasing cell cycle progression (22). Crosstalk between estrogen-bound ER
and Src drives mitogenic pathways that are frequently activated in
ovarian cancer (15). We observed
that activated Src was positively expressed in 20 out of 40
ER-positive ovarian cancer (50%). It was unclear whether ER
antagonist was beneficial for patients with ER+ ovarian
cancer. According to subgroup survival analysis on the basis of
p-Src status in ovarian cancer, the cohort with
ER+p-Src− possessed a propensity for
metastasis and an advantage for DFS at 3 years after ER antagonist
treatment as compared with ovarian cancer patients with
ER+p-Src+, indicating the possibility that
Src activation exerted the effects on underestimating therapeutic
efficacy by resistance to ER antagonist in patients with
ER+ ovarian cancer.
Src has potential to promote the metastatic and
survival effects of cancer cells, while Src acts to promote the
degradation of p27, a G1 arrestor of anti-estrogen, by
phosphorylation (11,22). p27 is essential for cell arrest by
anti-estrogen, and Src promotes p27 proteolysis to eliminate the
effect of p27 on cell arrest. Therefore, it was hypothesized that
Src inhibitor, saracatinib, could restore anti-estrogen in
resistant ER-positive ovarian cancers. In the present study, the
data demonstrated the therapeutic potential of combined saracatinib
and fulvestrant in ovarian cancer with resistance to ER antagonist.
As reported by Simpkins et al (3) 338 primary ovarian cancers confirmed
that 67% had detectable ER expression with p-Src (activated Src,
phospho-Y416-Src) and total Src. Supporting the relevance of Src
with ER, phosphorylated Src in ER-positive SKOV-3R expressed at a
higher level than that in SKOV-3. It was reported that estrogen
triggering ER signaling was potential to activate Src kinase in
breast epithelial cells (27,28).
Despite the presence of ERα and ERβ on the surface of ovarian
cancer cells, E2-stimulated SKOV-3R cell proliferation was
determined by ERα alone. In addition, it was observed for the first
time that estrogen rapidly activated Src, Src-ER interaction and
their co-localization in the perinuclear cytoplasm in ovarian
cancer. It was inferred that the rapid ER-Src interaction may be
linked to ER-mediated transcription, that was confirmed by the
observation that estrogen-stimulated Src activation was impeded by
saracatinib by downregulating ER expression at two ER-modified
genes: Fosl and c-Myc (3,4).
Notably, cell arrest was greater for combined
fulvestrant and saracatinib than either alone. Dual therapy
contributed to more antiproliferative efficacy compared to
monotherapy. Furthermore, a more significant growth arrest was
caused by combined blockade of Src and ER with a greater increase
in p27 than that with monotherapy in SKOV-3R cells. In SKOV-3R,
fulvestrant and saracatinib may synergistically block cell cycle by
upregulating p27 and activating p27-related signaling. The outcomes
were consistent with recent reports that Src blockade intensifies
anti-estrogen efficacy in breast cancers (29–31).
To grow and culture tumor cells from primary ovarian cancers may
allow future prediction of individualized use of targeted
therapies.
Saracatinib, Src kinase inhibitor, suppressed a
variety of Src family members (32). As compared with treatment of
saracatinib, Src silence failed to inhibit cell growth, indicating
other Src family members may be ascribed to drug efficacy. Although
neither fulvestrant alone nor shSrc inhibited cell cycle, Src
silence co-worked with ER blockade to efficiently hinder cell cycle
progression, suggesting that Src and ER led cell growth in
ER-positive ovarian cancer. Accordingly, Src and ER blockade
impaired tumor cell growth, not only via antiproliferation, but
also via autophagy. Autophagy is characterized as leading to type
II programmed cell death (33–35).
Fulvestrant alone was unable to efficiently induce autophagy in the
present study, while dual drugs yielded the greatest effect on
inducing autophagy relative to single drugs. Thus, autophagic cell
death was promoted by Src inhibition and augmented by the extra
addition of ER blockade. Src inhibitor-induced autophagy accounted
for saracatinib-mediated antitumor effect.
We have characterized a new model for the study of
anti-estrogen resistance in ovarian cancer. SKOV-3R was markedly
anti-estrogen resistant, but retained estrogen sensitivity in
vitro and in vivo. SKOV-3R xenografts required estrogen
for growth. Implantation of luciferase-tagged SKOV-3R allowed
highly sensitive and quantitative monitoring of ovarian tumor
growth. While short-term saracatinib was effective in vitro,
resistance rapidly emerged in vivo. Estrogen-driven Src
activation and upregulation of other Src family kinases may all
contribute to failure of saracatinib alone. It was noteworthy that,
when combined with fulvestrant, saracatinib resistance did not
occur in ovarian cancer SKOV-3R xenografts. While most targeted
therapies reduced tumor growth, combined saracatinib and
fulvestrant caused tumor regression. The late regression reflected
the induction of autophagy leading to loss of tumor viability. It
was highly likely that the greater antitumor effect of dual therapy
resulted from more effective inhibition of estrogen-activated gene
expression in order to facilitate a considerable number of cell
deaths.
Ovarian cancer is greatly unresponsive to hormonal
therapies. However, our results raised the possibility that ER and
Src blockade prevented or reversed anti-estrogen resistance in
ovarian cancer. Well-designed pre-clinical evaluation of dual drug
combination is deemed as necessary.
Acknowledgements
This study was supported by the Key Science and
Technology Program of Shaanxi Province (no. 2011K13-01-13).
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar
|
|
2
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2012. CA Cancer J Clin. 62:10–29. 2012. View Article : Google Scholar
|
|
3
|
Simpkins F, Hevia-Paez P, Sun J, et al:
Src inhibition with saracatinib reverses fulvestrant resistance in
ER-positive ovarian cancer models in vitro and in vivo. Clin Cancer
Res. 18:5911–5923. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Argenta PA, Um I, Kay C, et al: Predicting
response to the anti-estrogen fulvestrant in recurrent ovarian
cancer. Gynecol Oncol. 131:368–373. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Badia E, Docquier A, Busson M, et al:
Long-term treatment with the pure anti-estrogen fulvestrant durably
remodels estrogen signaling in BG-1 ovarian cancer cells. J Steroid
Biochem Mol Biol. 132:176–185. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bossard C, Busson M, Vindrieux D, et al:
Potential role of estrogen receptor beta as a tumor suppressor of
epithelial ovarian cancer. PLoS One. 7:e447872012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Halon A, Materna V, Drag-Zalesinska M, et
al: Estrogen receptor alpha expression in ovarian cancer predicts
longer overall survival. Pathol Oncol Res. 17:511–518. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kothari R, Argenta P, Fowler J, Carter J
and Shimp W: Antiestrogen therapy in recurrent ovarian cancer
resulting in 28 months of stable disease: a case report and review
of the literature. Arch Oncol. 18:32–35. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Smyth JF, Gourley C, Walker G, et al:
Antiestrogen therapy is active in selected ovarian cancer cases:
the use of letrozole in estrogen receptor-positive patients. Clin
Cancer Res. 13:3617–3622. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Park MA, Hwang KA, Lee HR, Yi BR, Jeung EB
and Choi KC: Benzophenone-1 stimulated the growth of BG-1 ovarian
cancer cells by cell cycle regulation via an estrogen receptor
alpha-mediated signaling pathway in cellular and xenograft mouse
models. Toxicology. 305:41–48. 2013. View Article : Google Scholar
|
|
11
|
Wang L, Wang G, Yang D, et al: Euphol
arrests breast cancer cells at the G1 phase through the modulation
of cyclin D1, p21 and p27 expression. Mol Med Rep. 8:1279–1285.
2013.PubMed/NCBI
|
|
12
|
Jiang D, Wang X, Liu X and Li F: Gene
delivery of cyclin-dependent kinase inhibitors
p21Waf1 and p27Kip1 suppresses
proliferation of MCF-7 breast cancer cells in vitro. Breast Cancer.
2013.[Epub ahead of print].
|
|
13
|
Psyrri A, Bamias A, Yu Z, et al:
Subcellular localization and protein levels of cyclin-dependent
kinase inhibitor p27 independently predict for survival in
epithelial ovarian cancer. Clin Cancer Res. 11:8384–8390. 2005.
View Article : Google Scholar
|
|
14
|
Hurteau JA, Allison BM, Brutkiewicz SA, et
al: Expression and subcellular localization of the cyclin-dependent
kinase inhibitor p27Kip1 in epithelial ovarian cancer.
Gynecol Oncol. 83:292–298. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xing D and Orsulic S: A genetically
defined mouse ovarian carcinoma model for the molecular
characterization of pathway-targeted therapy and tumor resistance.
Proc Natl Acad Sci USA. 102:6936–6941. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Integrated genomic analyses of ovarian
carcinoma. Nature. 474:609–615. 2011. View Article : Google Scholar
|
|
17
|
Huang YW, Chen C, Xu MM, Li JD, Xiao J and
Zhu XF: Expression of c-Src and phospho-Src in epithelial ovarian
carcinoma. Mol Cell Biochem. 376:73–79. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kim HS, Han HD, Armaiz-Pena GN, et al:
Functional roles of Src and Fgr in ovarian carcinoma.
Clin Cancer Res. 17:1713–1721. 2011.
|
|
19
|
Matsuo K, Nishimura M, Bottsford-Miller
JN, et al: Targeting SRC in mucinous ovarian carcinoma. Clin Cancer
Res. 17:5367–5378. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhao Y, Li X, Sun X, Zhang Y and Ren H:
EMT phenotype is induced by increased Src kinase activity via
Src-mediated caspase-8 phosphorylation. Cell Physiol Biochem.
29:341–352. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Castoria G, Giovannelli P, Lombardi M, et
al: Tyrosine phosphorylation of estradiol receptor by Src regulates
its hormone-dependent nuclear export and cell cycle progression in
breast cancer cells. Oncogene. 31:4868–4877. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chu I, Sun J, Arnaout A, et al: p27
phosphorylation by Src regulates inhibition of cyclin E-Cdk2. Cell.
128:281–294. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Serkies K, Sinacki M and Jassem J: The
role of hormonal factors and endocrine therapy in ovarian cancer.
Contemp Oncol. 17:14–19. 2013.PubMed/NCBI
|
|
24
|
Brenton JD and Stingl J: Stem cells:
anatomy of an ovarian cancer. Nature. 495:183–184. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Coates AS, Keshaviah A, Thürlimann B, et
al: Five years of letrozole compared with tamoxifen as initial
adjuvant therapy for postmenopausal women with endocrine-responsive
early breast cancer: update of study BIG 1–98. J Clin Oncol.
25:486–492. 2007.PubMed/NCBI
|
|
26
|
Regan MM, Neven P, Giobbie-Hurder A, et
al: Assessment of letrozole and tamoxifen alone and in sequence for
postmenopausal women with steroid hormone receptor-positive breast
cancer: the BIG 1–98 randomised clinical trial at 8.1 years median
follow-up. Lancet Oncol. 12:1101–1108. 2011.PubMed/NCBI
|
|
27
|
Jung J, Kim HY, Kim M, Sohn K, Kim M and
Lee K: Translationally controlled tumor protein induces human
breast epithelial cell transformation through the activation of
Src. Oncogene. 30:2264–2274. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hui AY, Meens JA, Schick C, et al: Src and
FAK mediate cell-matrix adhesion-dependent activation of Met during
transformation of breast epithelial cells. J Cell Biochem.
107:1168–1181. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nautiyal J, Yu Y, Aboukameel A, et al:
ErbB-inhibitory protein: a modified ectodomain of epidermal growth
factor receptor synergizes with dasatinib to inhibit growth of
breast cancer cells. Mol Cancer Ther. 9:1503–1514. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Moulder S, Yan K, Huang F, et al:
Development of candidate genomic markers to select breast cancer
patients for dasatinib therapy. Mol Cancer Ther. 9:1120–1127. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gucalp A, Sparano JA, Caravelli J, et al:
Phase II trial of saracatinib (AZD0530), an oral SRC-inhibitor for
the treatment of patients with hormone receptor-negative metastatic
breast cancer. Clin Breast Cancer. 11:306–311. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Arcaroli JJ, Touban BM, Tan AC, et al:
Gene array and fluorescence in situ hybridization biomarkers of
activity of saracatinib (AZD0530), a Src inhibitor, in a
preclinical model of colorectal cancer. Clin Cancer Res.
16:4165–4177. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Boya P, Reggiori F and Codogno P: Emerging
regulation and functions of autophagy. Nat Cell Biol. 15:713–720.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Choi AM, Ryter SW and Levine B: Autophagy
in human health and disease. N Engl J Med. 368:651–662. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Fimia GM, Corazzari M, Antonioli M and
Piacentini M: Ambra1 at the crossroad between autophagy and cell
death. Oncogene. 32:3311–3318. 2013. View Article : Google Scholar : PubMed/NCBI
|