Introduction
Allicin (diallyl thiosulfinate) is the major
component in garlic (Allium sativum) extract and it has
multiple antineoplastic and other pharmacological effects. The
antitumor activities of allicin include the inhibition of cell
proliferation and the induction of apoptosis in multiple types of
malignancies, including colon, breast and lung cancer (1–5).
Allicin has both lipid- and water-soluble components. Lipid-soluble
compounds of allicin include diallyl sulfide (DAS), diallyl
disulfide (DADS) and diallyl trisulfide (DATS), whereas
water-soluble components of allicin consist of S-allylcysteine
(SAC), S-allylmercaptocysteine (SAMC) and other components.
The functions of SAMC and other organic sulfur
compounds derived from garlic have been compared. Among the 10
garlic-derived compounds tested, SAMC, DADS and S-trityl-L-cysteine
were shown to markedly inhibit cell proliferation, cause G2-M
arrest and induce apoptosis (6).
Although DAS, DADS, DATS, SAC and SAMC are organic sulfur compounds
with similar functions, SAMC showed the most significant effect in
promoting apoptosis, including apoptosis in the colon cancer cell
lines, HT-29 and SW620 (4).
Studies have shown that allicin induces apoptosis
mainly through a mitogen-activated protein kinase (MAPK) signaling
cascade (4,6–9). The
MAPK signaling cascade possesses a bi-directional mode of action
for regulating tumor cell growth, proliferation and apoptosis
according to environmental conditions. This cascade includes
signal-related kinase (ERK), c-jun-N-terminal kinases (JNK) and the
p38 MAPK pathways. Shirin et al (4) and Xiao et al (10) demonstrated that, by activating JNK1
and caspase-3, SAMC inhibited proliferation and induced the
apoptosis of colon cancer cells (4,10). In
addition, Chen et al demonstrated that the three main MAPKs
(ERK, JNK and p38) are activated after treatment of human hepatoma
cells with DATS (11).
The ability to inhibit apoptosis via the MAPK
signaling cascade is shared by SP600125, SB203580 and PD98059,
which inhibit JNK, P38K and ERK, respectively (12–14).
It has been reported that the synergistic actions of these
inhibitors with allicin result in higher rates of apoptosis than
when these inhibitors act alone. Examples of this synergy include
SB203580 enhancement of DADS-induced apoptosis in hepatocellular
carcinoma (HepG2 cells) (13) and
more efficient induction of apoptosis when the inhibitors (SB203580
and PD98059) act cooperatively with allicin and SAMC in the
treatment of colon cancer (cell line SW480) versus the independent
actions of these agents (4).
The transforming growth factor β (TGF-β) signaling
pathway plays important roles in tumorigenesis (15–21)
and has extensive interactions with the MAPK signaling pathways
(4,6–9). In
addition to the TGF-β pathway, the cytokine TGF-β1 activates the
MAPK pathways. As a consequence, activated ERK, JNK and P38 can
phosphorylate Smad2/3 and Smad4 to facilitate their translocation
to the nucleus (22,23). Thus, it is possible that the TGF-β
signaling pathway participates in the allicin-induced apoptosis of
tumor cells. However, this has yet to be established.
The TGF-β signaling pathway in hepatocellular
carcinoma HepG2 cells has been extensively studied. However, the
roles of the TGF-β pathway in the inhibition of cell proliferation
or apoptosis induction have not been extensively studied (24). The TGF-β pathway may induce
apoptosis when the MAPK pathway is inhibited. Caja et al
reported that the TGF-β pathway did not induce apoptosis in HepG2
cells until the MEK/ERK pathway was repressed (25). The TGF-β pathway was found to be
inactivated in the SW620 colon cancer cell line due to the lack of
Smad4 protein (24), while
reactivation occurred after transfecting the cells with
Smad4. Based on these observations, we aimed to ascertain
whether the TGF-β signaling pathway is activated by SAMC to induce
apoptosis in HepG2 and SW620 cells. We then examined the specific
role of the TGF-β pathway in apoptosis induction by further
suppressing the MAPK pathway.
Materials and methods
Experimental reagents
SAMC was generously provided by Wakunaga
Pharmaceutical Company, Hiroshima, Japan. ERK inhibitor PD98059
(cat. no. 513000), JNK inhibitor 600125 (cat. no. 420119) and P38
inhibitor SB203580 (cat. no. 559389) were from Calbiochem. The
Annexin V-FITC Apoptotic Kit and the In Situ Cell Death Detection
Kit were purchased from BD and Roche, respectively. The following
primary antibodies were used for immunoblotting and were purchased
from Santa Cruz: anti-TGF-β1, anti-TβR1, anti-TβR2,
anti-phosphorylated Smad2/3, anti-Smad4, anti-Bim, anti-Bcl-2,
anti-Bax, anti-Bad, anti-caspase-3 and anti-caspase-9. Secondary
anti-rabbit and anti-goat antibodies were from Zhongshan Golden
Bridge Biotechnology. The following parallel experiments were
conducted using both the colon cancer cell line SW620 and the liver
cancer cell line HepG2. The SAMC solution was newly prepared before
each use.
Cell culture
The colon cancer cell line SW620 and the liver
cancer cell line HepG2 obtained from the American Type Culture
Collection (ATCC, Manassas, VA, USA) were maintained in Dulbecco’s
modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine
serum (FBS; Gibco) and incubated at 37°C. We maintained a total of
four sets of cultured cells: the control set received only DMEM,
whereas the other sets received SAMC (group S), MAPK inhibitors
(group I) and SAMC combined with the MAPK inhibitors (group I+S).
The MAPK inhibitors consisted of three inhibitors, each at the
concentration of 10 μg/ml, which blocked ERK (PD98059), JNK
(600125) and P38 (SB203580). All groups of cells were treated for 8
h. Specifically, the cells in group I+S were pretreated with a
combination of MAPK inhibitors for 30 min before receiving 800
μmol/l SAMC for an additional 8 h.
Apoptosis was detected using electron microscopy
(EM). After the cells had grown to 85% confluency in 100-ml Petri
dishes, they were treated with the MAPK inhibitors and/or SAMC for
8 h as described above. Next, the cells were trypsinized,
centrifuged and washed twice with PBS. The resulting cell pellets
were fixed with 2.5% glutaraldehyde for 24 h at 4°C, sectioned at
50 nm thickness, stained on copper screens and observed by EM. Ten
microscopic fields (×3,000) were randomly chosen for each sample,
and the apoptotic cells were counted.
Flow cytometry
Cells in an exponential growth stage were seeded in
6-well plates at a density of 5×106 cells/well and
incubated at 37°C until the cells reached 85–90% confluency.
Following an 8-h treatment with the MAPK inhibitors and/or SAMC,
the cells were trypsinized, washed twice with cold PBS and then
washed once with the binding buffer. Annexin V and PI staining were
performed according to the manufacturer’s instructions. The cells
were analyzed by flow cytometry within 2 h to detect apoptotic
cells.
Immunocytochemistry
Sterilized coverslips were placed in 24-well plates,
and 2×105 cells in an exponential growth stage were
seeded in each well onto the coverslips. At 85–90% confluency, the
cells received the same treatments (i.e., MAPK inhibitors and/or
SAMC) for 8 h as previously described. The cells were then fixed
with 4% newly prepared paraformaldehyde solution for 10 min at room
temperature, followed with two 10-min washes with PBS, and
incubated with 0.1% Triton X-100 on ice for 2 min before additional
PBS washes.
To detect cellular apoptosis, the samples were
incubated with 100 μl of TUNEL reaction solution and Tdt enzyme at
37°C for 1 h in a dark humid environment, while the negative
control received only 100 μl of the TUNEL label solution.
TUNEL-positive apoptotic cells were detected by fluorescence
microscopy. We randomly chose 10 microscopic fields (×100) and
counted the apoptotic cells. The rate of cellular apoptosis was
expressed as a percentage of the total cells.
To detect protein expression and phosphorylation,
the samples were incubated separately with a 1:50 dilution of
primary antibodies, including those of anti-TGF-β1, anti-TβR1,
anti-TβR2, anti-phosphorylated Smad2/3, anti-Smad4, anti-Bcl-2,
anti-Bax, anti-Bad, anti-caspase-3, anti-caspase-9 or with a 1:100
dilution of anti-Bim at 37°C for 1 h, followed by PBS washes and
incubation with the secondary antibody at room temperature for 20
min. The samples were then washed with PBS, stained with DAB and
hematoxylin dye according to the standard protocols. After gradient
dehydration, the samples were mounted with Neutral gum.
The immunohistochemistry results were assessed using
the following standards. Brown-yellow granules in the cytoplasm
indicated positive expression of TGF-β1, TβR1, TβR2 and Smad4;
brown particles in the nucleus and the cytoplasm indicated positive
expression of P-Smad2/3, Bim and Bax. For each sample, 10
microscopic fields were chosen (×100), and both positive and
negative cells were counted. Staining intensity was divided into
four grades according to the proportion of positive cells to the
total cell numbers: the number of positive cells <10% (−),
10–25% (+), 26–50% (++), >50% (+++).
Western blot analysis
Cultured cells were lysed with the T-PER tissue
protein extraction reagent in the presence of protease inhibitors
(Pierce, Rockford, IL, USA). Equal amounts of cellular proteins
were loaded into each well and resolved using 10% SDS-PAGE gels.
Nitrocellulose membrane blotting was then performed under standard
conditions. For immunoblotting, we used primary antibodies against
TGF-β1, TβR1, TβR2, phosphorylated Smad2/3, Smad4, Bcl-2, Bax, Bad,
caspase-3, caspase-9 and Bim. The primary antibody solution was
diluted at room temperature and incubated with the membrane on a
swing bed for 1 h. The membrane was then washed 3 times for 10 min
with TBS-T before adding a 1:2,000 dilution of the secondary
antibody, which was diluted in the blocking solution at room
temperature. The membrane was then incubated on a swing bed for 1 h
before three 10-min washes with TBS-T.
Statistical analysis
All data are reported as the mean ± SD. A one-way
analysis of variance was used to test the differences among three
or more groups (SAS 9.2 package). For all statistically significant
differences observed after performing one-way analysis of variance,
the appropriate multiple comparison tests were used to perform a
post-hoc analysis. Statistical significance was set at
P<0.05.
Results
Detection of SAMC-induced cell apoptosis
by EM
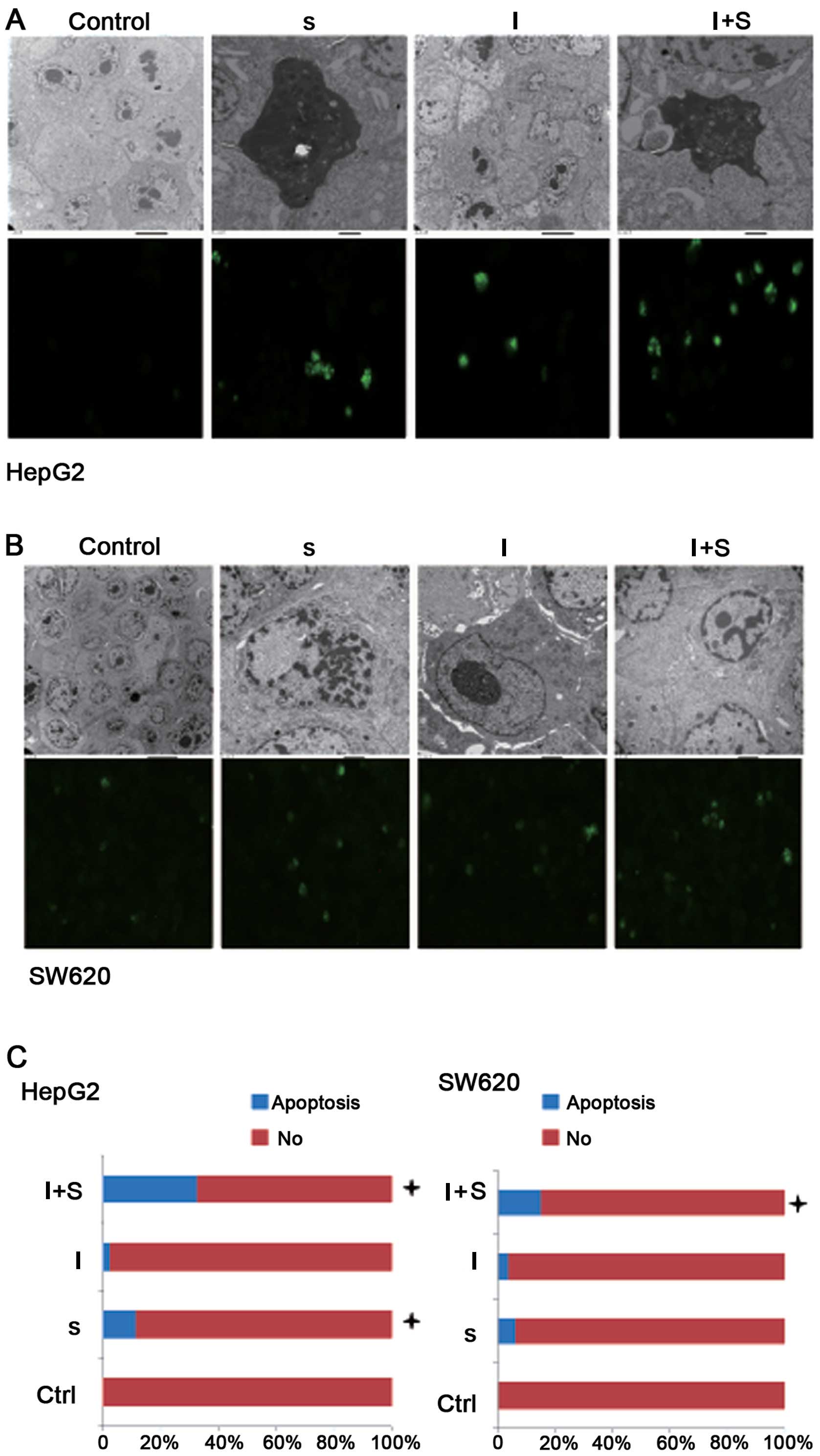
Apoptosis is the process of programmed cell death
that is characterized by cell shrinkage, nuclear condensation,
nuclear fragmentation and formation of apoptotic bodies (26). First, we observed the morphological
changes in tumor cells by EM to determine the impact of allicin,
MAPK inhibitors or the combined treatment of SAMC and MAPK
inhibitors on the tumor cells. The characteristic markers of the
control HepG2 and SW620 cells were as follows: surface rich in
microvilli; abundant cytoplasm, mitochondria and endoplasmic
reticulum, low electron density, large and irregular nuclei,
distinct nuclear membranes and large nucleoli near the nuclear
membranes (Fig. 1A and B). Cells
from group S were characterized as follows: decreased or absent
membrane microvilli, smaller cell size, concentrated cytoplasm with
increased electron density, expanded and vacuolized endoplasmic
reticulum, nuclear chromatin that was gathered at the nuclear
membranes and visible apoptotic bodies, which contained pyknotic
nuclear remains and organelles that were encapsulated by the cell
membranes. Compared with the control group, we detected apoptotic
cells and fewer proliferating cells in the inhibitor group. Evident
mitochondrial vacuolation and an increased number of apoptotic
cells were observed in group I+S compared with the other groups.
There were no significant differences among all the groups
(P>0.05).
TUNEL assay reveals the highest number of
apoptotic cells in group I+S
TUNEL assay results showed that the number of
apoptotic cells was increased (P<0.05) in the HepG2 cells from
group S compared with those from the control group. Group I+S had
the largest number of apoptotic cells when compared with the
control group, and the difference was statistically significant
(P<0.0001). There was no difference in the rate of apoptosis
between group I and the control group (P>0.05). The number of
apoptotic cells was increased in the SW620 cells from group S
compared with those from the control group (P>0.05). Group I+S
had an increased number of apoptotic cells compared with this
number in the control group, and the difference was statistically
significant (P<0.001). There was no significant difference in
the cellular apoptotic rate when comparing group I with the control
group (P>0.05) (Fig. 1A–C).
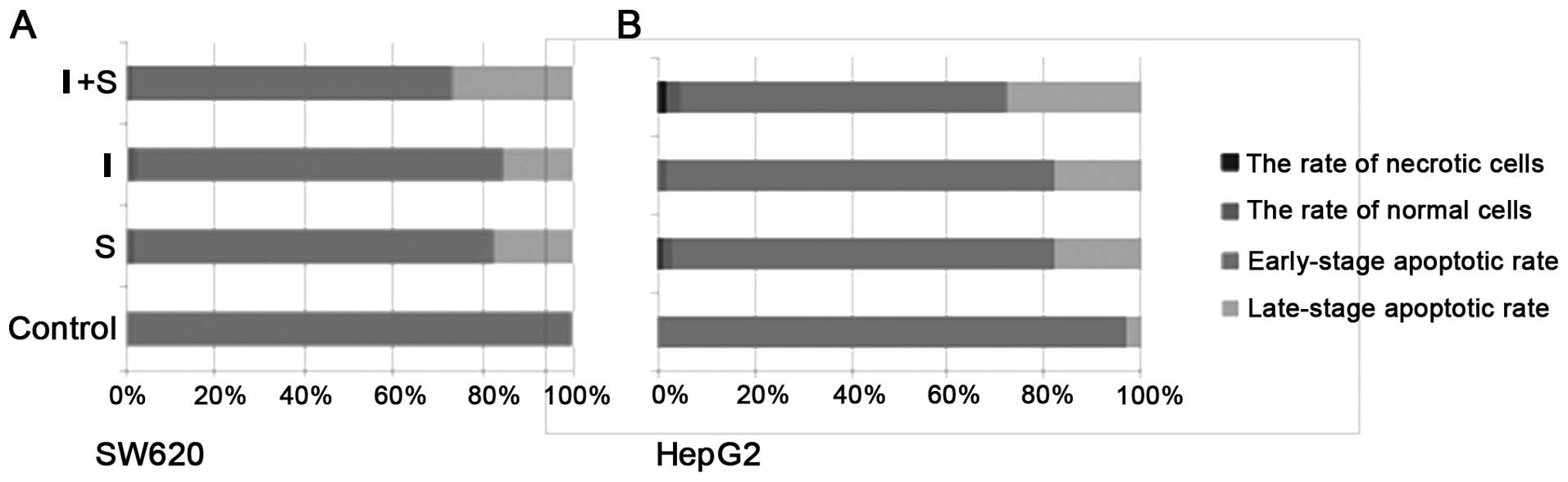
Flow cytometry results showed that cells
in groups S, I and I+S underwent early-stage apoptosis; group I+S
exhibited the most significant induction of apoptosis
In HepG2 cells, the late-stage apoptotic rates were
2.52, 1.66 and 3.12% in group S, group I and group I+S,
respectively, compared with the control group rate of 0.17%; the
results indicated that there was a significant increase in the
number of apoptotic cells following treatment (P<0.05) (Fig. 2). The early-stage apoptotic rates of
groups S, I, I+S and the control group were 18.03, 17.88, 27.74 and
2.87%, respectively; the number of apoptotic cells also increased
after treatment and the results indicated that there was a
significant increase in the number of apoptotic cells after
treatment (P<0.05). In the SW620 cells, the late-stage apoptotic
rates were 1.80, 1.33, 1.35 and 0.06% in groups S, I, I+S and the
control group, respectively; the number of apoptotic cells
increased after treatment and this increase was statistically
significant (P<0.05). The early-stage apoptotic rates were
17.80, 15.79 and 26.84% in groups S, I and I+S, respectively, when
compared with 0.37% of the control group; the number of apoptotic
cells increased and the difference was statistically significant
(P<0.05). The number of apoptotic cells increased in group I+S
and was statistically significant when compared with that of group
S (P<0.05).
Immunohistochemical results
In HepG2 cells from the group I+S, TGF-β1,
phosphorylated Smad2/3 and Smad4 and caspase-3 expression levels
were 1+ (Fig. 3A); whereas their
expression levels were negative or not evident and the differences
in the protein expression levels were not significant when
comparing group I+S with the other three groups (P>0.05). TβR2,
Bim, and caspase-9 expression levels were 2+ in group I+S while the
other groups were negative and 1+; the differences in the
expression levels of these proteins were statistically significant
when comparing group I+S with the control group (P<0.05). In the
SW620 cells from group S, TβR2 expression increased to 1+ as
compared with the control group but the difference did not achieve
statistical significance (P>0.05) (Fig. 3B). TGF-β1, TβR2, Smad4 and caspase-9
protein expression was increased in group I+S compared with the
control group, but the differences in protein levels did not
achieve statistical significance (P>0.05).
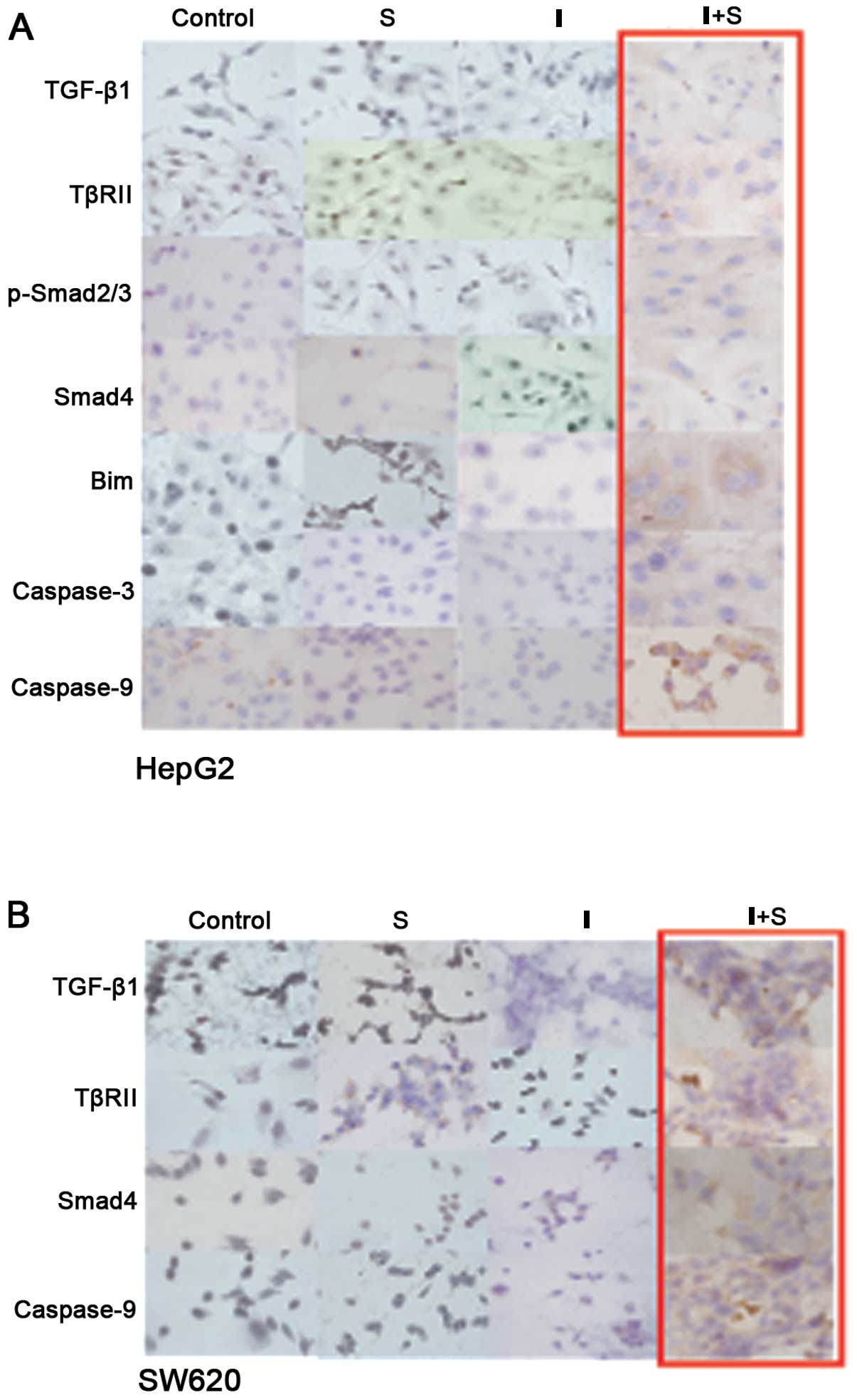 | Figure 3Immunocytochemical analysis
(magnification, ×400). (A) In the HepG2 cells from group I+S, the
expression levels of TGF-β1, p-Smad2/3, Smad4 and caspase-3 were
1+, the expression levels of TβRII, Bim and caspase-9 were 2+,
whereas the expression of the same proteins were negative or 1+ in
cells from the other groups. (B) In the SW620 cells, the expression
level of TβRII was 1+ in group S and was high when compared with
the level in the control group; the expression levels of TGF-β1,
TβRII, Smad4 and caspase-9 were increased in group I+S compared
with the level in the control group. |
Western blotting results
In the HepG2 cells, Smad4 expression increased and
Smad7 expression decreased in group S compared with the cells from
the control group (Fig. 4A and B).
The expression of TGF-β1 and Smad4 decreased while the Bcl-2
protein levels were elevated in group I as compared with the
control group. TGF-β1, TβRII, phosphorylated Smad2/3, Smad4, Smad7,
Bim, caspase-3 and caspase-9 expression increased while Bcl-2
decreased in the HepG2 cells from group I+S compared with those
from group S.
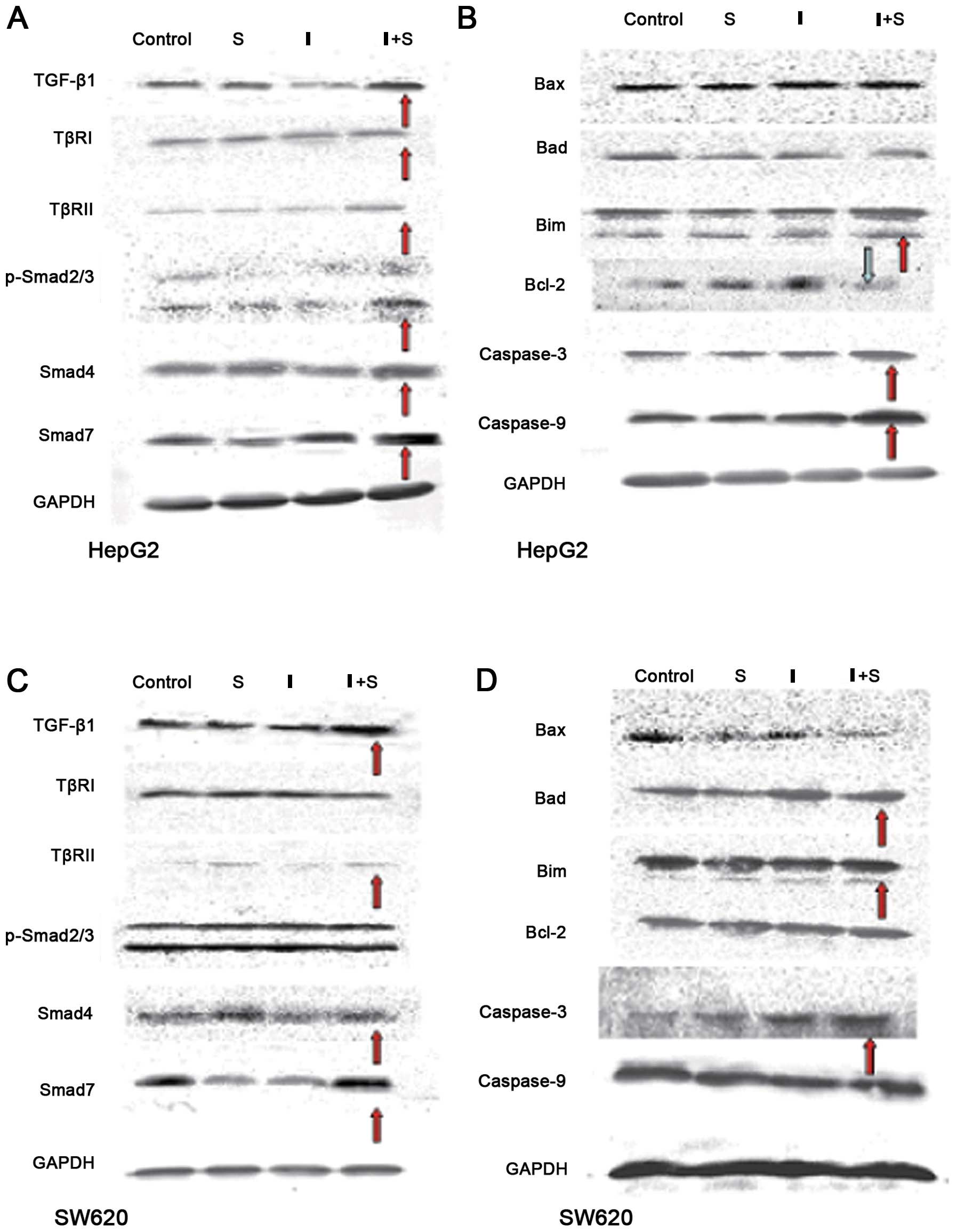 | Figure 4Western blot analysis. (A and B) In
the HepG2 cells, increased Smad4 expression and decreased Smad7
expression were detected in group S compared with the control
cells. The expression levels of TGF-β1, TβRII, p-Smad2/3, Smad4,
Smad7, Bim, caspase-3 and caspase-9 were increased while Bcl-2
expression was decreased in group I+S compared with group S. (C and
D) In the SW620 cells, TβRII, Smad4 and Bax expression levels were
increased while Smad7 expression was decreased in group S compared
with the control group. The expression levels of TGF-β1, Smad7,
Bax, Bad and caspase-9 were increased in group I+S as compared with
those in group S. |
In the SW620 cells from group S, the expression of
TβRII increased and Smad4, Bax and Smad7 expression decreased as
compared with the cells from the control group. Smad7 expression
was decreased in group I compared with that in the control group.
TGF-β1, Smad7, Bax, Bad and caspase-9 expression increased in group
I+S compared with group S (Fig. 4C and
D).
Discussion
SAMC-induced apoptosis of HepG-2 and
SW620 cells
Mechanisms of the suppression of cancer by allicin
include inhibition of cell proliferation, induction of cell cycle
arrest, induction of cancer cell differentiation and apoptosis
(2,4,10,
27–29), direct killing of cancer cells and
other functions. The inhibition of cell proliferation and the
induction of apoptosis are the most significant functions. When
HepG2 and SW620 cancer cells were treated with water-soluble
allicin SAMC, their apoptotic rates were increased compared with
the untreated cells. These results are consistent with previous
studies performed in SW480, SNU-1, HEL and OCIM-1 cells (2,6,10),
which indicate that SAMC induces apoptosis in multiple cancer
cells.
The MAPK signaling cascade is an important
intracellular signaling pathway that regulates cell growth, and its
inhibitors can induce cancer cell apoptosis (12–14).
In the present study, we treated HepG2 and SW620 cells with three
inhibitors of the MAPK pathway (JNK, ERK and P38MAPK) and each
inhibitor induced obvious signs of apoptosis. Therefore, the
activation of the intrinsic MAPK signaling pathway contributed to
the protection of the cancer cells and the inhibition of
apoptosis.
We subsequently combined the inhibitors of the MAPK
pathway with SAMC to treat the cancer cells. This combined
treatment resulted in significantly higher apoptotic rates than the
treatment with SAMC or the inhibitors alone (P<0.05). In this
case, higher apoptotic rates were observed in the HepG2 cells than
in the SW620 cells. These results indicated that the induction of
apoptosis by the MAPK inhibitors was synergistically enhanced with
SAMC. Previous studies have shown that the collaborative action of
inhibitors and allicin induced higher apoptotic rates than that of
each single factor (10,13). Our results also demonstrated that
the rate of SAMC-induced apoptosis was the highest when the MAPK
pathway was suppressed. Allicin induces apoptosis via the MAPK
pathway. Therefore, there remains the question of whether allicin
induces apoptosis via other pathways when the MAPK pathway is
blocked by inhibitors. To elucidate this issue, we examined the
TGF-β signaling pathway, which is closely related to the MAPK
pathway.
SAMC activation of the TGF-β signaling
pathway following suppression of the MAPK pathway
Intracellular signaling transduction composes a
complex network and there is communication between the TGF-β and
MAPK signaling pathways (30,31).
Perturbances of the TGF-β signaling pathway could lead to the
occurrence and development of various types of tumors (15–21).
For instance, the TGF-β pathway was found to be inactivated in 20
out of 38 cancer cell lines (24)
and there was also low or no expression of several proteins such as
TβRII and Smad4. Furthermore, the activity of the TGF-β pathway
could be partially recovered by the transfection of downregulated
genes. We observed that SAMC activates the TGF-β signaling pathway
and induces apoptosis in HepG2 cells with intact TGF-β pathways
(32) and also in SW620 cells with
perturbed TGF-β pathways (24).
SAMC activation of TGF-β1 with MAPK
pathway suppression
Our results indicated that SAMC alone was not able
to activate TGF-β1 in HepG2 and SW620 cells. This suggests that
SAMC may induce apoptosis via other signaling pathways, such as the
MAPK pathway (4,6–9). We
observed that the expression of TGF-β1 was decreased after the
treatment of MAPK inhibitors in HepG2 cells. These results are
consistent with those of previous studies where PD98059 and
SB203580 repressed the expression of TGF-β1 in mouse macrophages
(33). However, TGF-β1 could be
activated by SAMC when the MAPK pathway is inhibited with
concurrent upregulation of proteins in the TGF-β pathway.
Therefore, given the complex interactions among the various
intracellular signaling pathways, it is possible for SAMC to
promote the expression of TGF-β1 through pathways other than MAPK
in the presence of inhibitors.
The expression of many proteins in the TGF-β pathway
was increased in the HepG2 cells, whereas only TGF-β1, TβRII and
Smad4 proteins were upregulated in the SW620 cells. We believe this
upregulation occurred as the TGF-β pathway is intact in HepG2
cells, which enables SAMC to activate the TGF-β pathway when the
MAPK pathway is completely inhibited. Conversely, the TGF-β pathway
was blocked in the SW620 cells resulting in insufficient pathway
activation, despite using comparable treatments.
Re-expression or upregulation of
inactivated Smad4 by SAMC in cancer cells
Smad4 is a tumor-suppressor gene whose function is
to initiate TGF-β-induced tumorigenesis and metastasis. Miyaki
et al discovered that the inactivation of the Smad4
gene results in tumor development, invasion and metastasis, and the
stable transfection of Smad4 into SW620 cells could inhibit cancer
cell migration and metastasis while reducing the occurrence of
tumors (34). Our results showed
that the expression of Smad4 was increased following SAMC treatment
and Smad4 expression decreased when HepG2 cells were treated with
the MAPK inhibitors, whereas combined treatments increased Smad4
expression levels, when compared with the SAMC treatment alone. The
expression of Smad4 was correlated with the TGF-β1 levels,
suggesting that TGF-β1 initiates changes in cells with active TGF-β
signaling pathways that result in the expression of downstream
Smad4 proteins (35–37). In addition, we discovered that SAMC
induces the re-expression of inactivated Smad4 genes in
SW620 cells, despite the non-obvious changes in TGF-β1 levels. The
expression of Smad4 may be achieved by both TGF-β-dependent and
TGF-β-independent pathways. These results suggest that regulation
of Smad4 could occur by the TGF-β signaling pathway, the MAPK and
other pathways (38–40). Therefore, we propose that SAMC
induces expression of Smad4 through other mechanisms as well as the
regulation by SAMC of the TGF-β signaling pathway.
Dual functions of Smad7
Smad7 is considered to be an inhibitory protein in
the TGF-β signaling pathway. TGF-β1 may suppress apoptosis by
interacting with active TβRI to further activate Smad7, which
subsequently inhibits the phosphorylation of p-Smad and thus
suppresses the TGF-β signaling pathway (41,42).
However, our results showed that, when the MAPK signaling pathway
was inhibited by SAMC, there was an increase in the expression of
endogenous TGF-β1, Smad7 and other proteins that promote apoptosis.
Similarly, Landström et al discovered that treating human
prostate cancer cells with TGF-β1 or the ectopic overexpression of
Smad7 induced apoptosis (43), and
Mazars et al reported that Smad7 induced apoptosis in HepG2
cells (44). However, we speculated
that the induction of apoptosis by Smad7 is dependent on the cell
type. Regardless, the potential for Smad7 to both inhibit and
induce apoptosis is a novel insight into the characteristics of
Smad7.
SAMC activation of the
TGF-β-mitochondrial pathway for the induction of apoptosis
following MAPK pathway inhibition
Activation of the TGF-β pathway promotes apoptosis
initiation and progression by supporting pro-apoptotic factors
while the anti-apoptotic factors (such as Bcl-2 family proteins)
are destroyed. The induction of apoptosis varies with each cell
type. For example, in the breast cancer cell lines JygMC(A) and
JygMC(B), TGF-β upregulated the Bim pathway through Foxcl (45) while TGF-β downregulated the Bcl-2
pathway through Smad3 in the lung cancer cell line A549 (46); and TGF-β1 upregulated Bim and Bax in
osteoclast cells (47). During the
induction of apoptosis, changes in the anti-apoptotic Bcl-2 family
proteins and pro-apoptotic proteins such as Bax, Bak and Bim play
an important role. We examined the Bcl-2 family and caspase
proteins, which are located downstream of the TGF-β pathway. TGF-β1
activated the expression of p-Smad2/3, Smad4 and Bim proteins in
HepG2 cells, when the MAPK pathway was inhibited by SAMC treatment,
similarly to previous reports on WEHI231B lymphocytes (19,48).
The expression of Bcl-2 protein decreased as the expression of Bim
increased and Bim interacted with Bcl-2 to induce the caspase
cascade via a change in the conformation of the complex (49). When the expression of Bcl-2 was
reduced, the ratio of Bcl-2/Bax decreased leading to apoptosis
(50). Our results also
demonstrated that the cells entered apoptosis when the expression
levels of caspase-3 and caspase-9 were elevated, as evident by cell
shrinkage, chromatin condensation, karyopyknosis, nuclear
fragmentation and an increased rate of apoptosis.
Although the TGF-β pathway in HepG2 cells is intact,
cell proliferation and induction of apoptosis are not believed to
be inhibited (24). Our study
demonstrated that SAMC induced apoptosis by activating the TGF-β
signaling pathway when the MAPK pathway was suppressed.
Wen et al also noted that SB203580, when
combined with DADS, enhanced the DADS-induced apoptosis of HepG2
cells (13). In the present study,
we discovered that SAMC further induced cell apoptosis by
activating the TGF-β signaling cascade and subsequently increased
the expression levels of apoptotic protein after the ERK, JNK and
p38K pathways were inhibited.
In the SW620 cells, when the MAPK signaling cascade
was suppressed, SAMC mainly stimulated the expression of Bax and
Bad proteins, but not Bim. These results indicated that SAMC was
able to promote apoptosis by partially activating the TGF-β
cascade, thereby increasing the expression levels of Bax and
caspase-9 in SW620 cells, whose TGF-β pathway was otherwise
inactivated. These results are similar to a study performed by Lee
et al, who reported that SAMC induced the apoptosis of SUN-1
cells by activating Bax (2).
The TGF-β pathway is inactivated in many tumor cells
and tissues. Although the TGF-β signaling pathway can function
normally in most hepatic carcinoma cell lines, these cells do not
undergo apoptosis via this signaling pathway (24). Notably, after the MAPK pathway was
suppressed, SAMC further induced cell apoptosis by reactivating the
TGF-β pathway. The combined effects of the MAPK inhibitors and SAMC
included higher induction rates of tumor cell apoptosis when
compared with either treatment alone. These results help to
elucidate the underlying mechanism and the possible signal networks
that are implicated in allicin-induced tumor cell apoptosis. MAPK
inhibitors have been tested in clinical trials, and the clinical
use of SAMC has been approved in China. Our results support the
translational and clinical applications of combining MAPK inhibitor
and SAMC in cancer therapy.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (no. 81372785), the Natural Science
Foundation of Heilongjiang (nos. D200921 and QC2009C29), the
Opening Project of the Key Laboratory of Medical Genetics (Harbin
Medical University), Heilongjiang Higher Education Institutions. We
thank Wakunaga Pharmaceutical, Hiroshima, Japan, for the gift of
the garlic-derived compound SAMC to Professor Yong-Chuan Wong,
University of Hong Kong.
References
|
1
|
Hu H, Zhang XP, Wang YL, et al:
Identification of a novel function of Id-1 in mediating the
anticancer responses of SAMC, a water-soluble garlic derivative, in
human bladder cancer cells. Mol Med Rep. 4:9–16. 2011.PubMed/NCBI
|
|
2
|
Lee Y: Induction of apoptosis by
S-allylmercapto-L-cysteine, a biotransformed garlic derivative, on
a human gastric cancer cell line. Int J Mol Med. 21:765–770.
2008.PubMed/NCBI
|
|
3
|
Pinto JT, Lapsia S, Shah A, Santiago H and
Kim G: Antiproliferative effects of garlic-derived and other allium
related compounds. Adv Exp Med Biol. 492:83–106. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shirin H, Pinto JT, Kawabata Y, et al:
Antiproliferative effects of S-allylmercaptocysteine on colon
cancer cells when tested alone or in combination with sulindac
sulfide. Cancer Res. 61:725–731. 2001.PubMed/NCBI
|
|
5
|
Wu X, Kassie F and Mersch-Sundermann V:
Induction of apoptosis in tumor cells by naturally occurring
sulfur-containing compounds. Mutat Res. 589:81–102. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xiao D, Pinto JT, Gundersen GG and
Weinstein IB: Effects of a series of organosulfur compounds on
mitotic arrest and induction of apoptosis in colon cancer cells.
Mol Cancer Ther. 4:1388–1398. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Malki A, El-Saadani M and Sultan AS:
Garlic constituent diallyl trisulfide induced apoptosis in MCF7
human breast cancer cells. Cancer Biol Ther. 8:2175–2185. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wu XJ, Hu Y, Lamy E and Mersch-Sundermann
V: Apoptosis induction in human lung adenocarcinoma cells by
oil-soluble allyl sulfides: triggers, pathways, and modulators.
Environ Mol Mutagen. 50:266–275. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang W, Ha M, Gong Y, Xu Y, Dong N and
Yuan Y: Allicin induces apoptosis in gastric cancer cells through
activation of both extrinsic and intrinsic pathways. Oncol Rep.
24:1585–1592. 2010.PubMed/NCBI
|
|
10
|
Xiao D, Pinto JT, Soh JW, et al: Induction
of apoptosis by the garlic-derived compound S-allylmercaptocysteine
(SAMC) is associated with microtubule depolymerization and c-Jun
NH(2)-terminal kinase 1 activation. Cancer Res. 63:6825–6837.
2003.PubMed/NCBI
|
|
11
|
Chen C, Pung D, Leong V, et al: Induction
of detoxifying enzymes by garlic organosulfur compounds through
transcription factor Nrf2: effect of chemical structure and stress
signals. Free Radic Biol Med. 37:1578–1590. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bennett BL, Sasaki DT, Murray BW, et al:
SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase.
Proc Natl Acad Sci USA. 98:13681–13686. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wen J, Zhang Y, Chen X, Shen L, Li GC and
Xu M: Enhancement of diallyl disulfide-induced apoptosis by
inhibitors of MAPKs in human HepG2 hepatoma cells. Biochem
Pharmacol. 68:323–331. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xia HH, He H, De Wang J, et al: Induction
of apoptosis and cell cycle arrest by a specific c-Jun NH2-terminal
kinase (JNK) inhibitor, SP-600125, in gastrointestinal cancers.
Cancer Lett. 241:268–274. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Herzer K, Grosse-Wilde A, Krammer PH,
Galle PR and Kanzler S: Transforming growth factor-beta-mediated
tumor necrosis factor-related apoptosis-inducing ligand expression
and apoptosis in hepatoma cells requires functional cooperation
between Smad proteins and activator protein-1. Mol Cancer Res.
6:1169–1177. 2008. View Article : Google Scholar
|
|
16
|
Korchynskyi O, Landström M, Stoika R, et
al: Expression of Smad proteins in human colorectal cancer. Int J
Cancer. 82:197–202. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Perlman R, Schiemann WP, Brooks MW, Lodish
HF and Weinberg RA: TGF-beta-induced apoptosis is mediated by the
adapter protein Daxx that facilitates JNK activation. Nat Cell
Biol. 3:708–714. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Teraoka H, Sawada T, Yamashita Y, et al:
TGF-beta1 promotes liver metastasis of pancreatic cancer by
modulating the capacity of cellular invasion. Int J Oncol.
19:709–715. 2001.
|
|
19
|
Wildey GM, Patil S and Howe PH: Smad3
potentiates transforming growth factor beta (TGFbeta)-induced
apoptosis and expression of the BH3-only protein Bim in WEHI 231 B
lymphocytes. J Biol Chem. 278:18069–18077. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yang YA, Zhang GM, Feigenbaum L and Zhang
YE: Smad3 reduces susceptibility to hepatocarcinoma by sensitizing
hepatocytes to apoptosis through downregulation of Bcl-2. Cancer
Cell. 9:445–457. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yu J, Zhang L, Chen A, et al:
Identification of the gene transcription and apoptosis mediated by
TGF-beta-Smad2/3-Smad4 signaling. J Cell Physiol. 215:422–433.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chapnick DA, Warner L, Bernet J, Rao T and
Liu X: Partners in crime: the TGFβ and MAPK pathways in cancer
progression. Cell Biosci. 1:422011.PubMed/NCBI
|
|
23
|
Javelaud D and Mauviel A: Crosstalk
mechanisms between the mitogen-activated protein kinase pathways
and Smad signaling downstream of TGF-beta: implications for
carcinogenesis. Oncogene. 24:5742–5750. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ijichi H, Ikenoue T, Kato N, et al:
Systematic analysis of the TGF-beta-Smad signaling pathway in
gastrointestinal cancer cells. Biochem Biophys Res Commun.
289:350–357. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Caja L, Sancho P, Bertran E,
Iglesias-Serret D, Gil J and Fabregat I: Overactivation of the
MEK/ERK pathway in liver tumor cells confers resistance to
TGF-{beta}-induced cell death through impairing up-regulation of
the NADPH oxidase NOX4. Cancer Res. 69:7595–7602. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Harada K, Kawaguchi S, Supriatno,
Kawashima Y, Yoshida H and Sato M: S-1, an oral fluoropyrimidine
anti-cancer agent, enhanced radiosensitivity in a human oral cancer
cell line in vivo and in vitro: involvement possibility of
inhibition of survival signal, Akt/PKB. Cancer Lett. 226:161–168.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chu Q, Lee DT, Tsao SW, Wang X and Wong
YC: S-allylcysteine, a water-soluble garlic derivative, suppresses
the growth of a human androgen-independent prostate cancer
xenograft, CWR22R, under in vivo conditions. BJU Int. 99:925–932.
2007. View Article : Google Scholar
|
|
28
|
Howard EW, Lee DT, Chiu YT, Chua CW, Wang
X and Wong YC: Evidence of a novel docetaxel sensitizer,
garlic-derived S-allylmercaptocysteine, as a treatment option for
hormone refractory prostate cancer. Int J Cancer. 122:1941–1948.
2008. View Article : Google Scholar
|
|
29
|
Liang D, Qin Y, Zhao W, et al:
S-allylmercaptocysteine effectively inhibits the proliferation of
colorectal cancer cells under in vitro and in vivo conditions.
Cancer Lett. 310:69–76. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Guo X and Wang XF: Signaling cross-talk
between TGF-beta/BMP and other pathways. Cell Res. 19:71–88. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Xu J, Lamouille S and Derynck R:
TGF-beta-induced epithelial to mesenchymal transition. Cell Res.
19:156–172. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tong JL, Nie F, Ran ZH, et al:
Epigallocatechin gallate induces apoptosis in human hepatocellular
carcinoma HepG2 cells via TGF/Smad signaling pathway. Zhonghua
Zhong Liu Za Zhi. 31:646–650. 2009.(In Chinese).
|
|
33
|
Wang L, Hu GY, Shen H, Peng ZZ, Ning WB
and Tao LJ: Fluorofenidone inhibits TGF-beta1 induced CTGF via MAPK
pathways in mouse mesangial cells. Pharmazie. 64:680–684.
2009.PubMed/NCBI
|
|
34
|
Miyaki M, Iijima T, Konishi M, et al:
Higher frequency of Smad4 gene mutation in human colorectal cancer
with distant metastasis. Oncogene. 18:3098–3103. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Miyazono K, ten Dijke P and Heldin CH:
TGF-beta signaling by Smad proteins. Adv Immunol. 75:115–157. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Rahimi RA and Leof EB: TGF-beta signaling:
a tale of two responses. J Cell Biochem. 102:593–608. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shi Y and Massagué J: Mechanisms of
TGF-beta signaling from cell membrane to the nucleus. Cell.
113:685–700. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Derynck R and Zhang YE: Smad-dependent and
Smad-independent pathways in TGF-beta family signalling. Nature.
425:577–584. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lutz M and Knaus P: Integration of the
TGF-beta pathway into the cellular signalling network. Cell Signal.
14:977–988. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Massagué J and Chen YG: Controlling
TGF-beta signaling. Genes Dev. 14:627–644. 2000.PubMed/NCBI
|
|
41
|
Patil S, Wildey GM, Brown TL, Choy L,
Derynck R and Howe PH: Smad7 is induced by CD40 and protects WEHI
231 B-lymphocytes from transforming growth factor-beta -induced
growth inhibition and apoptosis. J Biol Chem. 275:38363–38370.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yamamura Y, Hua X, Bergelson S and Lodish
HF: Critical role of Smads and AP-1 complex in transforming growth
factor-beta-dependent apoptosis. J Biol Chem. 275:36295–36302.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Landström M, Heldin NE, Bu S, et al: Smad7
mediates apoptosis induced by transforming growth factor beta in
prostatic carcinoma cells. Curr Biol. 10:535–538. 2000.PubMed/NCBI
|
|
44
|
Mazars A, Lallemand F, Prunier C, et al:
Evidence for a role of the JNK cascade in Smad7-mediated apoptosis.
J Biol Chem. 276:36797–36803. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hoshino Y, Katsuno Y, Ehata S and Miyazono
K: Autocrine TGF-beta protects breast cancer cells from apoptosis
through reduction of BH3-only protein, Bim. J Biochem. 149:55–65.
2011. View Article : Google Scholar
|
|
46
|
Samanta D, Gonzalez AL, Nagathihalli N, Ye
F, Carbone DP and Datta PK: Smoking attenuates transforming growth
factor-beta-mediated tumor suppression function through
downregulation of Smad3 in lung cancer. Cancer Prev Res. 5:453–463.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Houde N, Chamoux E, Bisson M and Roux S:
Transforming growth factor-beta1 (TGF-beta1) induces human
osteoclast apoptosis by up-regulating Bim. J Biol Chem.
284:23397–23404. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ohgushi M, Kuroki S, Fukamachi H, et al:
Transforming growth factor beta-dependent sequential activation of
Smad, Bim, and caspase-9 mediates physiological apoptosis in
gastric epithelial cells. Mol Cell Biol. 25:10017–10028. 2005.
View Article : Google Scholar
|
|
49
|
O’Reilly LA, Cullen L, Visvader J, et al:
The proapoptotic BH3-only protein bim is expressed in
hematopoietic, epithelial, neuronal, and germ cells. Am J Pathol.
157:449–461. 2000.PubMed/NCBI
|
|
50
|
Park WS, Cho YG, Kim CJ, et al:
Hypermethylation of the RUNX3 gene in hepatocellular carcinoma. Exp
Mol Med. 37:276–281. 2005. View Article : Google Scholar : PubMed/NCBI
|