Introduction
The complement system is one of the defense
mechanisms against microorganisms and is also involved in various
immune and inflammatory diseases (1). In response to microbes, the complement
system is activated through any of the three pathways, eventually
causing cytolysis of pathogens. Emerging evidence suggests that the
complement system is activated in cancer tissues in human specimens
(2,3) and animal models (4,5).
Activation of the complement system may be involved in cancer
immune surveillance by its direct cytolytic effect (2) and the sensitization of cancer cells to
the immune effector cells via release of chemoattractants (6). However, cancer cells can evade
damaging complement attack by expressing either soluble or
membrane-associated complement regulators (7–9), such
as CD55, which protects cancer cells from complement-dependent
cytolysis (10,11) and anticancer immune responses
(12,13). It is unlikely that the complement
system works for cancer cell elimination.
Anaphylatoxin C5a is an N-terminal 74 amino acid
fragment of the α-chain of the complement fifth component (C5) and
acts as a leukocyte chemoattractant and inflammatory mediator
(14,15). A previous report that C5a recruited
myeloid-derived suppressor cells for inhibiting the antitumor
CD8+ T cell response (4)
suggests its indirect role in fostering cancer cells by protecting
them from cytotoxic T cells. C5a induces endothelial cell
chemotaxis and blood vessel formation (5), promoting neovascularization (16). Thus, C5a creates a favorable
microenvironment for cancer progression.
C5a activities are triggered by its binding to C5a
receptor (C5aR; CD88) which was originally identified in leukocyte
cell lines (17). We recently
demonstrated aberrant C5aR expression in cancer cells originated
from various organs and revealed enhancement of cancer cell
invasiveness via the C5a-C5aR system (18), indicating a supportive role of the
anaphylatoxin in cancer progression. However, C5a generation in
cancer tissues has not been fully elucidated. C5a generation
through activation of the complement system has been reported
(4), however cancer cell expression
of complement regulatory molecules (7–9)
suggests inefficient C5a generation through activation of the
complement system in response to cancer cells. In addition to
complement activation, thrombin (19) and proteases from bacteria (20) and phagocytes (21) can release C5a directly from C5.
Cancer cell-derived protease(s) may be capable of releasing C5a.
Therefore, we investigated C5a release from C5 by cancer cells as a
new C5a generation mechanism in the cancer tissues.
Materials and methods
Materials
Human C5, aprotinin, GM6001, and decanoyl-Arg-
Val-Lys-Arg-chloromethylketone were purchased from Calbiochem (San
Diego, CA, USA) and the carboxypeptidase N inhibitor
(DL-2-mercaptomethyl-3-guanidinoethylthiopropanoic acid) from Merck
(Darmstadt, Germany). Recombinant human C5a was purchased from EMD
Millipore (Billerica, MA, USA). Anti-human C5a goat antibody was
obtained from R&D Systems (Minneapolis, MN, USA). E-64,
pepstatin and phospholamidon were products of the Peptide Institute
(Minoh, Japan). Other chemicals were purchased from Wako Pure
Chemical Industries (Osaka, Japan).
Cells
Human bile duct cancer cell lines HuCCT1 and MEC,
and the human colon cancer cell line HCT15 were provided by the
Cell Resource Center for Biomedical Research Institute of
Development, Aging, and Cancer, Tohoku University (Sendai, Japan).
Human cholangiocarcinoma cell lines, SSp-25, RBE and YSCCC were
obtained from the Riken Cell Bank (Tsukuba, Japan). The human colon
cancer cell line HCT116 was a gift from Dr B. Vogelstein, Johns
Hopkins University. Cells were cultured in RPMI-1640 medium
supplemented with 10% fetal bovine serum (FBS), penicillin (40
U/ml), and streptomycin (40 μg/ml) and were maintained at
37°C in 5% CO2. HuCCT1 cells do not express C5aR but we
prepared HuCCT1 cells stably expressing C5aR (HuCCT1/C5aR) by
transfecting the plasmid carrying human C5aR cDNA (18).
Immunoblotting
To detect C5a released from human C5, cancer cells
(1×104 cells/100 μl) were cultured in serum-free
RPMI-1640 medium supplemented with C5 at the normal plasma
concentration (350 nM) at 37°C for 24 h. MEC and HuCCT1 cells
(5×104 cells/100 μl) were cultured in the medium
at 37°C for various periods. HuCCT1 (5×104 cells/100
μl) or MEC (5×104 cells/100 μl) cells were
cultured for 24 h at 37°C in serum-free RPMI-1640 medium and
supernatants were incubated at 37°C for 24 h in the presence of C5
(350 nM). To detect C5a generated from human plasma, citrated human
plasma was treated at 56°C for 30 min to immobilize the activation
reaction of the complement system and 100 μl of the plasma
supplemented with carboxypeptidase N inhibitor
DL-2-mercaptomethyl-3-guanidinoethylthiopropanoic acid (3
μM) was incubated with cancer cells (1×104) at
37°C. At various incubation periods, 2 μl of the plasma was
withdrawn. To characterize the C5a-releasing protease activity, C5
was incubated with HuCCT1 cells (5×104 cells/100
μl) in the presence of nontoxic doses of inhibitors specific
for cysteine- (E64, 10 μM), serine- (aprotinin 10
μg/ml), acid- (pepstatin 1 μM) or metallo-proteases
(GM, GM6001 5 μM; phospholamidon 10 μM). Twenty
microliters of the supernatant were withdrawn. To obtain
glycosylated C5a as a positive control, 5 ml of heparinized human
plasma supplemented with the carboxypeptidase N inhibitor (3
μM) was incubated with 1 unit of cobra venom factor (Quidel
Corporation, San Diego, CA, USA) at 37°C for 30 min; 2 μl of
the plasma was used (20). These
samples were analyzed by SDS-PAGE under reducing conditions using
15% polyacrylamide gels and transferred onto polyvinylidene
fluoride membranes. The membranes were incubated with anti-human
C5a goat IgG (x1,000 dilution), followed by HRP-conjugated
anti-goat IgG rabbit antibody. Bands were visualized by enhanced
chemiluminescence (Amersham Biosciences, Blauvelt, NY, USA).
Invasion assay
To assess invasion of HuCCT1/C5aR cells in
vitro, BioCoat Matrigel invasion chambers (22) (24-well plate, 8-μm pore) (BD
Biosciences, San Jose, CA, USA) were used. Five hundred microliters
of parental HuCCT1 cells (5.0×104) in the lower chamber
were incubated at 37°C for 24 h in serum-free RPMI-1640 medium
supplemented with or without C5 (350 nM). Then, HuCCT1/C5aR
(3.75×104) cell suspension in 0.75 ml of serum-free
RPMI-1640 medium supplemented with 2.6 μg/ml of anti-C5a IgG
or non-specific IgG was placed in the upper chamber and incubated
at 37°C for 24 h. Cells on the upper surface of the filter were
removed with a cotton wool swab, and cells that migrated to the
lower surface were fixed in 100% methanol and stained with 1%
toluidine blue. Migrated cells were counted in five low power
fields (x20). The invasion-enhancing effect was shown as the ratio
of cell invasion by samples vs. serum-free RPMI-1640 medium in the
lower chamber.
Statistical analyses
Statistical analyses were performed using the
unpaired Student’s t-test. Values are expressed as the means ± SD
and experiments were performed in triplicate, unless otherwise
stated.
Results
Release of C5a from C5 by cancer
cells
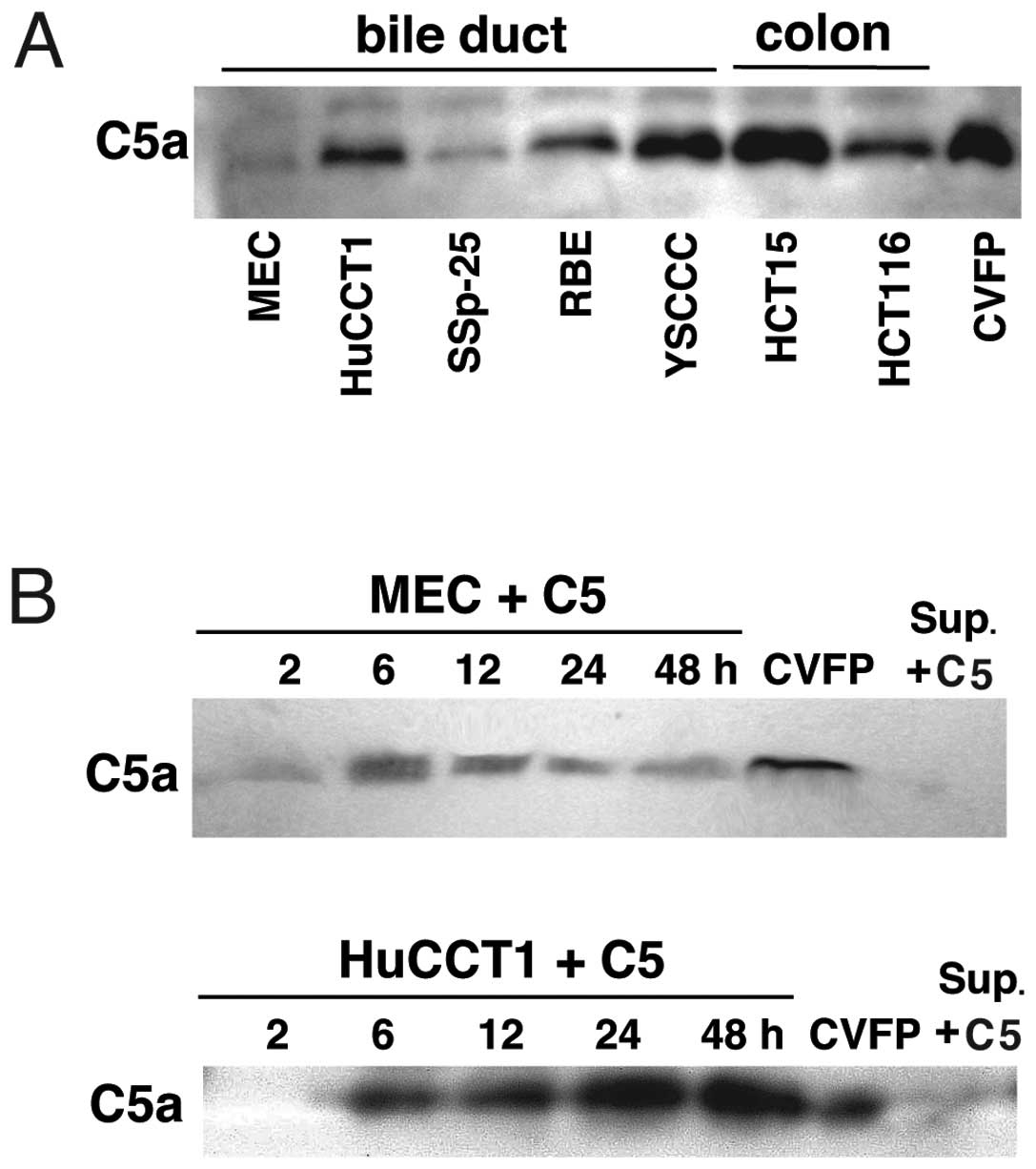
To explore cancer cell C5a release from C5, we
cultured cancer cells in RPMI-1640 medium supplemented with C5 and
examined the culture medium for C5a. All tested cancer cell lines
from bile ducts or colon released C5a from human C5 at its plasma
concentration (20) and the
C5a-releasing activity varied in cell lines (Fig. 1A). MEC, HCT15 and HCT116 cells, but
not the other cell lines, express C5aR (18), suggesting that cancer cells do not
always express C5aR together with the protease responsible for the
C5a release. By culturing MEC or HuCCT1 cells, C5a concentrations
in the C5-supplemented medium reached a maximum in 6–12 h and 24–48
h, respectively (Fig. 1B). Since
C5a was not detected in the C5-supplemented medium treated with the
culture supernatant of MEC or HuCCT1 cells (Fig. 1B), the C5a-releasing protease is
associated with cancer cells but is not secreted into the culture
medium. It is likely that a cancer cell membrane-bound protease
releases C5a from C5.
Invasion enhancement by cancer
cell-released C5a
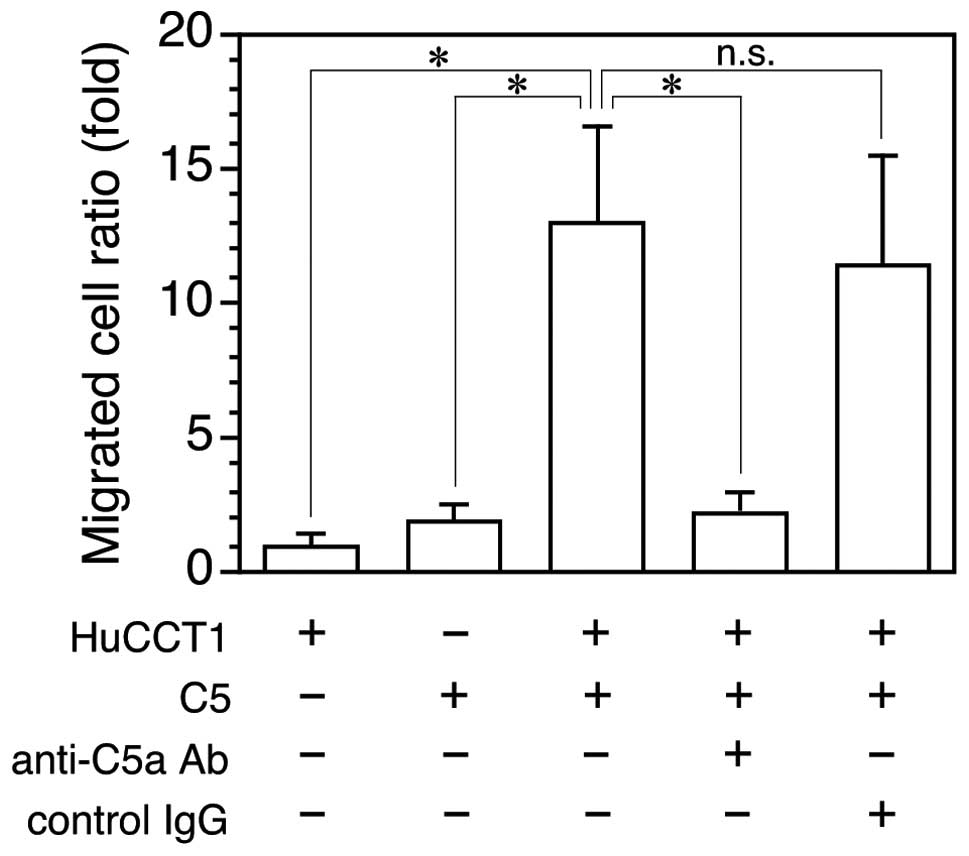
C5a enhances invasion of C5aR-expressing cancer
cells (18). To determine whether
cancer cell-released C5a is active for C5aR-expressing cancer
cells, the C5-supplemented medium in which HuCCT1 cells were
cultured was examined for invasion enhancing activity using
HuCCT1/C5aR cells. The C5-free cancer cell culture medium did not
affect HuCCT1/C5aR cell invasion but the C5a-supplemented culture
medium enhanced cancer cell invasion >10-fold, which was
equivalent to the activity of ~10 nM C5a (18) and was almost completely inhibited by
anti-C5a IgG, but not control IgG (Fig.
2). The result indicates that the cancer cell-released C5a is
active and sufficient to enhance C5aR-expressing cancer cell
invasion.
Inhibition of cancer cell C5a release by
protease inhibitors
To characterize the C5a-releasing protease, C5a
release from C5 by HuCCT1 cells was investigated in the presence of
various types of protease inhibitors. Aprotinin, a serine protease
inhibitor, inhibited C5a release by HuCCT1 cells but inhibitors
specific for cysteine, acid or metal proteases did not affect C5a
release by the cells (Fig. 3A),
indicating that the protease is serine-type. We previously reported
C5a release from human C5 by ASP (Aeromonas sobria serine
protease) (20) belonging to the
kexin subfamily (23). Similar to
other members of the subfamily, ASP cleaves peptide bonds at the
C-terminal side of paired basic amino acid residues (24) and proteases of such substrate
specificity are inhibited by decanoyl-Arg-Val-
Lys-Arg-chloromethylketone (dRVKRck) (25). Therefore, we examined effects of
dRVKRck on cancer cell C5a-releasing activity. This inhibitor
almost completely suppressed C5a release from C5 by HuCCT1 cells
(Fig. 3B), suggesting that the
cancer cell C5a-releasing protease has substrate specificity
similar to that of the kexin subfamily proteases.
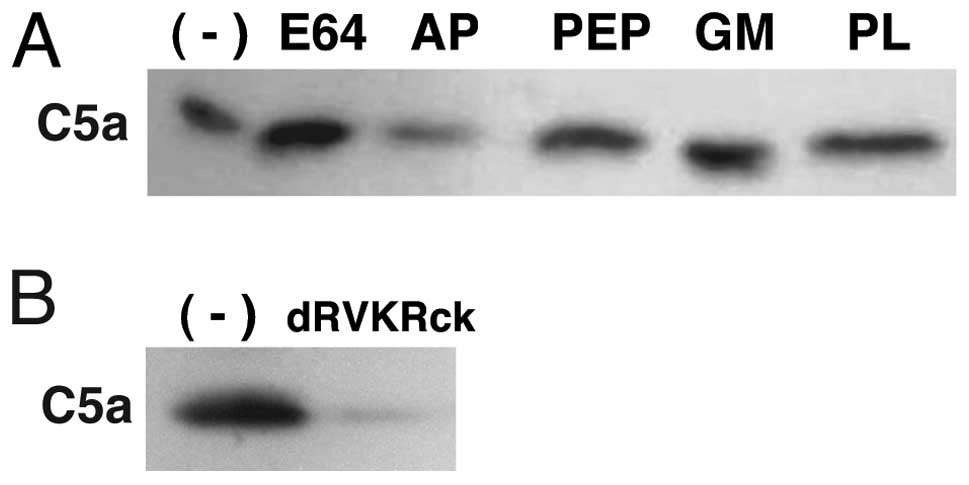 | Figure 3Inhibition of cancer cell C5a release
by protease inhibitors. (A) HuCCT1 cells were cultured in
C5-supplemented media for 24 h in the presence of either nontoxic
doses of inhibitors specific for cysteine- (E64, 10 μM),
serine- (AP, 10 μg/ml), acid- (PEP, 1 μM) or
metallo-proteases (GM, GM6001 5 μM; PL, 10 μM). (B)
HuCCT1 cells were cultured for 24 h in the C5 supplemented media in
the presence or absence (−) of dRVKRck (2 μM). C5a in
culture supernatants was detected by immunoblotting. AP, aprotinin;
PEP, pepstatin; PL, phospholamidon. |
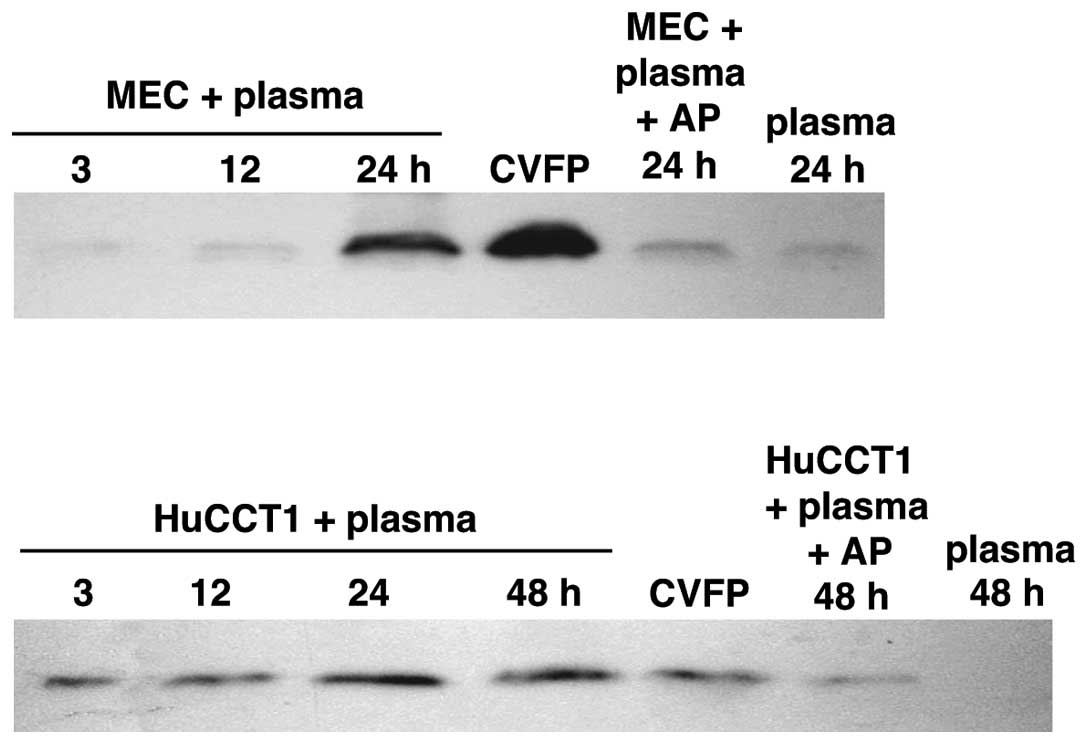
C5a release from immobilized plasma by
cancer cells
C5 is a plasma protein. Under physiological
conditions, plasma leaks from the blood stream and fills the
interstitial tissue space as lymph fluid. To address cancer cell
C5a release from C5 in the lymph fluid that is present in contact
with cancer cells, human plasma treated to immobilize the cascade
reaction of complement activation was incubated with MEC or HuCCT1
cells and then the plasma was examined for C5a. The two clones of
cancer cells released C5a from the plasma in an incubation
time-dependent manner and aprotinin inhibited the C5a release
mostly (Fig. 4), showing complement
activation-independent C5a release from C5 probably by the cancer
cell serine protease. No or negligible C5a was released from the
plasma incubated in the absence of cancer cells. Aprotinin at the
concentration used does not inhibit thrombin (26), excluding C5a release by thrombin.
The result suggests that the cancer cell protease can release C5a
from C5 in the lymph fluid in cancer tissues.
Discussion
In the present study, we demonstrated C5a release
directly from human C5 by cancer cells (Fig. 1A and B). This is a new mechanism of
C5a generation in the cancer microenvironment. Since activation of
the complement system is not involved in this mechanism, cancer
cell-associated complement regulators (7–11) do
not hinder the C5a release. Recently, C5 binding to cancer cells
cultured in serum-supplemented media and C5a production by washed
cancer cells cultured in serum-free conditions were shown by flow
cytometry and ELISA, respectively (5). However, the concentrations of the C5a
were <1 nM, the minimal concentration at which C5a enhances
cancer cell invasion (18). As the
molecular weight and activity of the C5a were not shown in the
present study, a possibility that the C5a antigens are derived from
non-functional fragments of cancer cell-bound C5, cannot be
excluded. The result that cancer cell culture medium not
supplemented with C5 did not enhance invasiveness of
C5aR-expressing cancer cells (Fig.
2) suggests the necessity of C5 in the release of significant
amounts of active C5a. In accordance with our result (Fig. 3), partial inhibition by serine
protease inhibitors suggested involvement of serine protease(s) in
the C5a production by mouse lung cancer cells, but localization of
the protease(s) was not determined (5). We demonstrated that the C5a antigen
released in the cancer cell culture medium supplemented with C5 had
a molecular weight identical to that of the C5a antigen released in
the cobra venom-treated human plasma (Fig. 1). Furthermore, the released C5a
exhibited cancer cell invasion enhancing activity that corresponds
to the activity of 10 nM C5a (18)
and its release requires both cancer cells and C5 (Fig. 2). As the C5a-releasing activity was
absent in the cancer cell culture medium (Fig. 1B), the protease that contributes to
the C5a release is possibly present on the cell membrane but is not
secreted into the medium. Collectively, the present study has
clearly shown that active C5a is released from C5 by a cancer cell
membrane-anchored protease and the C5a concentration is sufficient
to enhance invasion of C5aR-expressing cancer cells.
C5 is present in the interstitial fluid that is
plasma leaked from capillaries into the interstitial space under
physiological conditions. Plasma leakage is enhanced by leaky
vasculature in cancer tissues (27). Loose cell-to-cell contact of cancer
cells and change from the mass to free cells by
epithelial-mesenchymal transition facilitate interstitial fluid C5
access to the cell surface, which enables C5 cleavage by the
cell-membrane protease. Binding of the released C5a to C5aR on the
same cell and/or neighboring cells lessens C5a dilution by
diffusion and allows C5a to efficiently activate cancer cells. In
contrast, non-cancerous epithelial cells do not release C5a from C5
(5). Tight adhesion among
epithelial cells prevents interstitial fluid C5 from accessing to
the cell surface, which makes C5a release unlikely, even if the
cells possess the C5a-releasing protease. C5a release by cancer
cells from the complement-immobilized plasma (Fig. 4) indicates that C5 cleavage by the
protease can occur in cancer tissues. C5a release from human C5 at
its plasma concentration by all of the cancer cell lines examined,
including C5aR-expressing cells (Fig.
1A), suggests that various types of cancer cells may have this
activity and continuously release C5a. Thus, there may be a
self-activation circuit via the C5a-C5aR system in cancer cells
that express C5aR and the protease on the cell surface.
Almost complete inhibition of HuCCT1 cell C5a
release by dRVKRck (Fig. 3B) may
indicate that the cancer cell C5a-releasing protease belongs to the
kexin subfamily proteases (28). A
kexin subfamily serine protease ASP cleaves peptide bonds at the
carboxy-terminal side of -Ile-Glu-Gly-Arg- and -Leu-Ser-Thr-Arg-
more efficiently than those of paired basic residues
-Arg-Thr-Lys-Arg- and -Glu-Lys-Lys- (24). In fact, this bacterial protease
activates prothrombin (24) and
releases C5a from C5 (18), which
requires cleavage at the carboxy-terminal side of Arg residues of
the two substrates (24,29). Inhibition of the C5a-releasing
protease by aprotinin (Figs. 3 and
4) that inhibits trypsin (30) appears to be consistent with the
cancer cell protease activity to cleave proteins at the
carboxy-terminal side of Arg residues to release C5a. These results
support that the cancer cell C5a-releasing protease is possibly a
membrane-anchored kexin-like protease. Furin, a membrane-anchored
protease, is, thus far, an only kexin subfamily member of human
cell origin and is inhibited by dRVKRck (31). Although recombinant human furin
released C5a from C5, C5a-releasing activity of HuCCT1 cells did
not change by furin knockdown using siRNA. Other candidates may be
cell membrane-anchored trypsin-like serine proteases (32), but the antibody against a
representative protease matriptase did not affect the HuCCT1 cell
activity to release C5a. Further study is required to identify the
cancer cell C5a-releasing protease.
As macrophages, neutrophils and myeloid derived
suppressor cells express C5aR (4,14,15,17),
cancer cell-released C5a probably recruits these leukocytes to
cancer tissues in addition to activation of C5aR-expressing cancer
cells. The macrophages, known as tumor-associated macrophages
(TAMs), promote cancer development and metastasis by secreting
several factors associated with angiogenesis, tumor cell growth,
migration and invasion, such as MMP-9, VEGF and EGF (33–35).
Infiltrating neutrophils can play an important role in activating
angiogenesis in the tissue vasculature in carcinogenesis (36). C5a also recruit myeloid-derived
suppressor cells that suppress the antitumor CD8+ T
cell-mediated response, thereby augmenting tumor growth (4). Thus, the cancer cell protease-released
C5a could contribute to create a microenvironment favorable for
cancer progression by gathering such cancer cell-supporting
leukocytes to cancer tissues in addition to inducing
neovascularization (5,16,36).
C5a activities that help cancer progression
(4,5,18)
suggest that agents targeting the C5a-C5aR system, such as anti-C5a
antibody, anti-C5aR antibody and C5aR antagonists, can interrupt
cell activation via the system, inhibiting C5aR-expressing cancer
cell invasion, cancer cell-supporting leukocyte recruitment and
neovascularization. Thus, these agents are potentially available
for anticancer therapy. In the present study, we revealed cancer
cell membrane-bound protease-mediated C5a release as a new C5a
generation mechanism in cancer tissues. Inhibitors specific for the
cancer cell C5a-releasing protease suppress C5a release, thus may
also provide a useful therapeutic option for cancer treatment in
the future.
Acknowledgements
The authors thank Dr A. Irie for the technical
assistance. This study was supported by JSPS KAKENHI grant nos.
22590363 and 25460498 to T.I.
References
|
1
|
Ricklin D, Hajishengallis G, Yang K and
Lambris JD: Complement: a key system for immune surveillance and
homeostasis. Nat Immunol. 11:785–797. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Niculescu F, Rus HG, Retegan M and Vlaicu
R: Persistent complement activation on tumor cells in breast
cancer. Am J Pathol. 140:1039–1043. 1992.PubMed/NCBI
|
|
3
|
Bjørge L, Hakulinen J, Vintermyr OK, Jarva
H, Jensen TS, Iversen OE and Meri S: Ascitic complement system in
ovarian cancer. Br J Cancer. 92:895–905. 2005.
|
|
4
|
Markiewski MM, DeAngelis RA, Benencia F,
Ricklin-Lichtsteiner SK, Koutoulaki A, Gerard C, Coukos G and
Lambris JD: Modulation of the antitumor immune response by
complement. Nat Immunol. 9:1225–1235. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Corrales L, Ajona D, Rafail S, et al:
Anaphylatoxin C5a creates a favorable microenvironment for lung
cancer progression. J Immunol. 189:4674–4683. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Müller-Eberhard HJ: Molecular organization
and function of the complement system. Annu Rev Biochem.
57:321–347. 1988.
|
|
7
|
Yamakawa M, Yamada K, Tsuge T, Ohrui H,
Ogata T, Dobashi M and Imai Y: Protection of thyroid cancer cells
by complementregulatory factors. Cancer. 73:2808–2817. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Niehans GA, Cherwitz DL, Staley NA, Knapp
DJ and Dalmasso AP: Human carcinomas variably express the
complement inhibitory proteins CD46 (membrane cofactor protein),
CD55 (decay-accelerating factor), and CD59 (protectin). Am J
Pathol. 149:129–142. 1996.
|
|
9
|
Jurians K, Ziegler S, Garcia-Schüller H,
Kraus S, Bohana-Kashtan O, Fishelson Z and Kirschfink M: Complement
resistance of tumor cells: basal and induced mechanisms. Mol
Immunol. 36:929–939. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Morgan J, Spendlove I and Durrant L: The
role of CD55 in protecting the tumour environment from complement
attack. Tissue Antigens. 60:213–223. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gancz D and Fishelson Z: Cancer resistance
to complement-dependent cytotoxicity (CDC): problem-oriented
research and development. Mol Immunol. 46:2794–2800. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu J, Miwa T, Hilliard B, Chen Y, Lambris
JD, Wells AD and Song WC: The complement inhibitory protein DAF
(CD55) suppresses T cell immunity in vivo. J Exp Med. 201:567–577.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mikesch JH, Buerger H, Simon R and Brandt
B: Decay-accelerating factor (CD55): a versatile acting molecule in
human malignancies. Biochim Biophys Acta. 1766:42–52.
2006.PubMed/NCBI
|
|
14
|
Guo RF and Ward PA: Role of C5a in
inflammatory responses. Annu Rev Immunol. 23:821–852. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Markiewski MM and Lambris JD: The role of
complement in inflammatory diseases from behind the scenes into the
spotlight. Am J Pathol. 171:715–727. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nozaki M, Raisler BJ, Sakurai E, et al:
Drusen complement components C3a and C5a promote choroidal
neovascularization. Proc Natl Acad Sci USA. 103:2328–2333. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gerard NP and Gerard C: The chemotactic
receptor for human C5a anaphylatoxin. Nature. 349:614–617. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nitta H, Wada Y, Kawano Y, et al:
Enhancement of human cancer cell motility and invasiveness by
anaphylatoxin C5a via aberrantly expressed C5a receptor (CD88).
Clin Cancer Res. 19:2004–2013. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Huber-Lang M, Sarma JV, Zetoune FS, et al:
Generation of C5a in the absence of C3: a new complement activation
pathway. Nat Med. 12:682–687. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nitta H, Imamura T, Wada Y, Irie A,
Kobayashi H, Okamoto K and Baba H: Production of C5a by ASP, a
serine protease released from Aeromonas sobria. J Immunol.
181:3602–3608. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Huber-Lang M, Younkin EM, Sarma JV, et al:
Generation of C5a by phagocytic cells. Am J Pathol. 161:1849–1859.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Albini A, Iwamoto Y, Kleinman HK, Martin
GR, Aaronson SA, Kozlowski JM and McEwan RN: A rapid in vitro assay
for quantitating the invasive potential of tumor cells. Cancer Res.
47:3239–3245. 1987.PubMed/NCBI
|
|
23
|
Kobayashi H, Utsunomiya H, Yamanaka H, Sei
Y, Katunuma N, Okamoto K and Tsuge H: Structural basis for the
kexin-like serine protease from Aeromonas sobria as
sepsis-causing factor. J Biol Chem. 284:27655–27663. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nitta H, Kobayashi H, Irie A, Baba H,
Okamoto K and Imamura T: Activation of prothrombin by ASP, a serine
protease released from Aeromonas sobria. FEBS Lett.
581:5935–5939. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tian S and Jianhua W: Comparative study of
the binding pockets of mammalian proprotein convertases and its
implications for the design of specific small molecule inhibitors.
Int J Biol Sci. 6:89–95. 2010. View Article : Google Scholar
|
|
26
|
Pintigny D and Dachary-Prigent J:
Aprotinin can inhibit the proteolytic activity of thrombin. A
fluorescence and enzymatic study. Eur J Biochem. 207:89–95. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Maeda H, Fang J, Inutsuka T and Kitamoto
Y: Vascular permeability enhancement in solid tumor: various
factors, mechanisms involved and its implications. Int
Immunopharmacol. 3:319–328. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Angliker H, Wilkstrom P, Shaw E, Brenner C
and Fuller RS: The synthesis of inhibitors for processing
proteinases and their action on the Kex2 proteinase of yeast.
Biochem J. 293:75–81. 1993.PubMed/NCBI
|
|
29
|
DiScipio RG, Smith CA, Müller-Eberhard HJ
and Hugli TE: The activation of human complement C5 by a fluid
phase C5 convertase. J Biol Chem. 258:10629–10636. 1983.PubMed/NCBI
|
|
30
|
Antonini E, Ascenzi P, Bolognesi M, Gatti
G, Guameri M and Menegatti E: Interaction between serine
(pro)enzymes, and Kazal and Kunitz inhibitors. J Mol Biol.
165:543–558. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Angliker H: Synthesis of tight binding
inhibitors and their action on the proprotein-processing enzyme
furin. J Med Chem. 38:4014–4018. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Szabo R and Bugge TH: Membrane-anchored
serine proteases in vertebrate cell and developmental biology. Annu
Rev Cell Dev Biol. 27:213–235. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Condeelis J and Pollard JW: Macrophages:
obligate partners for tumor cell migration, invasion, and
metastasis. Cell. 124:263–266. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
van der Bij GJ, Oosterling SJ, Meijer S,
Beelen RH and van Egmond M: The role of macrophages in tumor
development. Cell Oncol. 27:203–213. 2005.PubMed/NCBI
|
|
35
|
Crowther M, Brown NJ, Bishop ET and Lewis
CE: Microenvironmental influence on macrophage regulation of
angiogenesis in wounds and malignant tumors. J Leukoc Biol.
70:478–490. 2001.PubMed/NCBI
|
|
36
|
Nozawa H, Chiu C and Hanahan D:
Infiltrating neutrophils mediate the initial angiogenic switch in a
mouse model of multistage carcinogenesis. Proc Natl Acad Sci USA.
103:12493–12498. 2006. View Article : Google Scholar : PubMed/NCBI
|