Introduction
Prostate cancer, formed in the gland of the male
reproductive system, is one of the leading causes of cancer-related
death in males. It is more common in developed countries compared
with developing countries (1).
According to an NIH report, more than two million men in the United
States are diagnosed with prostate cancer. Clinical surgery,
radiation therapy, chemotherapy and numerous other therapeutic
methods are used for the treatment of prostate cancer. However,
prostate cancer is still the second leading cause of cancer-related
death in men in the United States, and more than 29,000 men died of
prostate cancer in 2013. More efficient and specific therapeutic
methods are needed to be developed. Lentiviral-based gene
therapeutic methods have been approved more than a decade ago and
no apparent serious side effects have been reported (2).
The ubiquitin-proteasome pathway is a common pathway
for protein degradation in eukaryotes. It is involved in a wide
variety of cellular processes including apoptosis, cell cycle
control, cell adhesion and tumor growth (3). Inhibition of the ubiquitin-proteasome
pathway has emerged as a powerful strategy for anticancer therapy
(4–6). Several proteasome inhibitors have been
approved for the treatment of cancer clinically (4,5,7,8).
Ubiquitin associated protein 2-like (UBAP2L) was found to be
cofractionated with ubiquitin in the high-density fraction and it
is colocalized in the ubiquitin-containing aggregates after
proteasome inhibition (9), which
suggests that UBAP2L may be involved in the ubiquitin-proteasome
pathway. However, the function of UBAP2L in prostate cancer is
largely unknown.
In the present study, we assessed the expression of
UBAP2L in different human prostate carcinoma cell lines. Using
lentiviral-mediated RNAi, we successfully depleted endogenous
UBAP2L expression. The effects of UBAP2L inhibition on prostate
carcinoma cell proliferation, tumorigenesis and metastasis were
then investigated.
Materials and methods
Cell culture
Human prostate carcinoma cell lines, DU145, PC-3,
LN-Cap, 22RV1, and human embryonic kidney cell line 293T (HEK293T)
were obtained from the Cell Bank of the Chinese Academy of Science
(Shanghai, China). PC-3 cells were cultured in Ham’s/F-12 medium
(Gibco-BRL) supplemented with 10% fetal bovine serum (FBS). LN-Cap
and 22RV1 cells were cultured in ATCC-formulated RPMI-1640
supplemented with 10% FBS. DU145 cells were cultured in Ham’s/F-12
medium supplemented with 10% FBS and 1% non-essential amino acids
(NEAA). HEK293T cells were cultured in DMEM supplemented with 10%
FBS. Cells were cultured in a 5% CO2 incubator at
37°C.
Lentivirus packaging and infection
Short hairpin RNA (shRNA)
(5′-GCCAATACTGATGATAACTATCTCGAGATAGTTATCATCAGTATTGGCTTTTT-3′) was
designed to knock down UBAP2L (NM_001127320.1). The non-targeting
nucleotide sequence
(5′-CTAGCCCGGTTCTCCGAACGTGTCACGTATCTCGAGATACGTGACACGTTCGGAGAATTTTTTTAAT-3′)
was designed as the control. The shRNA oligos were ligated into the
lentivirus expression plasmid pFH-L (Shanghai Hollybio, China). To
package the shUBAP2L and shCon lentivirus, shRNA plasmids, envelope
plasmid pVSVG-I and packaging plasmid pCMVΔR8.92 (Shanghai
Hollybio, China) were transfected into HEK293T cells using
Lipofectamine 2000 (Invitrogen) according to the manufacturer’s
instructions. Three days after transfection, the
lentivirus-containing media were collected and ultra-centrifuged
for infection. PC-3 and DU145 cells were cultured in 6-well plates
at a density of 50,000 cells/well, respectively. The lentivirus was
added to the culture medium at an MOI of 40. Three days after
infection, the cells were observed under a microscope (10×
objective lens), and GFP-positive cell numbers were counted to
calculate the infection efficiency.
Real-time qRT-PCR
After being washed by ice-cold PBS, total-RNA was
extracted using TRIzol reagent (Invitrogen) following the
manufacturer’s instructions. M1705 M-MLV reverse transcriptase kit
(Promega) was used to synthesize cDNA. Real-time qRT-PCR was
performed to evaluate the expression level of UBAP2L, and β-actin
was used as an endogenous control. The primers used were UBAP2L,
forward, 5′-ACACAATCCCCATCACTGGT-3′ and reverse,
5′-CAGAGGAGAAGACGGAGGTG-3′; β-actin, forward,
5′-GTGGACATCCGCAAAGAC-3′ and reverse, 5′-AAAGGGTGTAACGCAACTA-3′.
Relative mRNA was determined by the formula 2−ΔΔCt (Ct,
cycle threshold).
Western blot analysis and
Pathscan® Intracellular Signaling Array
Five days after infection, prostate carcinoma cells
were washed twice with ice-cold PBS and lysed in 2X SDS sample
buffer (10 mM EDTA, 4% SDS, 10% glycine in 100 mM Tris-HCl buffer,
pH 6.8) for 1 h at 4°C. Total cell lysates were then centrifuged
(12,000 rpm, 15 min, 4°C), and the supernatants were employed for
further processing. The protein concentration was determined using
the BCA protein assay kit. Equal amounts of proteins (30 μg) were
loaded and separated on 10% SDS-PAGE gels and transferred onto PVDF
membranes (Millipore). Proteins were probed overnight at 4°C with
the primary antibodies UBAP2L (1:2,000; Abcam; cat. ab70319) or
GAPDH (1:50,000; Proteintech Group, Inc.; cat. 10494-1-AP),
followed by incubation with horseradish peroxidase-conjugated goat
anti-rabbit IgG (1:5,000; Santa Cruz; cat. sc-2054) or goat
anti-mouse IgG (1:5,000; Santa Cruz; cat. sc-2005) at room
temperature for 2 h. GAPDH was used as the internal standard.
To detect the activation of intracellular signaling,
Pathscan Intracellular Signaling Array was performed. In brief, 5
days after lentiviral infection, DU145 cells were collected and
lysed. Intracellular signaling was detected using the Pathscan
Intracellular Signaling Array Kit (Cell Signaling Technology)
following the manufacturer’s instructions.
MTT assay
After lentiviral infection for 4 days, PC-3 and
DU145 cells were seeded in 96-well plates at a density of 2,000 and
2,500 cells/well, respectively. At different time points, 20 μl of
5 mg/ml MTT [3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium
bromide] was added into each well. After incubation with MTT for 3
h, 100 μl of acidic isopropanol (10% SDS, 5% isopropanol, 0.01
mol/l HCI) was added. The formazan precipitate was then dissolved
by shaking for 10 min. The absorbance of each well was recorded at
a wavelength of 595 nm using a microplate reader.
Colony formation assay
After lentiviral infection for 4 days, PC-3 and
DU145 cells were seeded into 6-well plates at an initial density of
400 and 500 cells/well, respectively. The medium was changed at
three-day intervals. PC-3 and DU145 cells were washed with PBS and
fixed with 4% paraformaldehyde for 30 min at room temperature after
culture for 13 and 8 days, respectively. The fixed cells were then
stained with freshly prepared diluted crystal violet for 10 min,
washed with water and air-dried. The total number of colonies was
counted using a light microscope.
Cell cycle analysis
After lentiviral infection for 4 days, PC-3 and
DU145 cells were seeded into 6-cm dishes at a density of 200,000
cells/dish and cultured until the confluency of the cells reached
~80%. Cells were harvested, washed with ice-cold PBS and fixed
overnight at 75% ethanol at 4°C. Propidium iodide (PI) staining was
performed following the instructions provided in the kit (C1052;
Biyuntian). The fluorescence of PI in the cells was measured using
Cell Lab Quanta (Beckman Coulter).
Transwell migration assay
In brief, 3 days after infection, 70,000 PC-3 or
50,000 DU145 cells were seeded into the upper chamber of Transwell
plates (8.0-μm pore; Corning Costar) in 200 μl of serum-free
medium, and 500 μl of medium containing 10% FBS was added to the
lower chamber as a chemoattractant. After incubation for 24 h,
cells on the top surface of the membrane were removed. After
fixation, cells that migrated to the bottom surface of the membrane
were stained using crystal violet. Images were captured under a
light microscope (10× objective lens), and cell numbers were
counted.
Statistical analysis
All data are presented as the mean ± SD of three
independent experiments performed in triplicate. Statistical
analysis was performed based on a Student’s t-test. P<0.05 was
considered to indicate a statistically significant difference.
Results
UBAP2L expression in prostate carcinoma
cell lines
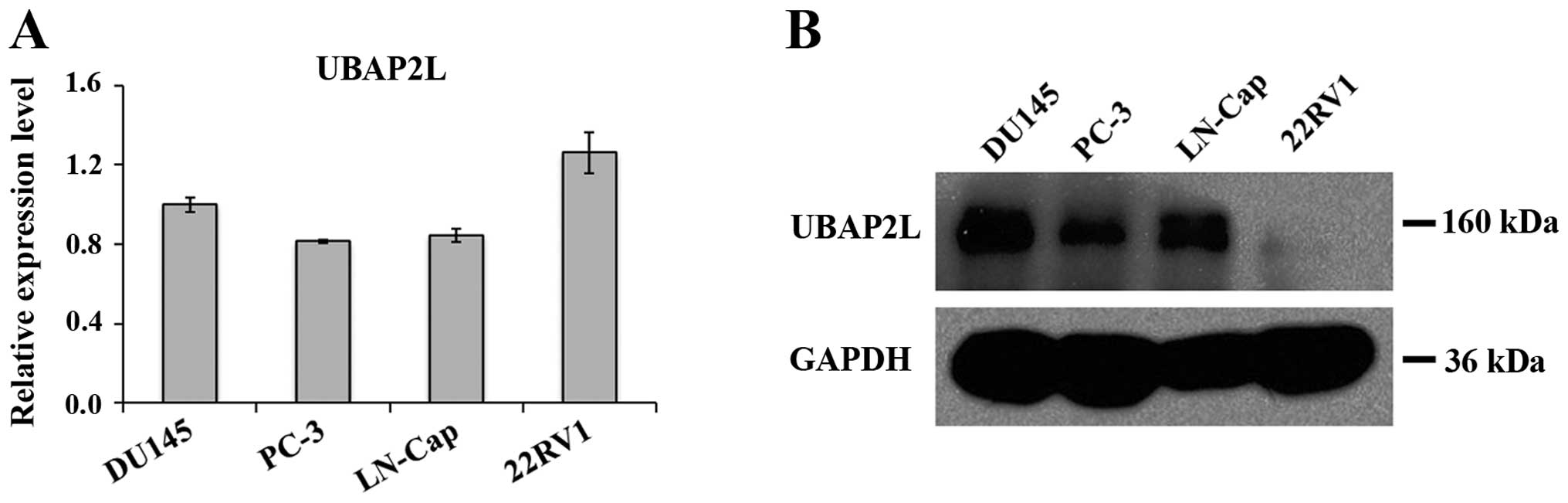
Expression levels of UBAP2L in prostate carcinoma
cells were analyzed by qRT-PCR and western blotting. As shown in
Fig. 1A, mRNA levels of UBAP2L were
detected in all 4 prostate carcinoma cell lines: DU145, PC-3,
LN-Cap and 22RV1. We found a strong immunoreactive band of UBAP2L
in the samples of DU145, PC-3 and LN-Cap cells, while a very faint
band was noted in the sample of 22RV1 cells (Fig. 1B). Therefore, PC-3 and DU145 cells
were used for the following loss-of-function investigation.
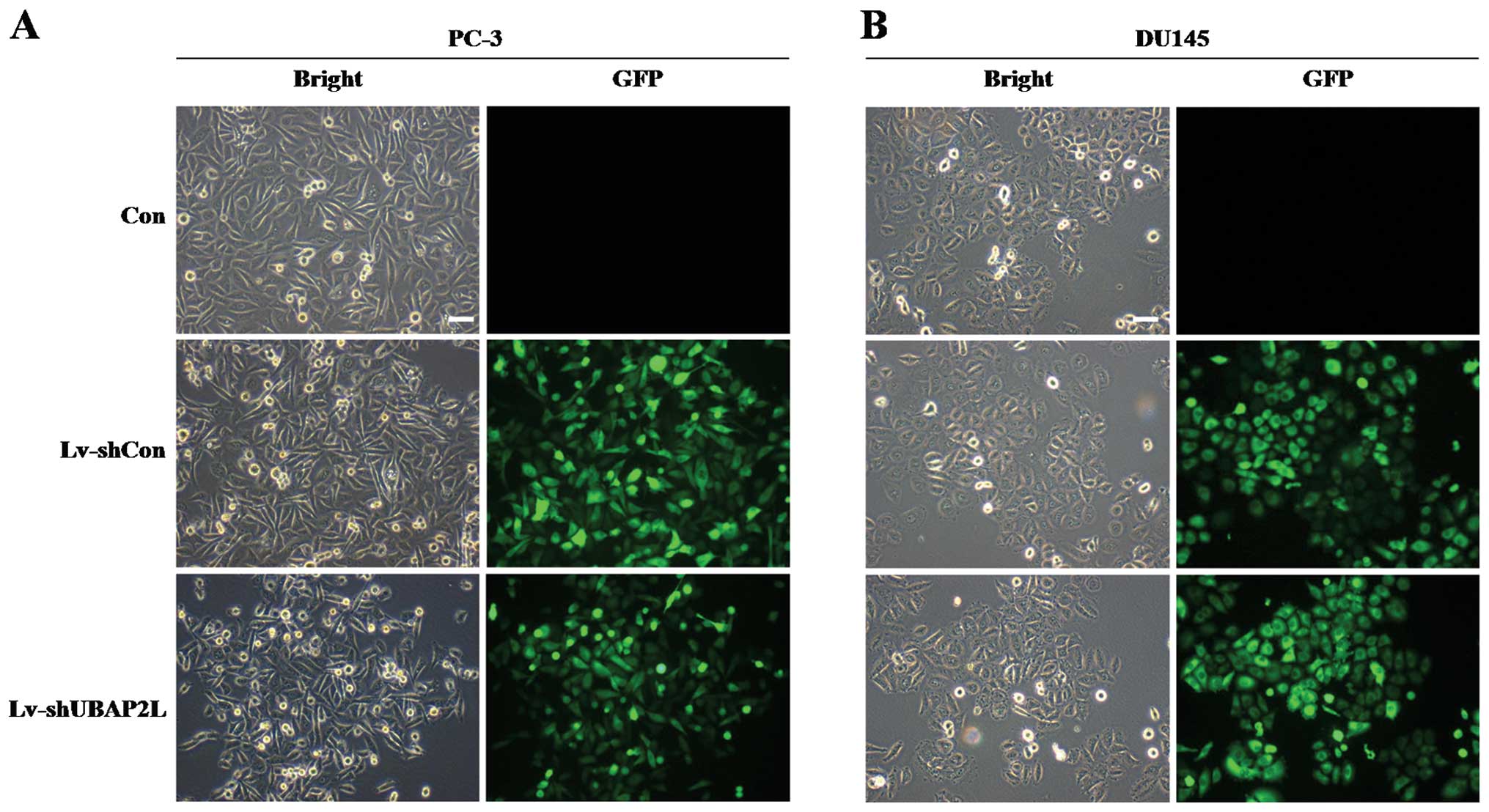
The lentivirus efficiently infects PC-3
and DU145 cells
To investigate the function of UBAP2L in prostate
carcinoma cells, we took advantage of lentiviral-mediated RNAi
technology. Firstly, the infection efficiency of the lentivirus was
evaluated. Four days after lentiviral infection, GFP was robustly
expressed in the PC-3 and DU145 cells (Fig. 2A and B). More than 90% cells were
infected by the recombinant lentivirus.
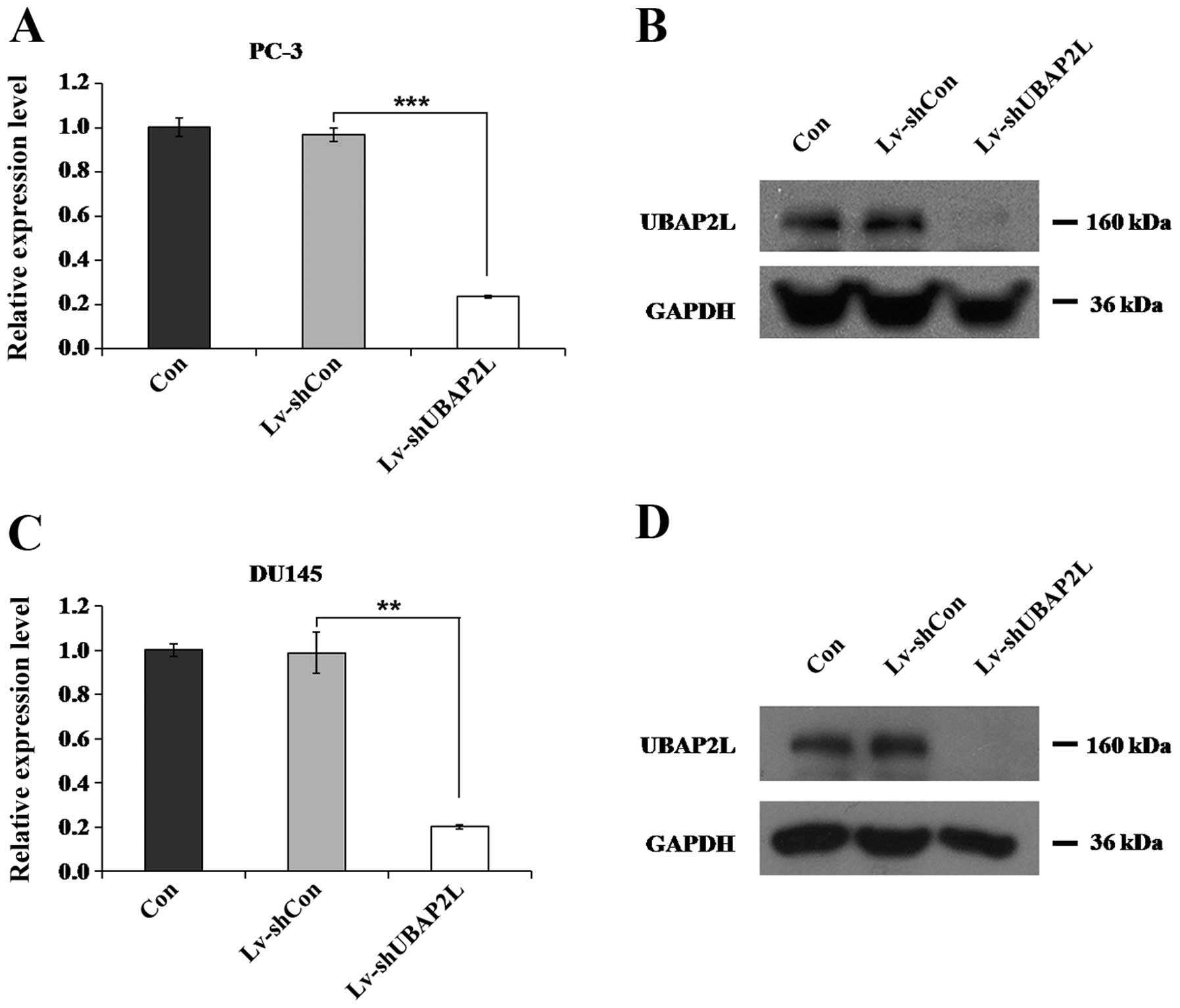
Lv-shUBAP2L infection efficiently reduces
endogenous UBAP2L expression
Four days after lentiviral infection, the expression
levels of UBAP2L in the PC-3 and DU145 cells were measured. Using
qRT-PCR, we found that the relative expression of UBAP2L was
markedly decreased after Lv-shUBAP2L infection, compared with the
control and the non-infected groups (Fig. 3A and C). The protein levels of
UBAP2L in the PC-3 and DU145 cells were also reduced after
Lv-shUBAP2L infection, as measured by western blotting (Fig. 3B and D).
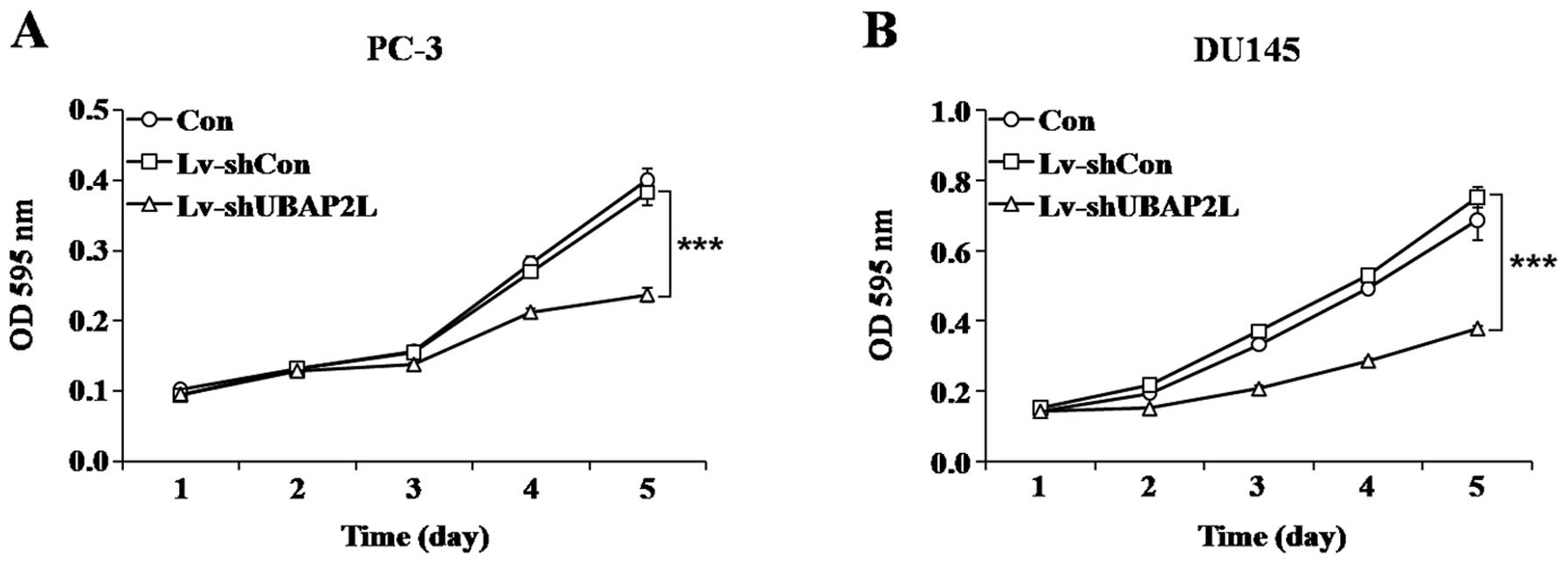
Knockdown of UBAP2L suppresses cell
proliferation and colony formation
To investigate whether UBAP2L is involved in the
proliferation and tumorigenesis of prostate carcinoma cells, MTT
and colony formation assays were performed. As shown by cell growth
curve, compared with the Lv-shCon-infected cells, the proliferation
of Lv-shUBAP2L-infected cells was significantly decreased. A 38.0%
reduction in the PC-3 cells (Fig.
4A) and a 49.8% reduction in the DU145 cells were noted
(Fig. 4B).
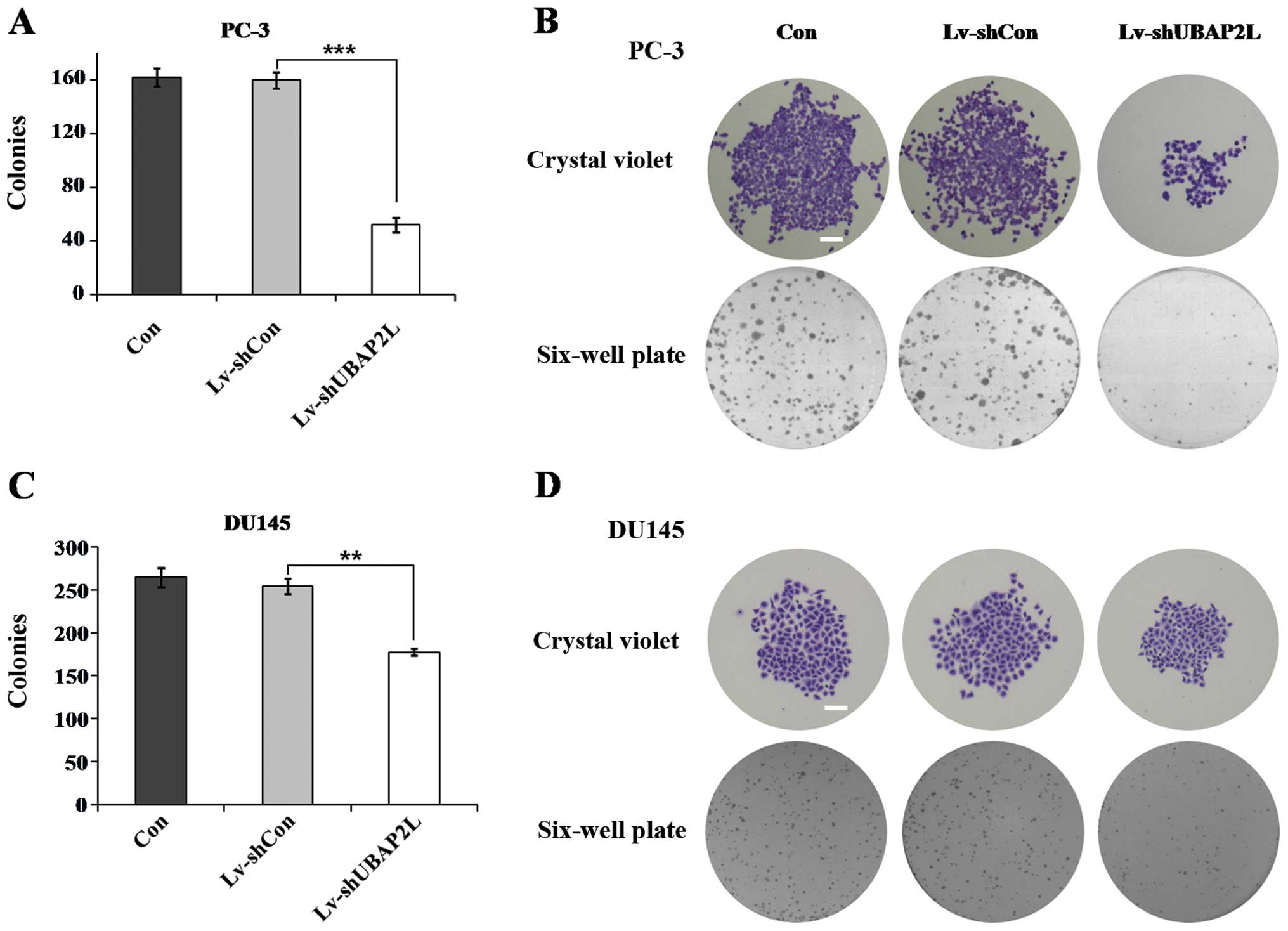
Colony formation assay was then carried out. As
shown in Fig. 5, the numbers of
colonies formed in the Lv-shUBAP2L groups (52.0±5.3 PC-3 cells and
177.7±4.2 DU145 cells) were markedly lower than the numbers of
colonies in the Lv-shCon groups (159.7±6.0 PC-3 cells and 254.7±9.1
DU145 cells). There was no significant difference in the number of
colonies between the Lv-shCon groups and the non-infected groups.
These results suggest that UBAP2L may play an important role in the
tumorigenesis of prostate carcinoma.
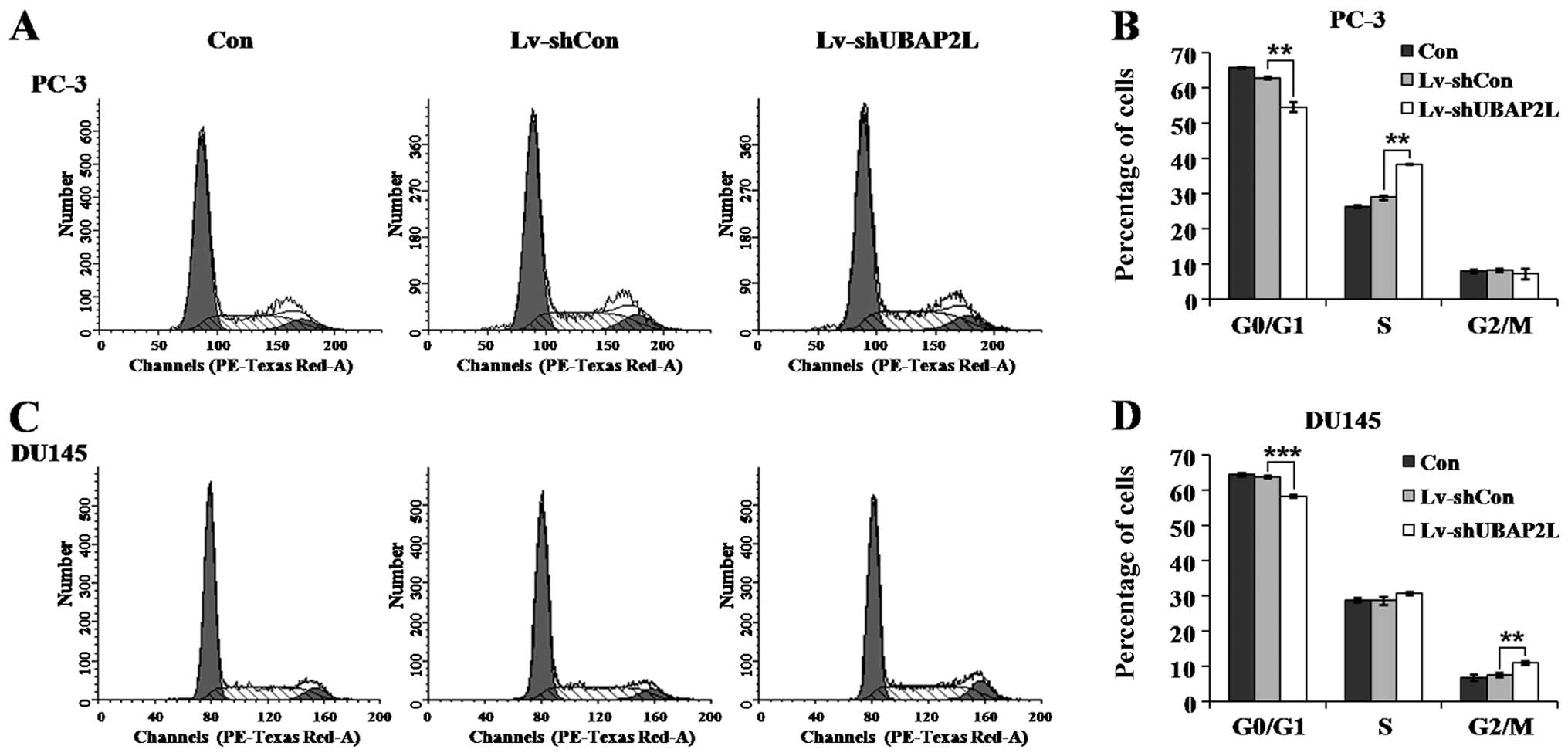
Knockdown of UBAP2L leads to cell cycle
arrest of prostate carcinoma cells
Flow cytometry was then carried out to detect
whether Lv-shUBAP2L affects the cell cycle progression of prostate
carcinoma cells. As shown in Fig. 6A
and B, downregulation of UBAP2L induced PC-3 cell accumulation
in the S phase compared with the control cells. The percentage of
cells in the S phase increased from ~28.88% in the Lv-shCon group
to 38.33% in the Lv-shUBAP2L group. In addition, UBAP2L knockdown
induced DU145 cell accumulation in the G2/M phase
compared with the control cells (Fig.
6C and D). The percentage of cells in the G2/M phase
increased from ~7.50% in the Lv-shCon group to 10.89% in the
Lv-shUBAP2L group. The cell population in the
G0/G1 phase was significantly decreased in
both PC-3 and DU145 cells following UBAP2L knockdown. These results
indicate that UBAP2L depletion inhibited cell proliferation by
inducing cell cycle arrest.
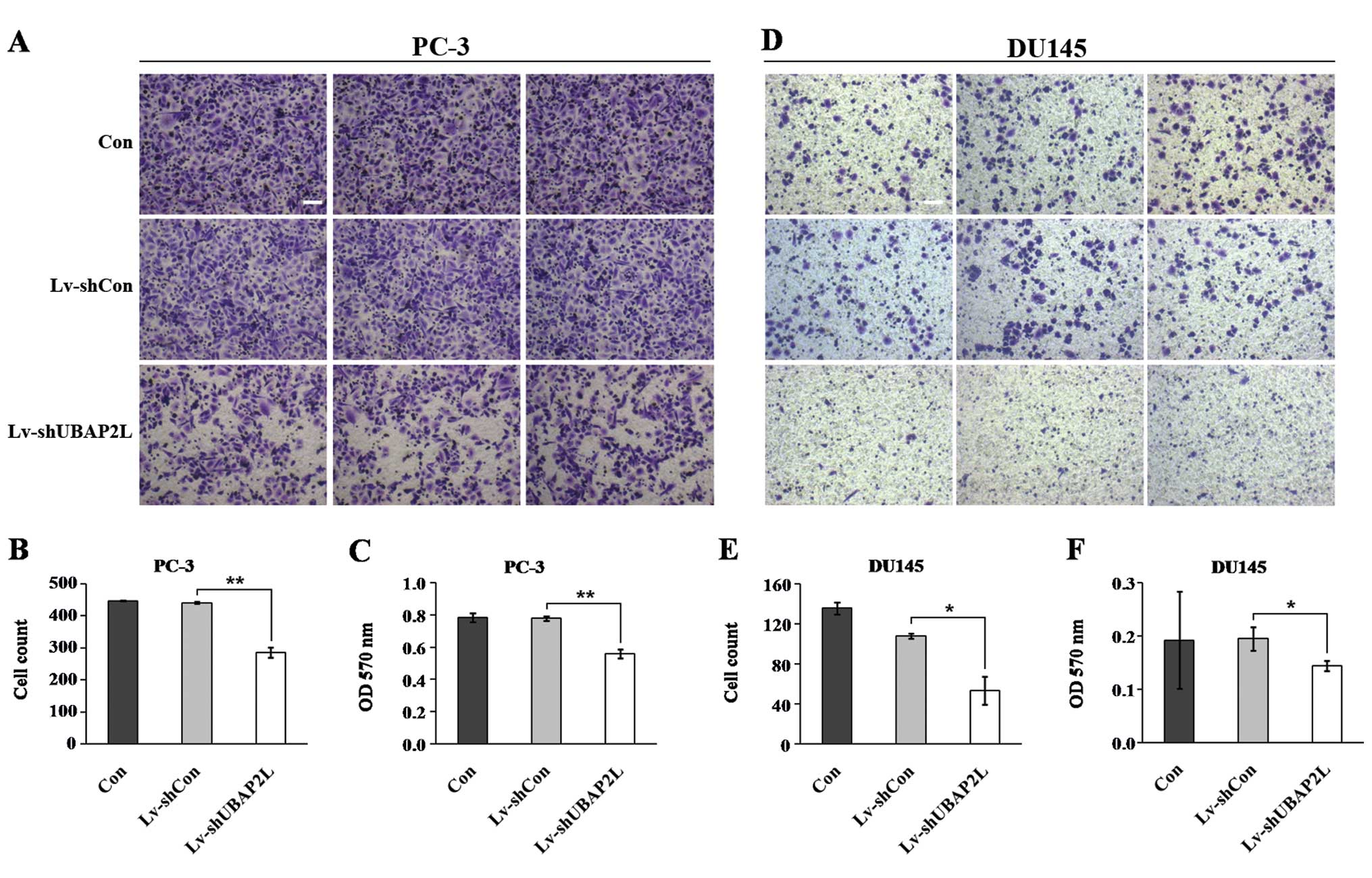
Knockdown of UBAP2L inhibits the
migration of prostate carcinoma cells
Cell migration is a critical step that occurs during
cancer progression. We next determined the effect of UBAP2L
knockdown in regulating prostate carcinoma cell migration by using
Transwell assay (Fig. 7A and D).
UBAP2L silencing markedly inhibited the migration of PC-3 and DU145
cells compared with the control cells. Fewer cells in the
Lv-shUBAP2L groups (285.7±15.2 PC-3 cells and 53.5±14.0 DU145
cells) migrated into the lower filter relative to cells in the
Lv-shCon groups (441.3±3.6 PC-3 cells and 108.0±2.8 DU145 cells)
(Fig. 7B and E). Moreover, the
crystal violet staining intensity was significantly lower in the
Lv-shUBAP2L groups than in the Lv-shCon and non-infected groups
(Fig. 7C and F). The results
suggest that UBAP2L is important for cell migration and may be a
key player in prostate cancer metastasis.
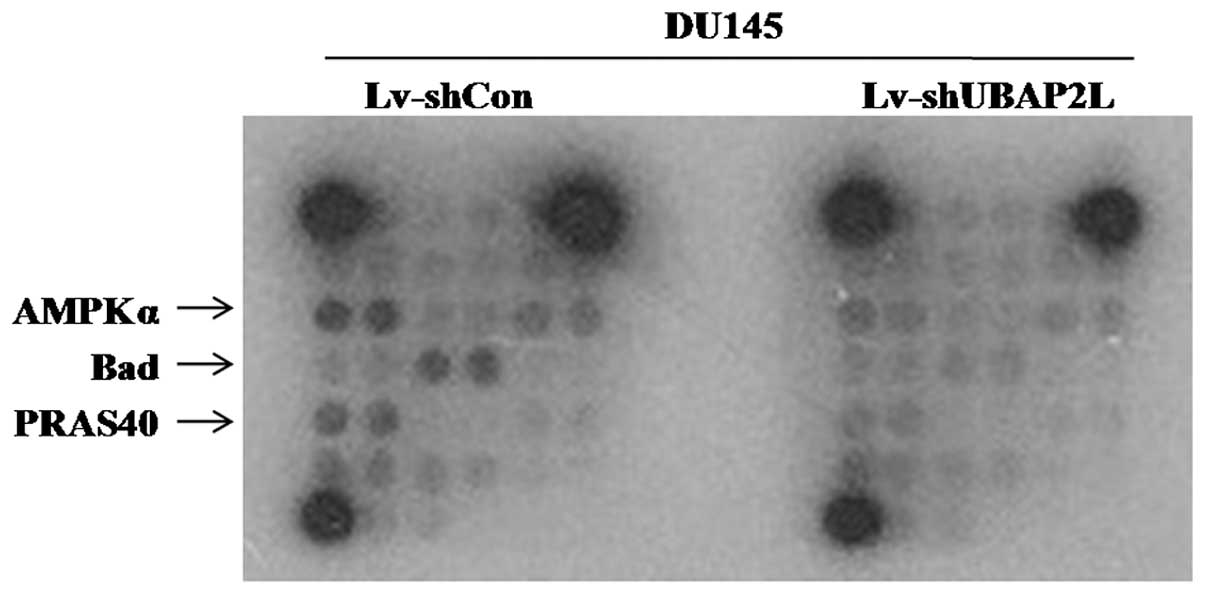
Knockdown of UBAP2L blocks AMPKa, Bad and
PRAS40 signaling pathways
To investigate the regulatory mechanism of UBAP2L in
the tumorigenesis and metastasis of prostate carcinoma, multiple
signaling pathways were analyzed in DU145 cells after UBAP2L
knockdown using PathScan Intracellular Signaling Array kit.
Compared with the Lv-shCon group, the amounts of phosphorylated
AMP-activated protein kinase α (AMPKα), Bad and PRAS40 were
obviously reduced in the Lv-shUBAP2L group (Fig. 8). Previous studies suggest that the
AMPKα, Bad and PRAS40 signaling pathways are crucial for carcinoma
cell proliferation and cell cycle control (10–12).
Our data revealed that UBAP2L depletion induced proliferation
inhibition and cell cycle arrest via the AMPKα, Bad and PRAS40
signaling pathways in prostate cancer.
Discussion
A previous study suggested that UBAP2L may be a
component of the ubiquitin-proteasome complex (9). Inhibition of the ubiquitin-proteasome
system is a promising anticancer strategy, and several proteasome
inhibitors have been approved to be used in the clinic (4–6). In
the present study, we found that UBAP2L was highly expressed in
different prostate carcinoma cell lines. Taking advantage of
lentiviral-mediated RNAi, we disrupted the expression of UBAP2L
in vitro. We found that UBAP2L depletion inhibited the
proliferation and colony formation of prostate carcinoma cells.
Cell cycle analysis showed that knockdown of UBAP2L led to PC-3
cell cycle arrest in the S phase, and DU145 cell cycle arrest in
the G2/M phase. This difference in cell cycle arrest
could be explained by the specific cell type. Moreover, UBAP2L
knockdown inhibited the migration of prostate carcinoma cells. To
investigate the molecular mechanisms of induced growth and
migration inhibition following UBAP2L depletion, cell cycle-related
signaling pathways were further analyzed using Pathscan
Intracellular Signaling Array Kit. We found that UBAP2L depletion
obviously inhibited the activation of AMPKα, Bad and PRAS40.
AMPK, a serine-threonine kinase, is a heterotrimeric
complex of catalytic α-subunits and regulatory β- and γ-subunits
with multiple isoforms (13). AMPK
is an energy sensor that is activated by phosphorylation at Thr172
in response to elevated AMP levels. AMPK regulates fatty acid
metabolism, as well as modulates protein synthesis and cell growth.
Although many studies support the tumor-suppressive role of AMPK,
emerging evidence suggests that the metabolic checkpoint function
of AMPK might be overridden by stress or oncogenic signals so that
tumor cells use AMPK activation as a survival strategy to gain a
growth advantage (10). These
findings underscore the complexity in the cellular function of AMPK
in maintaining energy homeostasis under physiological versus
pathological conditions. Previously, Smith et al
demonstrated that increased BAD expression promotes prostate cancer
cell proliferation (12). The BAD
phosphorylation status plays a major role in apoptosis regulation
by serving as a convergence point of several anti-apoptotic
signaling pathways, including constitutively active PI3K (14). BAD phosphorylation at serines 112
and 136 (based on the mouse sequence) (15,16),
facilitates interaction with 14-3-3 chaperones, whereas
phosphorylation at S155 within the BH3 domain disrupts binding to
BCL-XL or BCL-2 (17). As a result,
phosphorylation inactivates the pro-apoptotic function of BAD by
preventing interaction with BCL-2 and BCL-XL. PRAS40 is an mTOR
binding protein that has complex effects on cell metabolism. PRAS40
regulates protein synthesis and the cell cycle (11). Phosphorylation of PRAS40 at Thr246
by Akt relieves PRAS40 inhibition of TORC1 (18). Our results suggest that UBAP2L may
promote prostate carcinoma cell growth through activation of AMPKα,
Bad and PRAS40, which induce cell proliferation and cell cycle
transition. However, the mechanisms involved in the regulation of
the AMPKα, Bad and PRAS40 signaling pathways by UBAP2L remain
unknown. One explanation is that, as a potential component of
proteasome, UBAP2L might directly regulate the degradation of
AMPKa, Bad and PRAS40. Another possibility is that UBAP2L may
regulates the expression of several other proteins, which regulate
the phosphorylation of AMPKα, Bad and PRAS40. Collectively, UBAP2L
may mediate tumorigenesis and metastasis via regulation of the
AMPKα, Bad and PRAS40 signaling pathways.
In conclusion, we demonstrated for the first time
that UBAP2L is crucial for the growth and migration of prostate
carcinoma cells. UBAP2L silencing by RNAi may be a promising novel
therapeutic method for prostate cancer.
References
|
1
|
Baade PD, Youlden DR and Krnjacki LJ:
International epidemiology of prostate cancer: geographical
distribution and secular trends. Mol Nutr Food Res. 53:171–184.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
McGarrity GJ, Hoyah G, Winemiller A, et
al: Patient monitoring and follow-up in lentiviral clinical trials.
J Gene Med. 15:78–82. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen FZ and Zhao XK: Ubiquitin-proteasome
pathway and prostate cancer. Onkologie. 36:592–596. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Iqbal M, Messina McLaughlin PA, Dunn D, et
al: Proteasome inhibitors for cancer therapy. Bioorg Med Chem.
20:2362–2368. 2012. View Article : Google Scholar
|
|
5
|
Crawford LJ, Walker B and Irvine AE:
Proteasome inhibitors in cancer therapy. J Cell Commun Signal.
5:101–110. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rastogi N and Mishra DP: Therapeutic
targeting of cancer cell cycle using proteasome inhibitors. Cell
Div. 7:262012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
San-Miguel JF, Richardson PG, Gunther A,
et al: Phase Ib study of panobinostat and bortezomib in relapsed or
relapsed and refractory multiple myeloma. J Clin Oncol.
31:3696–3703. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
O’Brien ME, Gaafar RM, Popat S, et al:
Phase II study of first-line bortezomib and cisplatin in malignant
pleural mesothelioma and prospective validation of progression free
survival rate as a primary end-point for mesothelioma clinical
trials (European Organisation for Research and Treatment of Cancer
08052). Eur J Cancer. 49:2815–2822. 2013.
|
|
9
|
Wilde IB, Brack M, Winget JM and Mayor T:
Proteomic characterization of aggregating proteins after the
inhibition of the ubiquitin proteasome system. J Proteome Res.
10:1062–1072. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chuang HC, Chou CC, Kulp SK and Chen CS:
AMPK as a potential anticancer target - friend or foe? Curr Pharm
Des. 20:2607–2618. 2013.PubMed/NCBI
|
|
11
|
Kazi AA and Lang CH: PRAS40 regulates
protein synthesis and cell cycle in C2C12 myoblasts. Mol Med.
16:359–371. 2010.PubMed/NCBI
|
|
12
|
Smith AJ, Karpova Y, D’Agostino R Jr,
Willingham M and Kulik G: Expression of the Bcl-2 protein BAD
promotes prostate cancer growth. PLoS One. 4:e62242009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li J, Coven DL, Miller EJ, et al:
Activation of AMPK alpha- and gamma-isoform complexes in the intact
ischemic rat heart. Am J Physiol Heart Circ Physiol.
291:H1927–H1934. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sastry KS, Smith AJ, Karpova Y, Datta SR
and Kulik G: Diverse antiapoptotic signaling pathways activated by
vasoactive intestinal polypeptide, epidermal growth factor, and
phosphatidylinositol 3-kinase in prostate cancer cells converge on
BAD. J Biol Chem. 281:20891–20901. 2006. View Article : Google Scholar
|
|
15
|
Datta SR, Katsov A, Hu L, et al: 14-3-3
proteins and survival kinases cooperate to inactivate BAD by BH3
domain phosphorylation. Mol Cell. 6:41–51. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fang X, Yu S, Eder A, et al: Regulation of
BAD phosphorylation at serine 112 by the Ras-mitogen-activated
protein kinase pathway. Oncogene. 18:6635–6640. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tan Y, Demeter MR, Ruan H and Comb MJ: BAD
Ser-155 phosphorylation regulates BAD/Bcl-XL interaction and cell
survival. J Biol Chem. 275:25865–25869. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sancak Y, Thoreen CC, Peterson TR, et al:
PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein
kinase. Mol Cell. 25:903–915. 2007. View Article : Google Scholar : PubMed/NCBI
|