Introduction
Lung cancer is characterized by uncontrolled cell
growth in the lungs, and is by far the leading cause of
cancer-related mortality in both men and women in the USA (1). Based on the relative size of the
cancer cells as examined under a microscope, lung cancer can be
classified as non-small cell lung cancer (NSCLC) or small cell lung
cancer (SCLC) (2). The etiological
factors for lung cancer are complex and it is believed that lung
cancer, similar to other types of cancer, is initiated by
activation of oncogenes and inactivation of tumor suppressor genes
(3). Current understanding of the
molecular mechanism of lung cancer showed that the genetic
mutations contribute to the oncogenesis and development of cancer.
Mutated oncogenes, such as epidermal growth factor receptor (EGFR),
c-myc, K-ras, anaplastic lymphoma kinase (ALK) and
phosphatidylinositol 3-kinases (PI3K), have been found to
contribute to the formation of NSCLC (4–7).
Cancerous inhibitor of PP2A (CIP2A) is a newly
characterized oncogenic protein. CIP2A was initially identified as
a tumor-associated autoantigen in gastric and liver cancer
(8). The high frequency of
autoantibodies to CIP2A detected in the sera of cancer patients
makes it a promising candidate for biomarker development. The
expression of CIP2A was only present in embryonic development but
was silenced from all tissues soon after birth. The function of
CIP2A was not discovered until 2007 during the search of protein
phosphatase 2A (PP2A) interacting proteins in human cervical cancer
cell line HeLa (9). PP2A complexes
function by inhibiting activity of several important oncogenic
signaling pathways as tumor suppressors (10). The PP2A holoenzyme consists of the
scaffold A subunit, various regulatory B subunits and the catalytic
C subunit (11–13). Junttila et al found that
CIP2A was co-purified with the A subunit of PP2A (9). The interaction between CIP2A and PP2A
led to the discovery of the inhibitory effect of CIP2A to the
phosphatase activity of PP2A. Therefore, the finding that CIP2A is
an endogenous PP2A inhibitor may greatly expand our understanding
of cellular transformation in cancer progression.
Previous studies have shown that the overexpression
of CIP2A is detected in various types of tumors, from solid tumors
to hematological malignancies, including breast, gastric, head and
neck, lung cancer and leukemia, and subsequently it was known as an
oncofetal protein (14–17). Additional functions of CIP2A in
cancer progression are still under investigation. A recent report
showed that CIP2A could protect the hepatocellular carcinoma (HCC)
cell lines from bortezomib-induced apoptosis by inhibiting
phosphor-AKT-associated PP2A phosphatase activity (18). The knockdown of CIP2A via siRNA thus
sensitizes HCC cell lines to bortezomib treatment. The present
study suggests that CIP2A is able to regulate the phosphorylation
of protein kinase B (PKB)/AKT in response to the treatment of
chemotherapeutic drugs and this protein may have more functions
unknown in tumorigenesis. However, whether the interplay between
CIP2A and phospho-AKT plays a role in cancer progression, and how
CIP2A facilitates AKT-mediated resistance to chemotherapy, remains
unresolved.
In the present study, we sought to elucidate the
association between CIP2A and the AKT signaling pathway in
regulating cell proliferation. The findings obtained from this
study will expand the understanding of protein-substrate
interaction and therefore direct the cancer drug design. At the
same time, we focused on the mechanism of AKT phosphorylation to
further investigate how CIP2A is involved in the PI3K/AKT/mTOR
pathway, which is critical to cancer progression.
Materials and methods
Cell culture and transfections
The three lung cancer cell lines, NCI-H838,
NCI-H1299 and NCI-H460 were purchased from American Type Culture
Collection (ATCC, Manassas, VA, USA). All cell lines were cultured
in RPMI-1640 medium containing 10% fetal bovine serum (FBS) (both
from Life Technologies, Carlsbad, CA, USA) at 37°C with 5%
CO2.
Transient transfection of pcDNA3.1 control or
pcDNA3.1 containing CIP2A cDNA (a gift from Dr Westermarck,
Finland) into the H1299 lung cancer cell line was performed with
Lipofectamine LTX (Life Technologies) according to the
manufacturer’s protocol.
Production of lentivirus encoding CIP2A
short hairpin RNA
Five CIP2A short hairpin RNAs, ligated in pLKO.1
vector, were obtained from Open Biosystems (Huntsville, AL, USA).
The knockdown efficiency of these shRNAs was evaluated in H1299
cells by western blotting and two of them showing the higher
knockdown efficiency were chosen to produce lentivirus. Lentivirus
was produced by co-transfection of pLKO.1 control (ligated with
scramble sequence) or other pLKO.1-derived vector with pMD2.G and
pCMV-VSVG (both from Addgene, Cambridge, MA, USA) into HEK293T
packaging cell lines. The supernatants containing lentivirus of
HEK293T were harvested at 36 and 72 h post-transfection.
Supernatants were pooled, centrifuged to remove cells and then
filtered through a 0.45 μm low protein binding filter. Cells were
plated in monolayer at different densities and infected with
lentivirus constructs using 8 ng/ml polybrene (Sigma, St. Louis,
MO, USA). The stable cell lines were selected in the presence of 1
μg/ml puromycin (Sigma) for two weeks.
Cell proliferation assay
To compare cell proliferation in cells with CIP2A
knockdown, the MTT assay was performed. The shRNA for CIP2A
lentivirus-transduced and control shRNA-transduced cells was plated
quadruplicate in 96-well plates at a density of
3×103/100 μl medium for each well. After a 3-day
culture, 15 μl of the dye solution was added to each well and
incubated at 37°C for another 4 h. At the end of the incubation,
100 μl stop solution was added to each well and colorimetric
absorbance was read in the SpectraMax Plus (Molecular Devices) at
570 nm. Cell free medium was used as mock control.
PP2A phosphatase activity assay
Measurement of PP2A phosphatase activity was
performed by using the RediPlate™ 96 EnzChek®
Serine/Threonine Phosphatase Assay kit (Life Technologies). Cell
lysates were prepared by using low detergent buffer (1% Nonidet
P-40, 10 mM HEPES, 150 mM NaCl, 10% glycerol, 1 mM PMSF, and
complete protease inhibitor cocktail). A total of 50 μl cell
lysates were incubated with 1X PP2A phosphatase reaction buffer for
30 min at 37°C. Fluorescence intensity was measured using
excitation at 355 nm and emission at 485 nm. The fluorescence
intensity was normalized to the expression level of PP2A catalytic
domain.
Western blot analysis
Cells were plated in 6-well tissue culture plates at
80% confluence and incubated overnight. Cell lysates were obtained
from transduced cells using cold radioimmunoprecipitation assay
buffer [20 mmol/l Tris-HCl (pH 8.0), 100 mmol/l NaCl, 10% glycerol,
1% NP40, 0.5% sodium deoxycholate]. Twenty micrograms of protein
mixture were separated on 10% SDS-PAGE gels and wet transferred to
nitrocellulose membrane (GE Healthcare Life Sciences) and then
blocked for 1 h at room temperature in TBS-T buffer [50 mmol/l
Tris-HCl (pH 7.5), 150 mmol/l NaCl, 0.1% Tween-20] containing 5%
non-fat milk. Membranes were then incubated overnight at 4°C or 1 h
at room temperature with the respective primary antibodies:
anti-CIP2A (1:500), and anti-actin (1:1,000; Santa Cruz
Biotechnology, Santa Cruz, CA, USA), phosphor-mTOR (1:1,000), mTOR
(1:1,000), phospho-AKT (S473) (1:1,000), (Cell Signaling
Technology, Danvers, MA, USA). Anti-mouse or anti-rabbit secondary
antibody-conjugated to horseradish peroxidase was used to visualize
the stained bands with an enhanced chemiluminescence visualization
kit (both from Santa Cruz Biotechnology).
Statistical analysis
All values from in vitro assays are expressed
as means ± SD or SEM of at least three independent experiments or
replicates. p-values were calculated with the two-tailed Student’s
t-test. A p-value <0.05 was considered to indicate a
statistically significant difference.
Results
CIP2A regulates lung cancer cell
proliferation
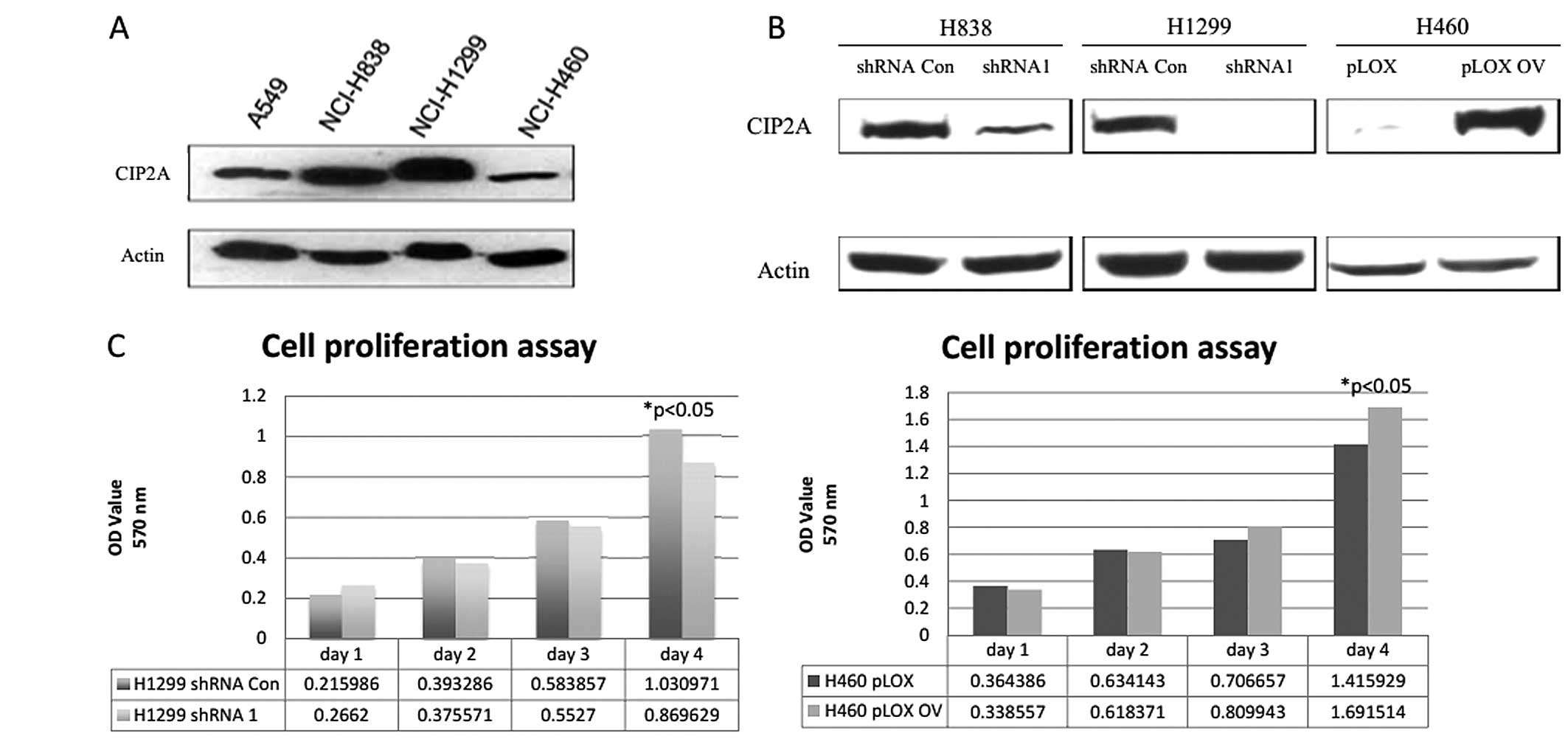
To establish the role of CIP2A in the cell
proliferation of lung cancer cells, we first examined the
expression of CIP2A in four different lung cancer cell lines, A549,
NCI-H838 (H838), NCI-H1299 (H1299) and NCI-H460 (H460). The four
NSCLC cell lines expressed apparently different levels of
endogenous CIP2A (Fig. 1A). We
generated five cell lines with wild-type CIP2A (pcDNA3.1 and shRNA
control), deficient CIP2A via stable transfection of two
independent shRNAs (CIP2A shRNA1 and CIP2A shRNA2) or overexpressed
CIP2A (pcDNA3.1+CIP2A) via transient transfection. The CIP2A
expression level in knockdown or overexpression cell lines was
confirmed by western blotting (Fig.
1B). The knockdown of CIP2A caused the decreased cell
proliferation while over-expression of CIP2A led to the increased
cell proliferation rate (Fig. 1C).
These data suggested the function of CIP2A in promoting lung cancer
cell proliferation.
CIP2A regulates cell proliferation
through epidermal growth factor (EGF)-stimulated AKT signaling
pathway
One of the most important growth factors associated
with lung cancer is the EGF (19).
Upon EGF binding to its receptor, the downstream MAPKs are
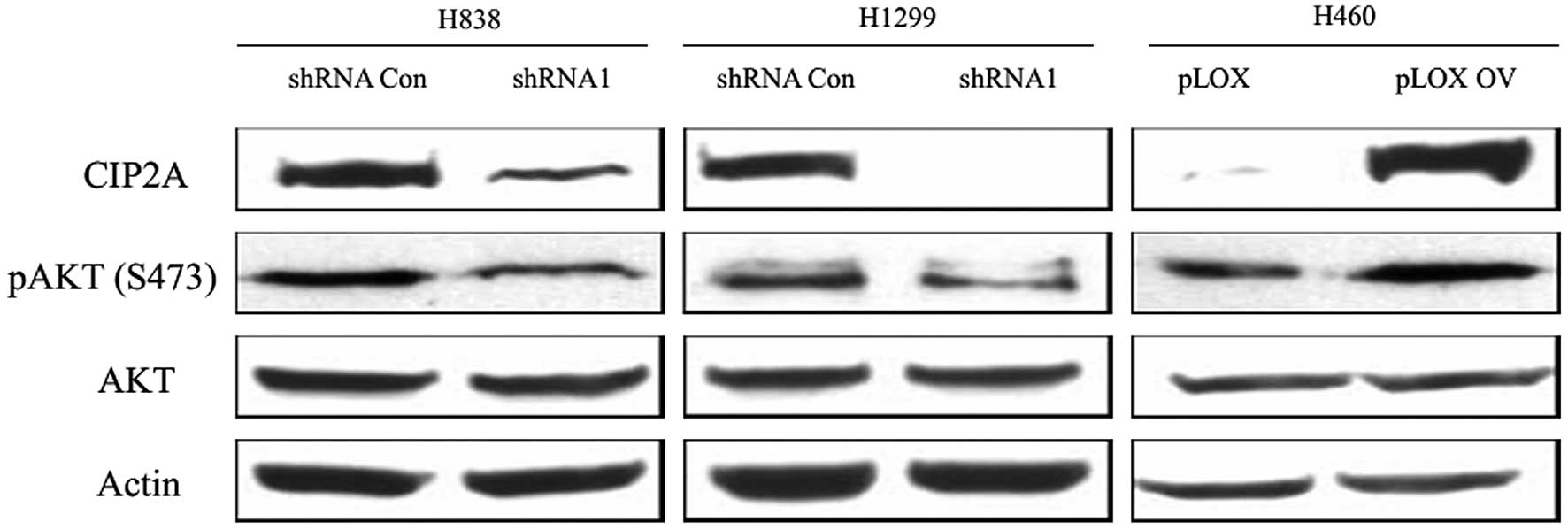
activated leading ultimately to cell proliferation. CIP2A can
decrease AKT associated PP2A phosphatase activity. In response to
EGF stimulation, AKT became phosphorylated at S473, a residue
indicated in enzymatic activity and substrate specificity. We
either depleted CIP2A using shRNA or elevated CIP2A expression
level via ectopic overexpression in these three cell lines. As
shown in Fig. 2, the knockdown of
CIP2A in H1299 and H838 caused the hypophosphorylation of AKT while
overexpression of CIP2A in H460 caused the hyperphosphorylation of
AKT. The total amount of AKT was unaffected by the level of CIP2A.
Therefore, CIP2A regulates AKT phosphorylation in lung cancer
cells.
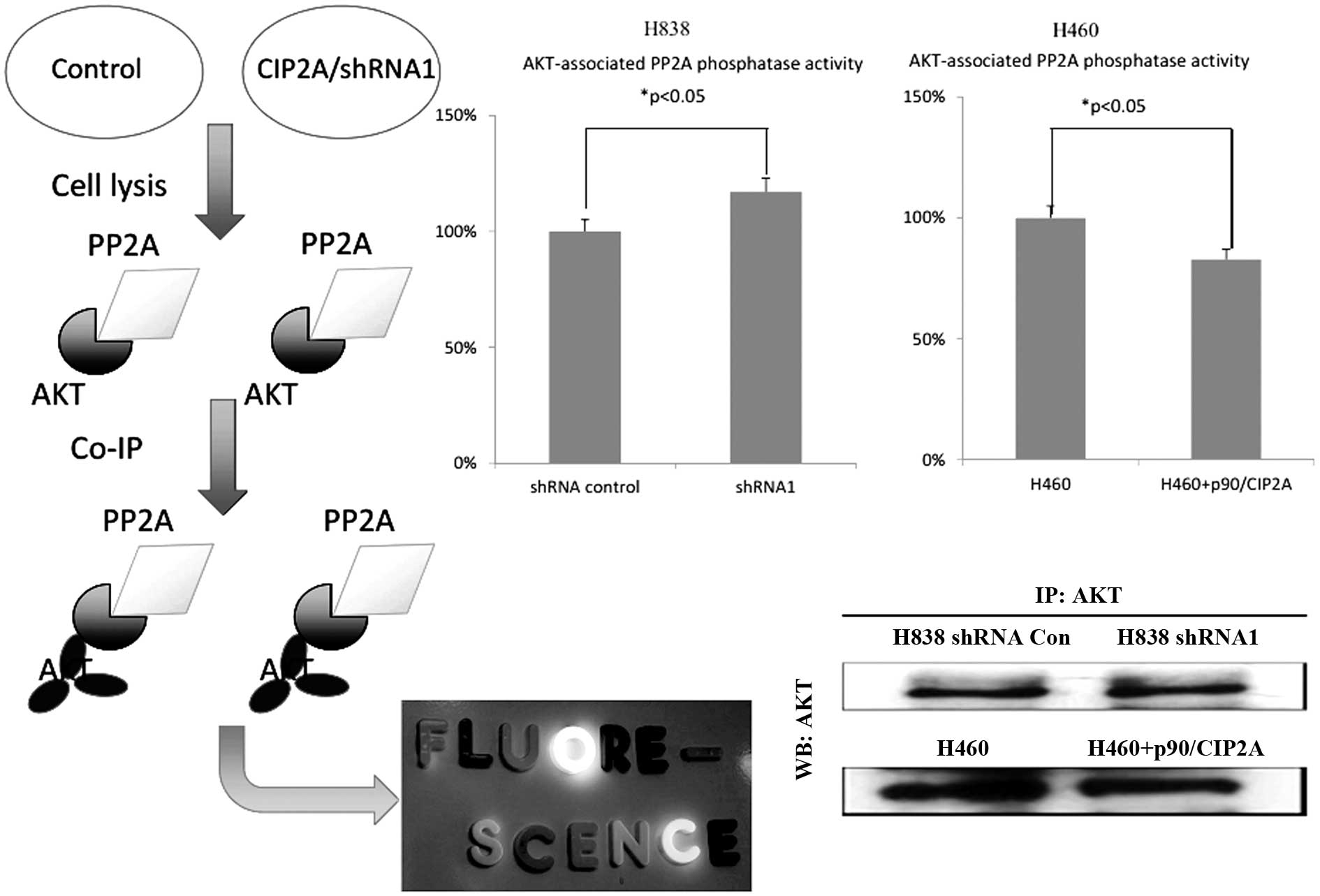
To investigate whether the downregulated AKT
phosphorylation is due to the elevated PP2A phosphatase activity,
we analyzed the AKT-associated PP2A phosphatase activity by
performing the PP2A phosphatase activity assay (Fig. 3). The knockdown of CIP2A increased
the AKT-associated PP2A phosphatase activity, while overexpression
of CIP2A downregulated AKT-associated PP2A phosphatase activity.
The results indicated that the regulation of CIP2A on
AKT-phosphorylation is associated with PP2A phosphatase
activity.
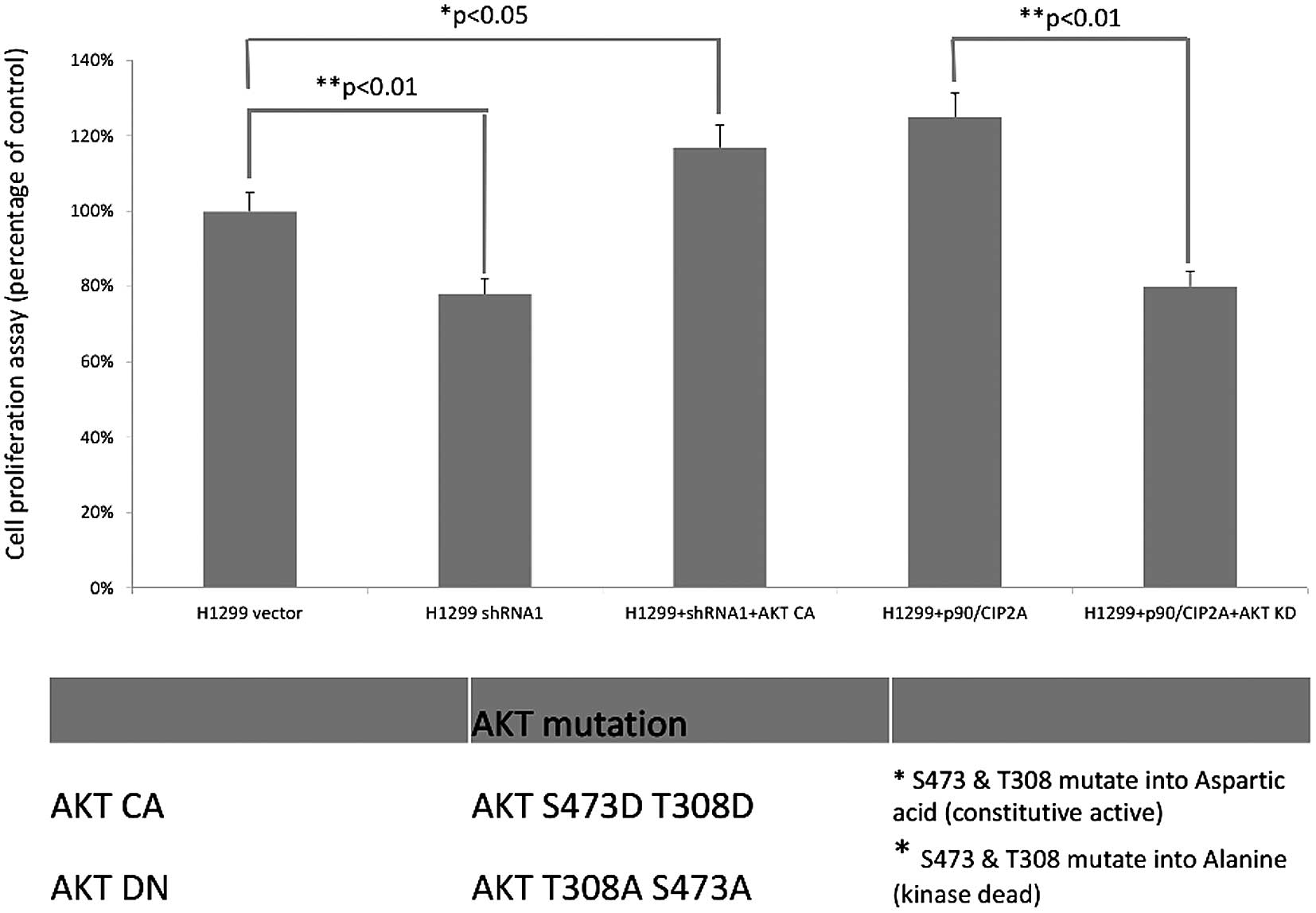
Since AKT is an important regulator in cancer cell
proliferation and tumor cell growth, we tested whether
CIP2A-mediated cell proliferation can be partly attributed to the
AKT phosphorylation. In the H1299 cell line, the knockdown of CIP2A
decreased cell proliferation to 78%. However, the introduction of
constitutively active AKT (AKT CA) rescued the decreased cell
proliferation to 119%. Furthermore, the expression of
dominant-negative AKT (AKT KD) therefore decreased the cell
proliferation imposed by the CIP2A overexpression (123 vs. 81%).
The introduction of these two AKT mutations provided direct
evidence that CIP2A promoted cell proliferation through the
regulation of AKT phosphorylation (Fig.
4).
CIP2A regulates mTOR phosphorylation and
its downstream effectors
Mammalian target of rapamycin (mTOR) is a downstream
target of AKT signaling pathway, which transmits the signaling to
promote protein synthesis and cell growth. mTOR functions by two
different complexes, mTOR complex 1 (mTORC1) and complex 2
(mTORC2). mTORC1 plays various roles in regulating cell growth,
proliferation and survival. It phosphorylates two downstream
factors S6 kinase and eukaryotic initiation factor 4E-binding
protein 1 (4E-BP1). Functions of mTORC2 have not been well studied,
but it is suggested to be part of downstream branch in the PI3K/AKT
pathway (20).
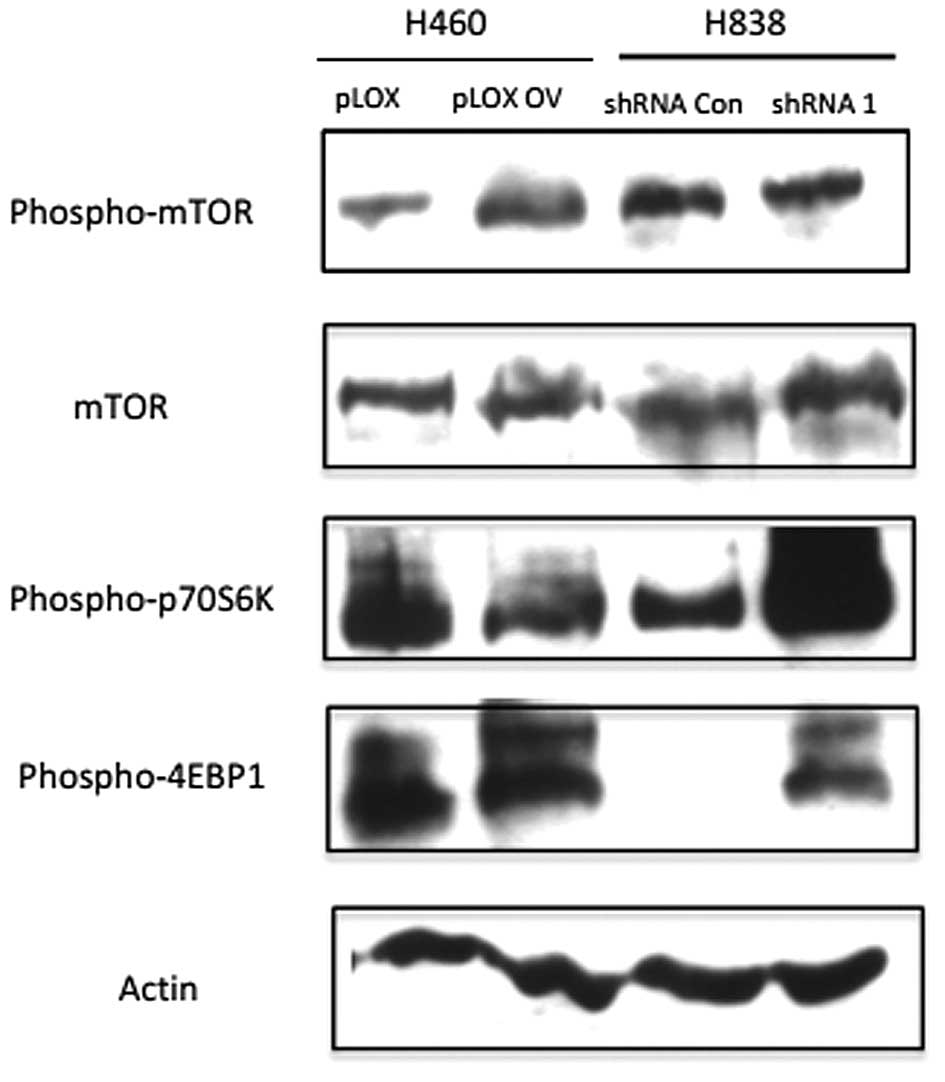
To test whether CIP2A-regulated AKT phosphorylation
would affect this signaling axis, we measured the phosphorylation
of mTOR at Serine 2441 (S2441). The knockdown of CIP2A in H838
decreased the phosphorylation of mTOR while the overexpression
caused the increased phosphorylation of mTOR (Fig. 5). These data suggested that CIP2A
influenced the mTOR phosphorylation.
Since the two downstream substrates, eukaryotic
initiation factor 4E-BP1 and P70 ribosomal S6 kinase 1 (P70S6K1),
play important roles in cell activity, we also detected whether
CIP2A is associated with any of mTOR downstream effectors. We found
that depletion of CIP2A upregulated phosphorylation of both P70S6K1
and 4E-BP1.
Discussion
Lung cancer causes the most cancer-related deaths in
both men and women in the USA (3).
Early diagnosis of lung cancer is technically difficult, since
there are few noticeable symptoms in the patients at the onset of
the disease (3). Although early
diagnosis cannot guarantee recovery, appropriate treatment can be
administered to prevent the advancement of this disease (8). Therefore, understanding the mechanisms
underlying lung cancer tumorigenesis is key for the development of
therapeutic targets.
Cancerous inhibitor of PP2A (CIP2A) was originally
identified as a tumor-associated autoantigen in gastric and liver
cancer (9). Previous studies have
indicated that CIP2A is an oncoprotein to promote cancer cell
proliferation through inhibition of c-myc associated PP2A
phosphatase activity (10,13). The expression of CIP2A is tightly
restricted to embryonic stage but often re-expressed in lung cancer
tissues. Studies in our laboratory have also demonstrated that
autoantibodies against this protein appear in high frequency in
sera from lung cancer patients. The expression of this protein also
has high frequency in lung cancer tissues. We proposed that CIP2A
may be a potential biomarker to be incorporated into existing
biomarker arrays for lung cancer diagnosis.
The functions of CIP2A in lung cancer have not been
fully understood yet and its clinical relevance has not yet been
established. According to our studies, we found the possible
interaction between CIP2A and AKT signaling pathway, which may help
to reveal the mechanism for CIP2A-promoted cancer progression. In
the present study, we firstly attempted to address the relationship
between CIP2A and AKT phosphorylation. The positive correlation
between CIP2A and AKT phosphorylation in our results clearly
demonstrated the possible regulation of AKT by CIP2A through
growth-factor stimulation. This is not unexpected since previous
studies had shown the role of CIP2A in promoting cell survival
through downregulating AKT-associated PP2A phosphatase activity.
Our studies not only confirmed the regulation under different
treatments but also indicated an alternative way by which CIP2A
promotes cancer progression, which is demonstrated by the altered
cell proliferation by the introduction of mutant AKT (AKT CA and
AKT DN). However, whether CIP2A physically interacts with AKT has
not yet been explored.
Resistance to rapamycin of cancer cells is the major
concern for the clinical use of this drug to treat cancer (21). Our preliminary data showed that
CIP2A regulates the phosphorylation status of mTOR, the target of
rapamycin and finally affects the sensitivity of cancer cells to
the treatment; however, how this process is actually executed has
yet to be determined. mTOR is the major component of the two
complexes, mammalian target of rapamycin complex 1 and mammalian
target of rapamycin complex 2 (mTORC1 and mTORC2). mTORC1 is a
direct target of AKT while mTORC2 is the upstream kinase of AKT
(22). mTORC1 is composed of mTOR,
MLST8 and PRAS40, two of which, mTOR and PRAS40, are the targets of
AKT. Therefore, CIP2A-mediated rapamycin sensitivity may be
dependent on the regulation of mTOR, PRAS40 or both, which need
further studies to be confirmed. In addition, our data also suggest
that CIP2A would affect the phosphorylation of downstream targets
of mTOR.
Collectively, our studies not only establish the
correlation between CIP2A and AKT phosphorylation but also reveal
novel functions of CIP2A in promoting AKT signaling cascade.
Although further studies are required to increase the resolution
for the identification of CIP2A-targeted AKT substrates as well as
the role of CIP2A in chemoresistance, based on our preliminary
data, we consider CIP2A a promising target for future drug design
since it is an effective regulator to restrain cancer cell growth.
The temporal expression of CIP2A will enable researchers to design
specific drugs to target CIP2A, which may result in fewer
side-effects. In addition, restoration of PP2A phosphatase activity
is another therapeutic strategy in cancer treatment. Therefore,
suppressing the expression of CIP2A may be an indirect way to
reverse tumorigenic phenotypes and to treat cancer patients more
efficiently.
Acknowledgements
The authors thank Dr Edward K.L. Chan (The
University of Florida, Gainesville, FL, USA) for his assistance and
support to this study. This study was supported by a grant
(SC1CA166016) from the National Institutes of Health (NIH). We
would also like to thank the Border Biomedical Research Center
(BBRC) Core facilities at The University of Texas at El Paso
(UTEP), funded by NIH grant (5G12MD007592), for their
assistance.
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar
|
|
2
|
Travis WD, Travis LB and Devesa SS: Lung
cancer. Cancer. 75(Suppl 1): 191–202. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fong KM, Sekido Y, Gazdar AF and Minna JD:
Lung cancer. 9: Molecular biology of lung cancer: clinical
implications. Thorax. 58:892–900. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Aviel-Ronen S, Blackhall FH, Shepherd FA
and Tsao MS: K-ras mutations in non-small-cell lung
carcinoma: a review. Clin Lung Cancer. 8:30–38. 2006.
|
|
5
|
Haura EB, Camidge DR, Reckamp K, et al:
Molecular origins of lung cancer: prospects for personalized
prevention and therapy. J Thorac Oncol. 5(Suppl 3): S207–S213.
2010. View Article : Google Scholar
|
|
6
|
Huff V: Wilms’ tumours: about tumour
suppressor genes, an oncogene and a chameleon gene. Nat Rev Cancer.
11:111–121. 2011.
|
|
7
|
Shackelford DB and Shaw RJ: The LKB1-AMPK
pathway: metabolism and growth control in tumour suppression. Nat
Rev Cancer. 9:563–575. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Soo Hoo L, Zhang JY and Chan EK: Cloning
and characterization of a novel 90 kDa ‘companion’ auto-antigen of
p62 overexpressed in cancer. Oncogene. 21:5006–5015. 2002.
|
|
9
|
Junttila MR, Puustinen P, Niemelä M, et
al: CIP2A inhibits PP2A in human malignancies. Cell. 130:51–62.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Khanna A, Pimanda JE and Westermarck J:
Cancerous inhibitor of protein phosphatase 2A, an emerging human
oncoprotein and a potential cancer therapy target. Cancer Res.
73:6548–6553. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Boehm JS, Hession MT, Bulmer SE and Hahn
WC: Transformation of human and murine fibroblasts without viral
oncoproteins. Mol Cell Biol. 25:6464–6474. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Drayton S, Rowe J, Jones R, et al: Tumor
suppressor p16INK4adetermines sensitivity of human cells
to transformation by cooperating cellular oncogenes. Cancer Cell.
4:301–310. 2003.
|
|
13
|
Westermarck J and Hahn WC: Multiple
pathways regulated by the tumor suppressor PP2A in transformation.
Trends Mol Med. 14:152–160. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Côme C, Laine A, Chanrion M, et al: CIP2A
is associated with human breast cancer aggressivity. Clin Cancer
Res. 15:5092–5100. 2009.PubMed/NCBI
|
|
15
|
Dong QZ, Wang Y, Dong XJ, et al: CIP2A is
overexpressed in non-small cell lung cancer and correlates with
poor prognosis. Ann Surg Oncol. 18:857–865. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Huang LP, Adelson ME, Mordechai E and
Trama JP: CIP2A expression is elevated in cervical cancer. Cancer
Biomark. 8:309–317. 2010.PubMed/NCBI
|
|
17
|
Vaarala MH, Väisänen MR and Ristimäki A:
CIP2A expression is increased in prostate cancer. J Exp Clin Cancer
Res. 29:1362010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen KF, Liu CY, Lin YC, et al: CIP2A
mediates effects of bortezomib on phospho-Akt and apoptosis in
hepatocellular carcinoma cells. Oncogene. 29:6257–6266. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Beckebaum S, Iacob S, Klein CG, et al:
Assessment of allograft fibrosis by transient elastography and
noninvasive biomarker scoring systems in liver transplant patients.
Transplantation. 89:983–993. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ren XS, Sato Y, Harada K, et al:
Activation of the PI3K/mTOR pathway is involved in cystic
proliferation of cholangiocytes of the PCK rat. PLoS One.
9:e876602014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Alayev A and Holz MK: mTOR signaling for
biological control and cancer. J Cell Physiol. 228:1658–1664. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kim DH, Sarbassov DD, Ali SM, et al: mTOR
interacts with raptor to form a nutrient-sensitive complex that
signals to the cell growth machinery. Cell. 110:163–175. 2002.
View Article : Google Scholar : PubMed/NCBI
|