Introduction
Breast cancer is one of the most common types of
cancer in industrial countries. Epidemiological studies indicate
that the incidence and mortality of breast cancer is up to 5-fold
higher in Western countries than in some Asian countries, and Asian
migrants to the USA eventually acquire the breast cancer incidence
of their host country, suggesting the importance of environmental
factors (1). Among the
environmental factors, it is clear that dietary exposure influences
breast cancer risk, and particularly dietary intake of fat may play
an important role in the genesis of breast cancer (2–5). Not
only the amount of fat consumption, but also the type of dietary
fat may have different effects on breast carcinogenesis. Long-chain
polyunsaturated fatty acids (PUFAs) have different effects on
mammary tumorigenesis based on the double bond position (6–8). The
first double bond located at the 3rd, 6th or 9th carbon from the
terminal methyl group of a fatty acid is called an n-3, n-6 or n-9
series fatty acid, respectively. Epidemiological studies and
preclinical data indicate a positive association between dietary
n-6 PUFA and breast cancer risk, while n-3 PUFA possesses
chemopreventive properties (9,10). n-3
PUFAs such as eicosapentaenoic acid (EPA; 20:5n-3) suppress human
breast cancer cell growth (11).
Among n-3 PUFAs, docosahexaenoic acid (DHA; 22:6n-2) suppresses
mammary carcinogenesis more effectively than EPA (7). In contrast, n-6 PUFAs such as linoleic
acid (LA; 18:2n-6) and arachidonic acid (AA; 20:4n-6) promote the
growth of human breast cancer cells (11,12).
The typical American high-fat diet contains high levels of LA
(13). Olive oil rich in oleic acid
(OA; 18:1n-9) suppresses mammary carcinogenesis (14). However, it is difficult to determine
if the effects of OA on experimental carcinogenesis are due to
specific biochemical properties of OA or simply due to the
substitution for n-6 PUFA (15).
The role of n-9 PUFA in relation to breast cancer has not been
studied in detail.
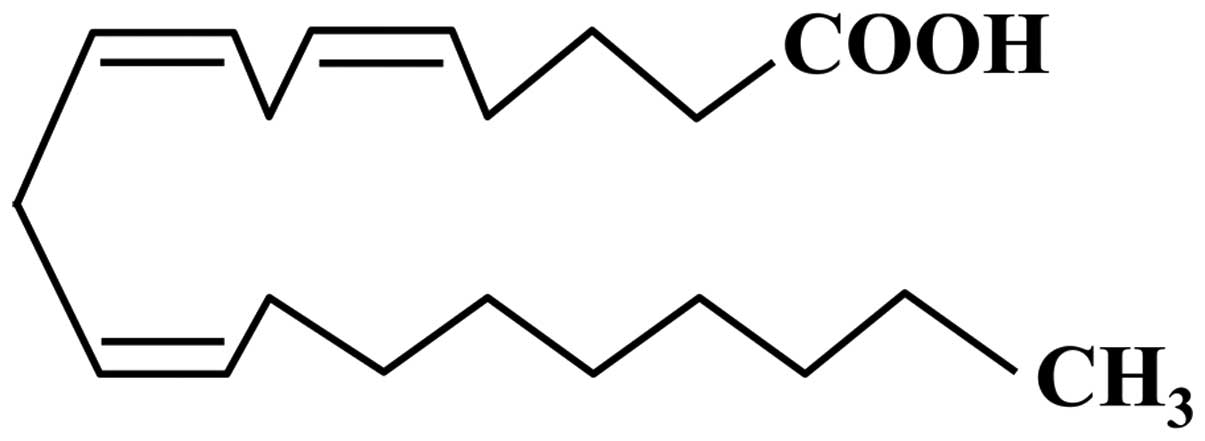
Mead acid (MA; also referred to as
5,8,11-eicosatrienoic acid), which was first characterized by James
F. Mead, is a carboxylic acid with a 20-carbon chain and three
methylene-interrupted cis double bonds, in which the first
double bond is located at the ninth carbon from the terminal methyl
group of a fatty acid (20:3n-9; Fig.
1). MA is a minor constituent of plasma and tissue in adult
mammals. Elongation and desaturation of OA take place to form MA
when n-6 and n-3 essential fatty acids, particularly LA, are
deficient. Therefore, MA elevation in the blood is an indication of
essential fatty acid deficiency. MA is found in large quantities in
cartilage; MA decreases osteoblastic activity for the maintenance
of cartilage to prevent ossification and suppresses angiogenesis to
maintain avascular status (16,17).
Angiogenesis plays an important role in the growth of breast cancer
(18), and anti-angiogenic agents
[inhibitors of vascular endothelial growth factor (VEGF)] and the
VEGF receptor (VEGFR) may be promising targets for breast cancer
control (19,20). The loss of cell adhesion is related
to cancer invasion and metastasis. MA is related to the expression
of the cell-cell adhesion molecule E-cadherin in human cancer cell
lines including breast cancer cells (21,22).
E-cadherin-mediated signaling can influence invasive and metastatic
behavior (23,24). VEGF and/or E-cadherin signaling may
modulate breast cancer growth at the primary site, and invasion and
metastasis in the MA-rich condition. In addition, leukotriene
B4 (LTB4) enhances tumor growth in human
cancer cells including breast cancer cells (25,26).
Dietary supplementation with MA suppresses LTB4 in rats
(27,28). Moreover, a nested case-control study
revealed an inverse association between MA and breast cancer risk
as well as overall cancer risk (29); in the present study, neither the
n-6/n-3 ratio nor AA intake correlated with breast cancer risk.
Collectively, MA may exert cancer preventive properties. However,
MA shows different effects on different types of cancer cells
(21,22). The present study was designed to
explore the effect of MA on KPL-1 human breast cancer cell growth
and metastasis. The mechanisms of action were investigated based on
VEGF and E-cadherin signaling and the modulation of fatty acid
composition.
Materials and methods
Cell line
KPL-1 is a human breast cancer cell line established
from the malignant effusion of a breast cancer patient (30). This cell line was derived from a
patient with recurrent breast cancer that appeared during
postsurgical adjuvant chemoendocrine therapy including tamoxifen
and medroxy-progesterone acetate. The KPL-1 cell line is estrogen
receptor (ER)-positive, progesterone receptor (PgR)-negative, and
human epidermal growth factor receptor-2 (HER2)-negative (luminal A
subtype) (31). Xenograft athymic
mice injected with KPL-1 cells often develop lymph node metastasis
(30). KPL-1 cells were cultured in
Dulbecco’s modified Eagle’s medium (DMEM; Sigma, St. Louis, MO,
USA) with 10% fetal bovine serum (FBS; Gibco-BRL, Grand Island, NY,
USA) in 5% CO2/95% humidified air at 37°C.
MA preparation
MA used for in vitro experiments was
purchased from Sigma and dissolved to 5 mg/500 μl (32.6 mM) in
ethanol and stored at −30°C. MA was diluted in DMEM containing 10%
FBS to achieve final concentrations of 16.25, 32.5, 75, 150 and 300
μM.
MTT assay
KPL-1 cells were seeded at 2×103
cells/well in 96-well plates in DMEM with 10% FBS. The cells were
treated with ethanol (final concentration, 0.9%) or the indicated
concentration of MA for up to 72 h. Cell proliferation was
monitored by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl
tetrazolium bromide (MTT) assay as previously described (32), and IC50 value was
calculated.
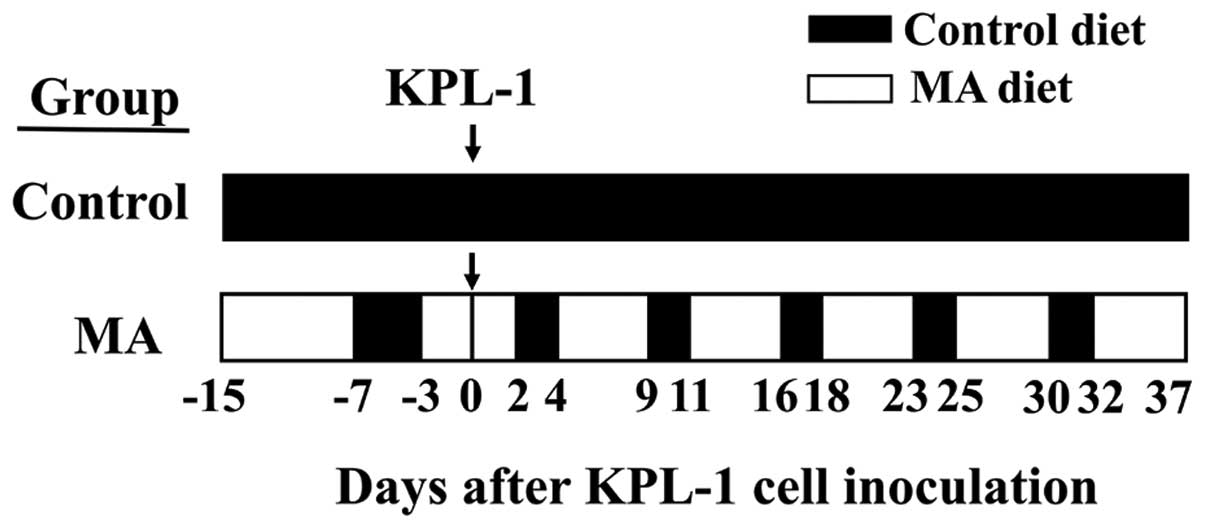
Animal experiments
Animals were housed in groups of four or five in
plastic cages with paper bedding (Paper Clean, SLC, Hamamatsu,
Japan) in a specific pathogen-free room maintained at 22±2°C and
60±10% relative humidity with a 12-h light/dark cycle (lights on at
8:00 AM and lights off at 8:00 PM). The mice were randomly divided
into two groups, the control diet group (n=15) and the MA diet
group (n=10). Both experimental diets were modifications of the
AIN-76 diet (33) and contained the
same amount of nutrients but had different fatty acid compositions
(Table I). The MA diet contained 5%
SUNTGM33 (a kind gift from Suntory Wellness, Tokyo, Japan), which
contains 48.0% MA (Table II).
SUNTGM33 is a microbial oil obtained by fungal fermentation
(34). Olive oil purchased from
Nacalai Tesque (Kyoto, Japan) was used for the control diet. The
fatty acid compositions of SUNTGM33 and olive oil are listed in
Table II. Each experimental diet
was formulated by Oriental Yeast (Tokyo, Japan). Four-week old
female athymic BALB/c mice purchased from Charles River Japan
(Kyoto, Japan) were fed either a control or MA diet starting at 4
weeks of age. In comparison to the control diet, the MA diet for 8
days starting at 4 weeks of age did not cause weight gain.
Therefore, the MA diet group was switched to the control diet for 4
successive days to accelerate weight gain; thereafter, the MA diet
group was treated with a cyclic feeding regime of 5 days on the MA
diet followed by 2 days on the control diet throughout the
experiment. The control group was treated with the control diet
throughout the experiment. Schematic representation of the in
vivo experiment is shown in Fig.
2. At 6 weeks of age, both groups were inoculated with
2.5×105 KPL-1 cells in 100 μl DMEM supplemented with 10%
FBS into the thoracic mammary fat pad (MA diet group was on the
third day of MA diet). During the experiment, the dose of diet
ingested, body weight and locally growing tumor volumes were
measured once per week. Tumor volume was calculated by using the
standard formula: width2 × length × 0.5. Thirty-seven
days after tumor cell inoculation (fifth day on MA diet), body
weight was measured, and bromodeoxyuridine (BrdU; Invitrogen,
Camarillo, CA, USA) was injected (50 mg/kg animal body weight) via
the abdominal cavity, blood was sampled and then animals were
sacrificed by exsanguination from aortic transection. At autopsy,
all organs were examined macroscopically, and the primary tumors
and local axillary lymph nodes were examined histologically; half
of the primary tumor was snap frozen and used for fatty acid
analysis. Tissues were fixed in 10% neutral buffered formalin,
embedded in paraffin, and stained with hematoxylin and eosin
(H&E). The study protocol and animal procedures were approved
by the Animal Care and Use Committee of Kansai Medical University
(permit no. 13-060). Throughout the experiments, animals were cared
for in accordance with the Guidelines for Animal Experimentation of
Kansai Medical University.
 | Table IComposition of experimental
diets. |
Table I
Composition of experimental
diets.
| MA diet | Control diet |
|---|
| Casein | 20 | 20 |
| DL-methionine | 0.3 | 0.3 |
| Cornstarch | 43 | 43 |
| α-cornstarch | 12 | 12 |
| Sucrose | 10 | 10 |
| Cellulose | 5 | 5 |
| AIN-76 mineral
mix | 3.5 | 3.5 |
| AIN-76 vitamin
mix | 1 | 1 |
| Choline
bitartrate | 0.2 | 0.2 |
| SUNTGM33 | 5 | 0 |
| Olive oil | 0 | 5 |
| Table IIFatty acid composition of SUNTGM33
and olive oil. |
Table II
Fatty acid composition of SUNTGM33
and olive oil.
| Fatty acid
composition (%) | SUNTGM33 | Olive oil |
|---|
| Myristic acid
(14:0) | 0.12 | 0.00 |
| Palmitic acid
(16:0) | 3.35 | 10.88 |
| Palmitoleic
(16:1n-7) | 0.00 | 0.85 |
| Stearic acid
(18:0) | 4.96 | 3.32 |
| Oleic acid
(18:1n-9) | 25.10 | 74.71 |
| Vaccenic acid
(18:1n-7) | 0.14 | 1.89 |
| Linoleic acid
(18:2n-6) | 0.42 | 6.90 |
| α-Linolenic acid
(18:3n-3) | 0.00 | 0.68 |
| Arachidic acid
(20:0) | 0.00 | 0.46 |
| Gondoic acid
(20:1n-9) | 4.95 | 0.31 |
| Mead acid
(20:3n-9) | 48.02 | 0.00 |
| Arachidonic acid
(20:4n-6) | 1.23 | 0.00 |
| Eicosapentaenoic
acid (20:5n-3) | 0.47 | 0.00 |
| Behenic acid
(22:0) | 2.15 | 0.00 |
| Lignoceric acid
(24:0) | 5.34 | 0.00 |
| Nervonic acid
(24:1n-9) | 2.42 | 0.00 |
| Total n-9 | 80.49 | 75.02 |
| Total n-3 | 0.47 | 0.68 |
| Total n-6 | 1.65 | 6.90 |
| n-6/n-3 | 3.53 | 10.19 |
| Total SFA | 15.91 | 14.66 |
| Total MUFA | 32.61 | 77.76 |
| Total PUFA | 50.15 | 7.58 |
Fatty acid analysis
The fatty acid composition of the total phospholipid
fraction of serum was determined. Total lipids were extracted by
the method of Bligh and Dyer (35).
The total phospholipid fraction was separated by thin-layer
chromatography. For an internal standard, 1,2-diheptadecanoyl-
sn-glycero-3-phosphocholine (Avanti Polar Lipids, Inc., Alabaster,
AL, USA) was added. Total phospholipid fractions were
transmethylated with HCl-methanol and then the fatty acid
composition was analyzed by gas chromatography (GC-2014; Shimadzu
Corporation, Kyoto, Japan) with a capillary column DB-225 (0.25 mm
× 30 m × 0.25 μM; J&M Scientific, Folsom, CA, USA). The entire
system was controlled with gas chromatography software (GCsolution;
Shimadzu Corporation). The fatty acid composition of the total
lipid fraction of KPL-1 tumors was determined. In brief, frozen
tumor tissues were thawed, minced and homogenized three times in 8
ml chloroform-methanol (2:1) by a polytron homogenizer (Kinematica,
Lucerne, Switzerland) for 10 sec. The fatty acid analysis of total
lipids in the tumor was performed by the same method as previously
mentioned (35,36).
Microvessel density and cell
kinetics
Microvessel density of primary KPL-1 tumors was
evaluated by anti-CD34 antiserum (Abcam, Cambridge, UK). The cell
kinetics (cell proliferation and cell death) in primary KPL-1
tumors were evaluated. Cell proliferation was evaluated by
anti-BrdU antibody (clone B44, 1:50; Becton-Dickinson, Franklin
Lakes, NJ, USA) by using an LSAB staining kit (Dako, Glostrup,
Denmark). Cell death was evaluated by anti-phospho-histone H2A.X
(γ-H2AX) antibody (Ser139, 1:100; Cell Signaling, Danvers, MA,
USA), an immunomarker of the DNA damage response. Each slide was
scanned with a high-resolution digital slide scanner (NanoZoomer
2.0 Digital Pathology; Hamamatsu Photonics, Hamamatsu, Japan) to
prepare digital images. The ndpi image files were opened in color
mode with NDP.view software (Hamamatsu Photonics). The images were
changed to jpeg files at ×40 magnification in five randomly
selected areas within each tumor that were used to analyze
immunohistochemical staining (37–39).
CD34, VEGF, VEGFR1, VEGFR2 and E-cadherin
immunohistochemistry
Cell blocks were prepared from KPL-1 cells cultured
with or without 214.2 μM MA for 72 h (IC50 value for 72
h). Cell blocks were prepared from cultured KPL-1 cells; cells were
centrifuged at 1,000 rpm for 5 min, and cell pellets were fixed in
10% neutral buffer formalin and embedded in paraffin. Antisera used
to detect CD34, VEGF, VEGFR1, VEGFR2 and E-cadherin were anti-CD34
antiserum (1:20; Abcam), anti-VEGF antiserum (1:50), anti-VEGFR1
antiserum (1:500) (both from Santa Cruz Biotechnology, Santa Cruz,
CA, USA) and anti-VEGFR2 antiserum (1:400) (Cell Signaling
Technology, Danvers, MA, USA), respectively, by using an LSAB
staining kit (Dako), and anti-E-cadherin antibody (NCH38 ready to
use) by using Histofine MAX-PO (both from Nichirei Biosciences,
Tokyo, Japan) according to the manufacturer’s instructions. The
reaction products were visualized with 3-3′-diaminobenzidine
tetrahydrochloride. CD34 immunohistochemistry was applied for
angiogenesis evaluation in KPL-1 tumor grown in athymic mice, and
VEGF, VEGFR1, VEGFR2 and E-cadherin immunohistochemistry,
respectively, were applied in cultured KPL-1 cells in cell
pellets.
Quantitation of VEGF, VEGFR1, VEGFR2 and
E-cadherin
KPL-1 cells cultured with or without 214.2 μM MA for
72 h (IC50 value for 72 h) were sampled. VEGF was
quantified in the culture supernatant, and VEGFR1, VEGFR2 and
E-cadherin were measured in the cell lysate. For cell lysate
preparation, after washing cells with PBS (−), cell pellets were
homogenized with RIPA buffer (Wako, Osaka, Japan), and the cell
lysate was centrifuged at 14,000 rpm for 15 min at 4°C to obtain
supernatant containing cell lysate. Protein concentrations were
measured by the DC protein assay kit (Bio-Rad, Hercules, CA, USA).
VEGF, VEGFR1, VEGFR2 and E-cadherin levels were measured by
enzyme-linked immunoabsorbent assay (ELISA) with a human VEGF assay
kit (IL-226; IBL, Fujioka, Japan), VEGFR1 human ELISA kit
(ab119613), VEGFR2 human ELISA kit (ab100665) (both from Abcam),
and E-cadherin human ELISA kit (R&D Systems, Minneapolis, MN,
USA), respectively, according to the manufacturers’ protocols. Each
protein standard or protein purified from each group was incubated
overnight at 4°C against VEGF or E-cadherin; 90 min at 37°C against
VEGFR1; or 150 min at room temperature against VEGFR2. After the
final colors were developed with the addition of each color
reagent, absorbance was measured at 450 nm. Cell sample
concentration was calculated from a standard curve and corrected
for protein concentration. The limits for detection for VEGF,
VEGFR1, VEGFR2 and E-cadherin were 8, 156, 70 and 39 pg/ml,
respectively.
Statistical analysis
Values are expressed as the means ± standard error
of the mean (SEM). Body weight, tumor volume, tumor weight, fatty
acid composition, CD34-positive area and the number of BrdU- and
γ-H2AX- positive cells per 1 mm2 among the groups were
analyzed by t-test. The incidence of metastasis was analyzed with
the χ2 test.
Results
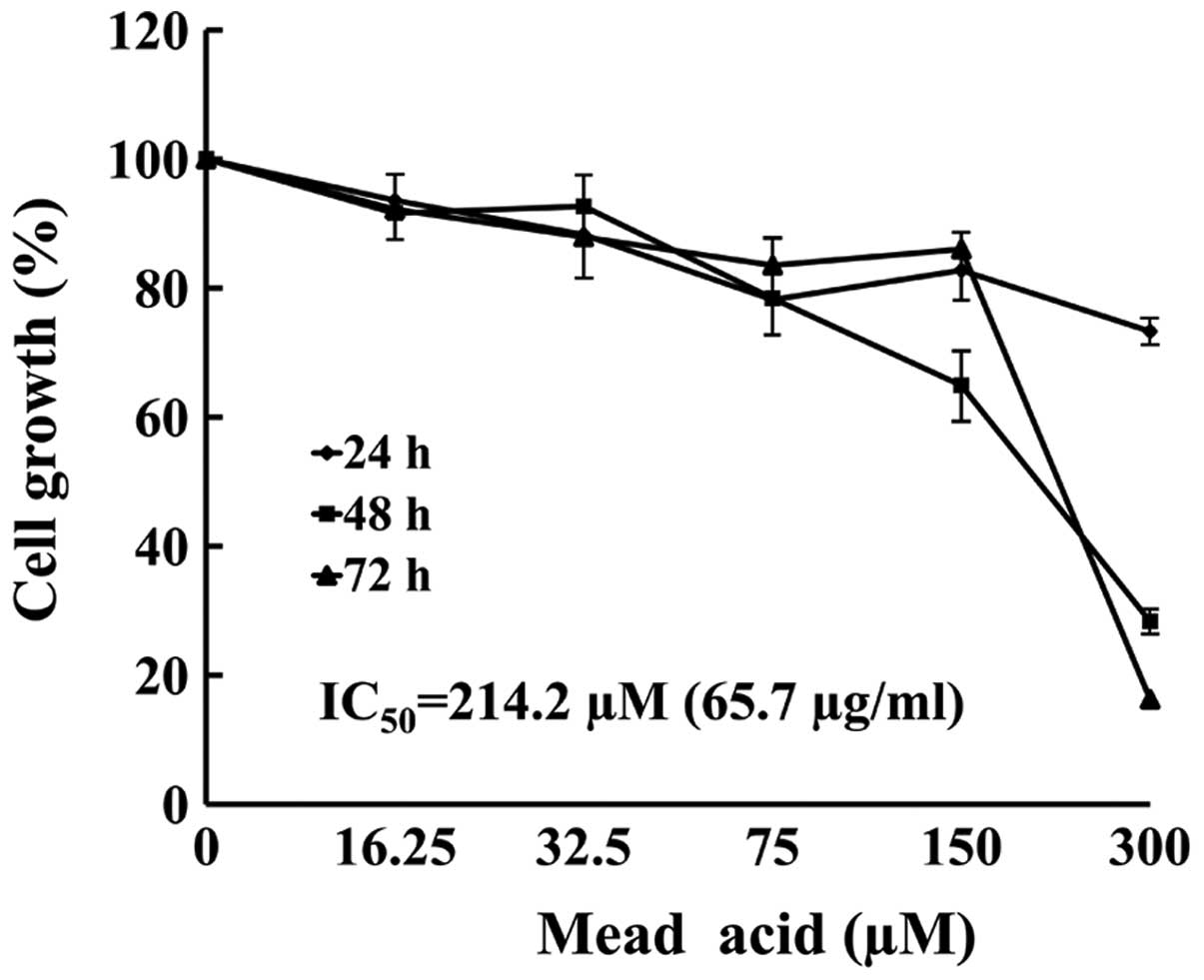
KPL-1 cell growth inhibition in
vitro
The KPL-1 cells were treated with 5 concentrations
(16.25–300 μM) of MA for up to 72 h. The MTT assay revealed that MA
induced growth inhibition in a dose- and time-dependent manner
(Fig. 3). The IC50 value
of MA against KPL-1 cells was 214.2 μM (65.7 μg/ml) for a 72-h
treatment.
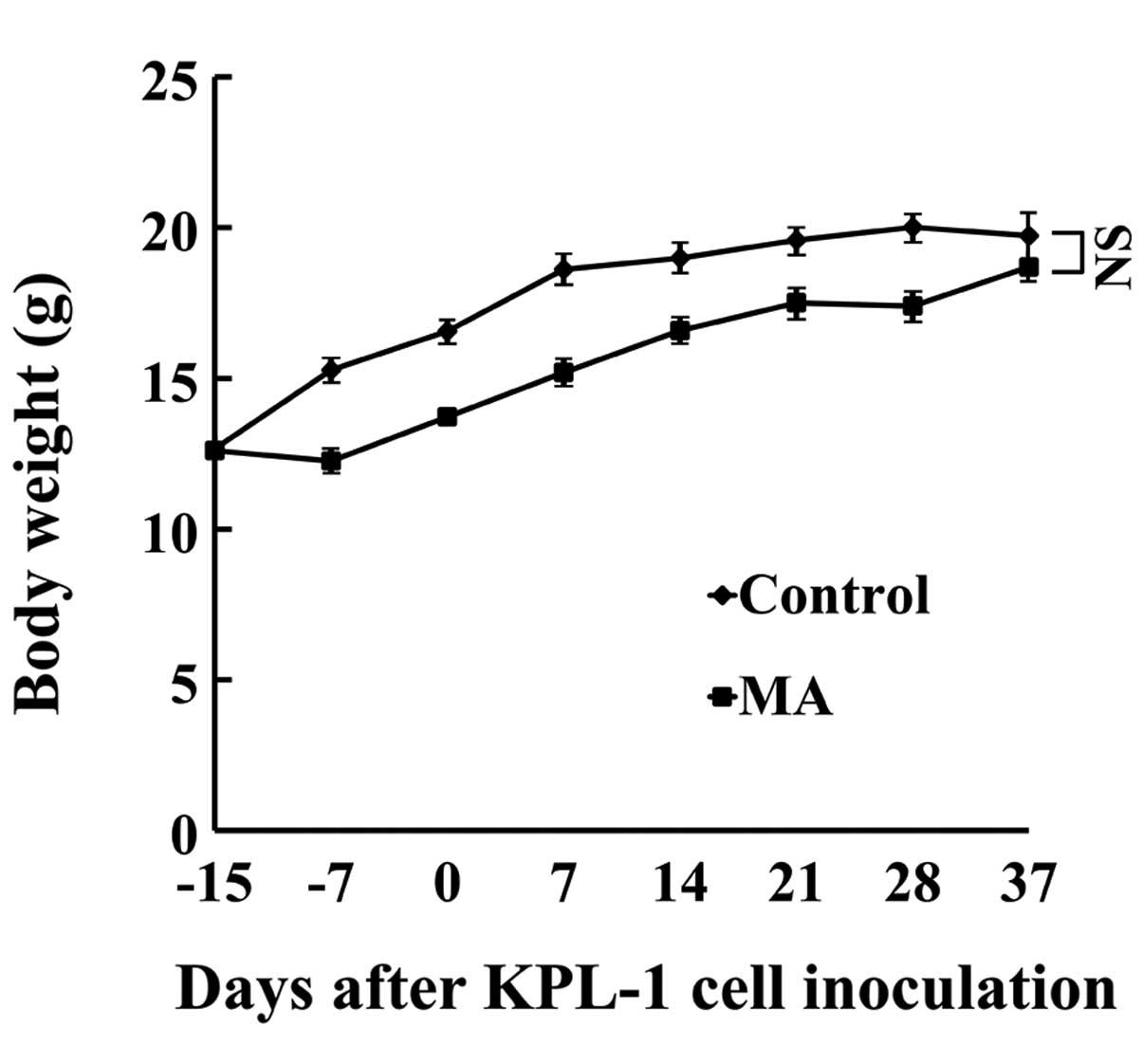
Host animals
During the experiment, the daily dose of MA
ingestion was 63–80 mg/mouse/day (mean, 74 mg/mouse/day). Body
weight gain in the MA group was significantly smaller during the
experiment but at the end of the experiment (eating the respective
diet for 8 weeks), the difference in body weight between the MA and
control diet group was not statistically significant (Fig. 4). No organs or tissues were
macroscopically abnormal. Throughout the experiment, the MA diet
group ingested less food than the control diet group (average,
92.1%).
Primary KPL-1 tumor growth and
metastasis
Locally growing KPL-1 tumors in the MA diet group
grew slower than in the control diet group. The final tumor volume
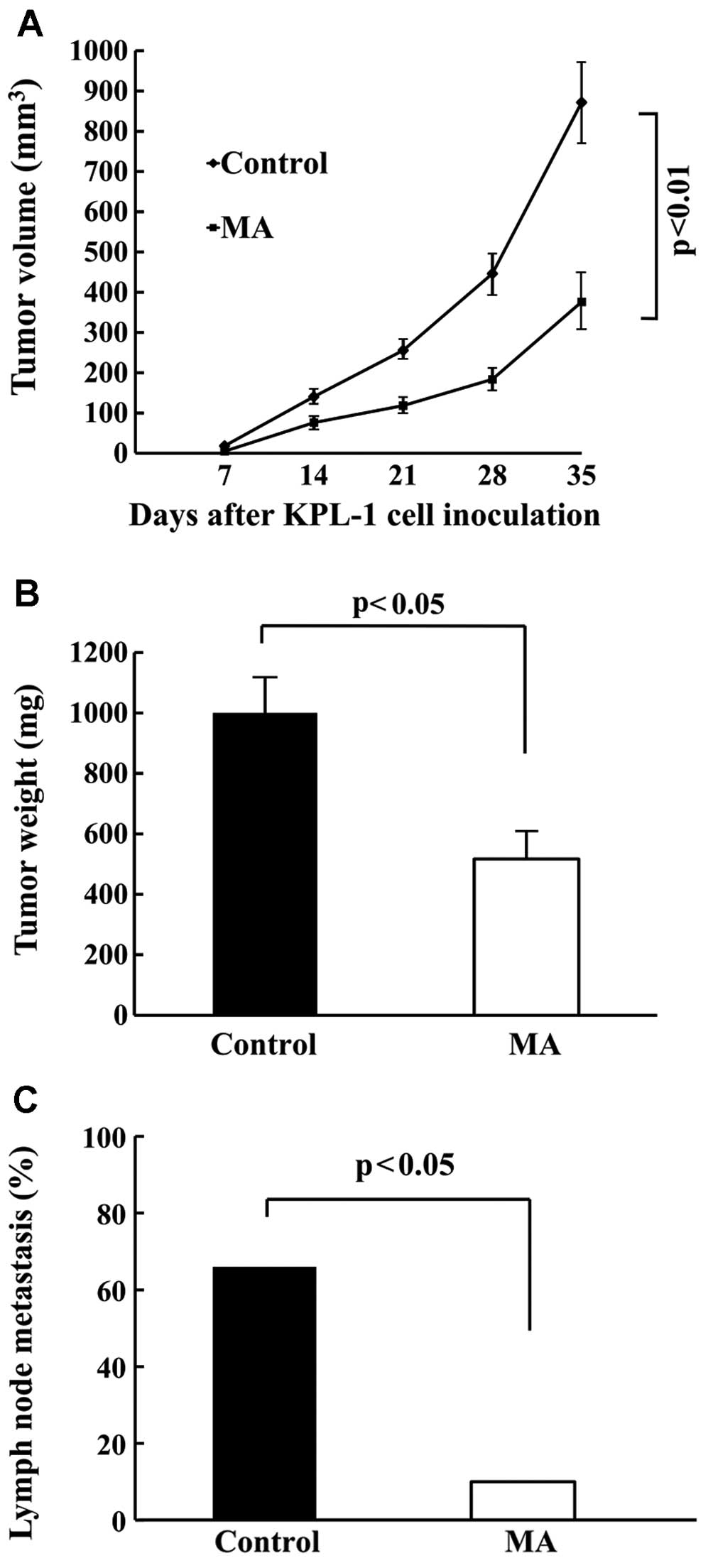
was significantly smaller (Fig. 5A)
and the final tumor weight was significantly lighter (Fig. 5B) in the MA diet group as compared
to the control diet group (p<0.01 and p<0.05, respectively).
Tumor volume two days before the termination of the experiment was
872±103 and 376±66 mm3, and the final tumor weight was
1,000±116 and 517±84 mg in the control and MA group, respectively.
Regional (axillary) lymph node metastasis was found in both groups
(Fig. 5C). Similarly, as compared
with the control diet group, metastasis was significantly
suppressed in the MA diet group (66.7%: 10/15 vs. 10%: 1/10).
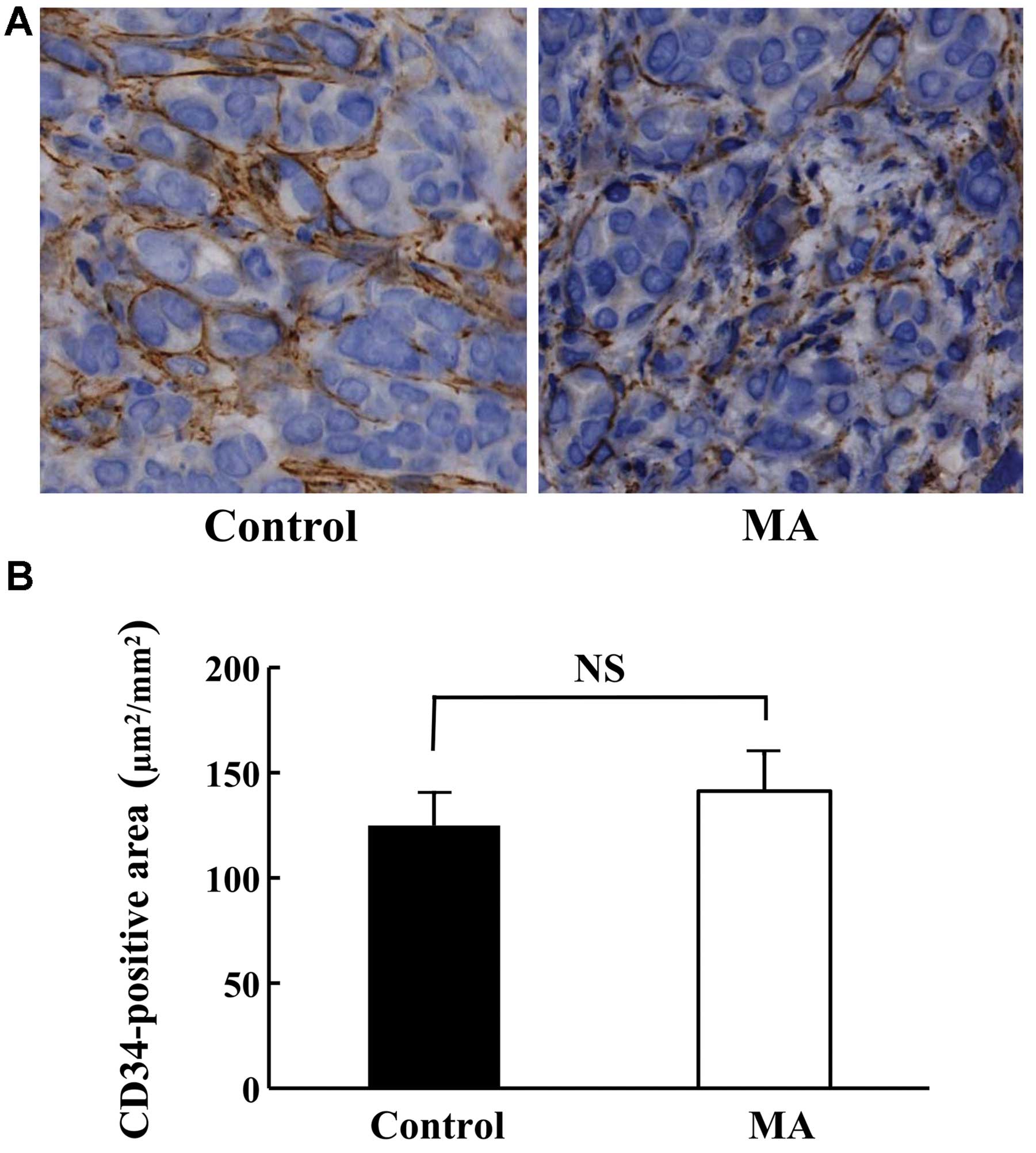
Morphology and angiogenesis of KPL-1
tumor
Although the growth of KPL-1 tumors was suppressed
in the MA diet group, the morphology was comparable in both groups
(data not shown). The angiogenesis of KPL-1 tumors at the primary
site (Fig. 6A) as well as the
CD34-stained area was compatible between the control and MA diet
groups (Fig. 6B).
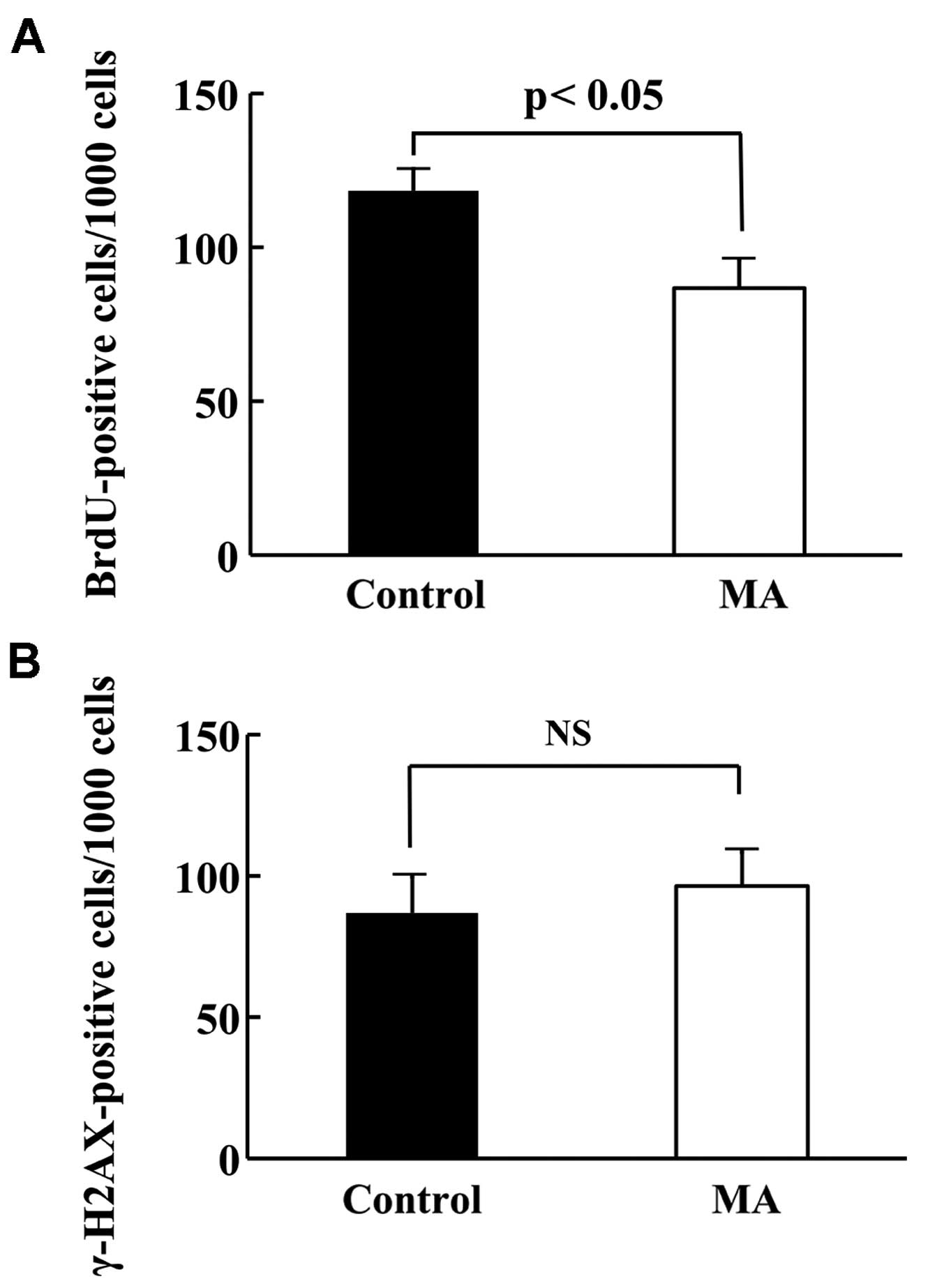
Proliferation and apoptotic ratio of
KPL-1 tumor
To compare the cell kinetics of KPL-1 tumors (cell
proliferation and cell death), the number of BrdU-positive cells
and γ-H2AX-positive cells per 1,000 cells in primary tumors from
the control and MA diet groups of mice was compared. The
proliferation and apoptotic ratios are shown in Fig. 7A and B, respectively. The
proliferation ratio in the control and MA diet groups was 11.8±0.7
and 8.7±0.9%, respectively (p<0.05), while the apoptotic ratio
in the control and MA diet groups was 8.7±1.3 and 9.6±1.2% (not
statistically significant).
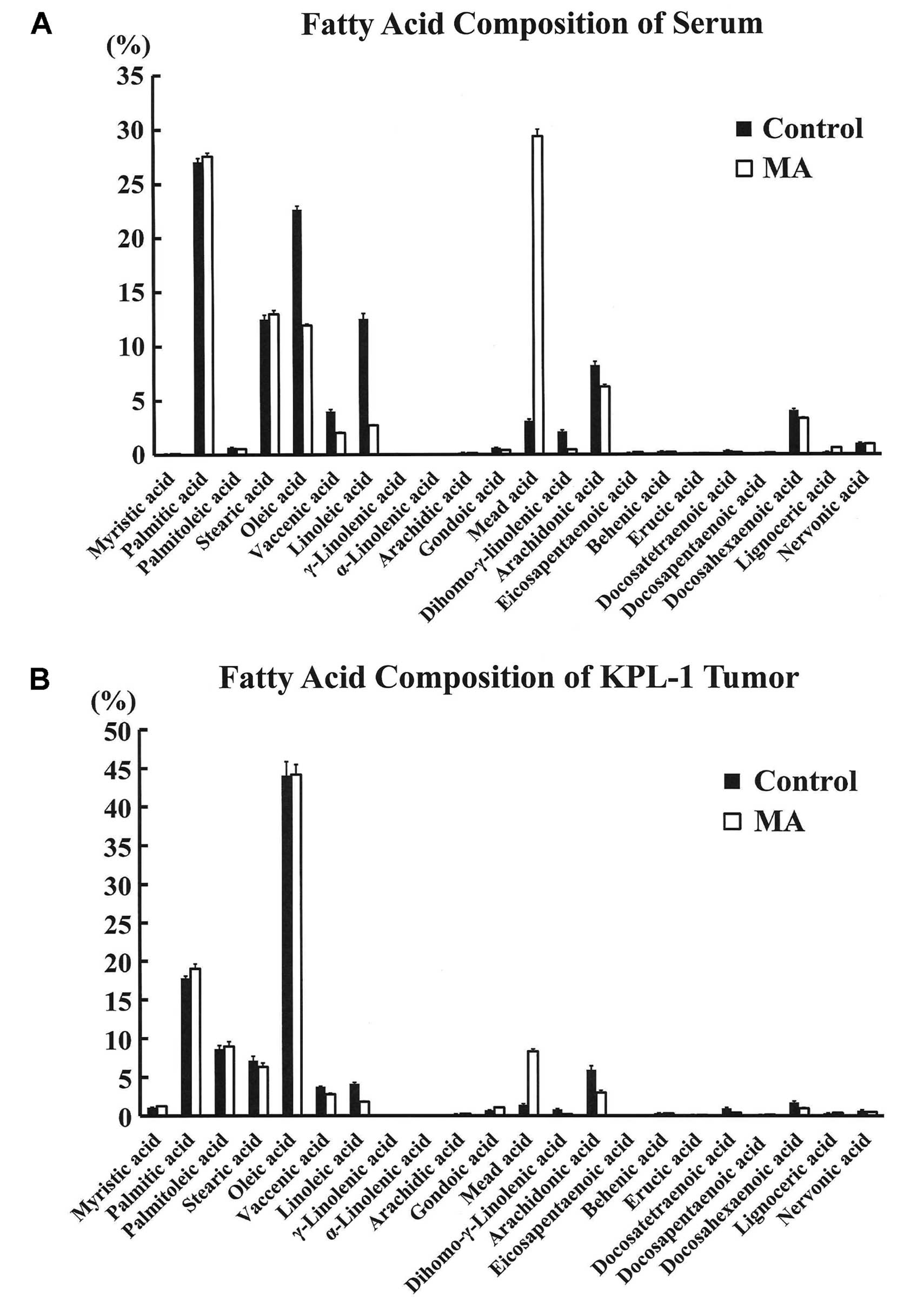
Fatty acid analysis
Fatty acid composition of sera and KPL-1 tumors in
the control and MA diet group is shown in Fig. 8A and B, respectively. In the sera,
the concentration of MA was significantly higher in the MA diet
group as compared to controls (237.2±17.6 vs. 23.8±1.1 μg/ml). In
contrast, LA and AA were significantly lower in the MA diet group
as compared with controls. In the KPL-1 tumor, the fatty acid
profile was generally similar to that of the fatty acid composition
in the serum. However, the serum OA composition was significantly
higher in the control diet group, as MA was replaced by OA in this
diet, while the OA concentration in KPL-1 tumors was comparable.
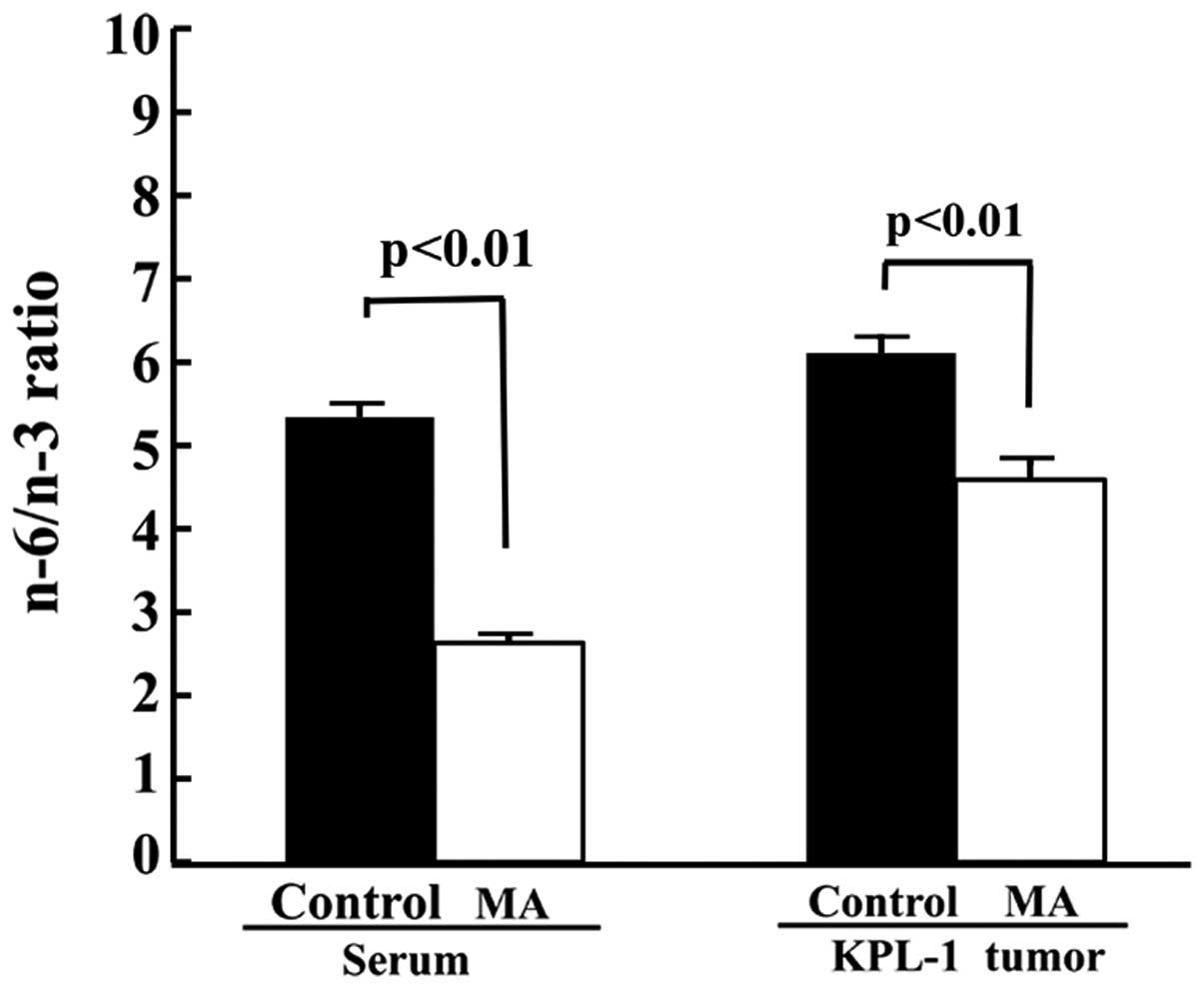
The MA diet significantly decreased the n-6/n-3 ratio in the sera
and the tumors (Fig. 9).
Expression of VEGF, VEGFR1, VEGFR2 and
E-cadherin in KPL-1 cells in culture
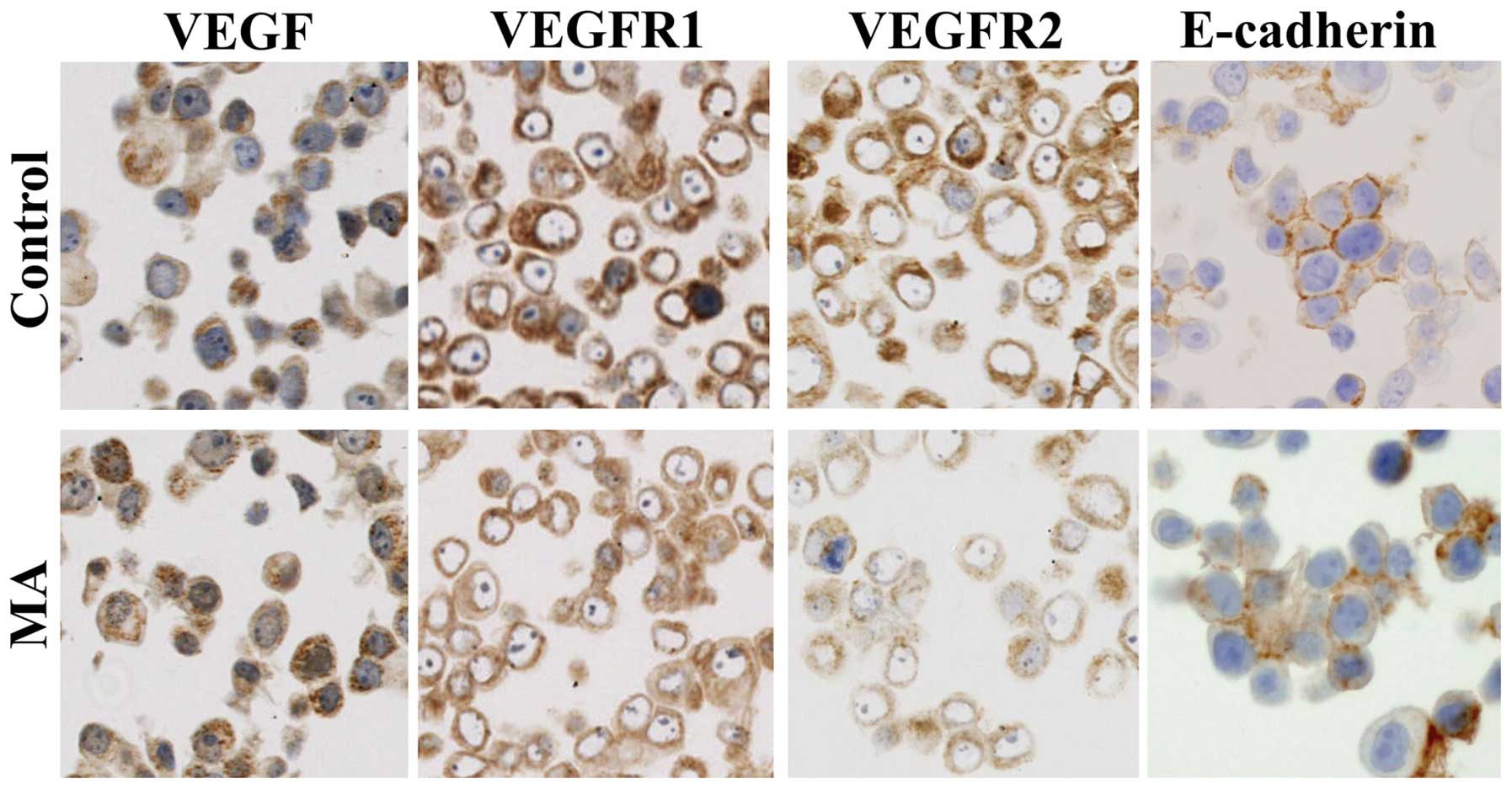
KPL-1 cells strongly expressed VEGF, VEGFR1 and
VEGFR2 in the cytoplasm and E-cadherin at the cell surface.
Although VEGF expression was unchanged, VEGFR1 and VEGFR2
expression tended to diminish and E-cadherin expression tended to
increase in KPL-1 cells treated with the IC50 dose of MA
for 72 h (214.2 μM) (Fig. 10).
Quantification of VEGF, VEGFR1, VEGFR2
and E-cadherin levels in KPL-1 cells in culture
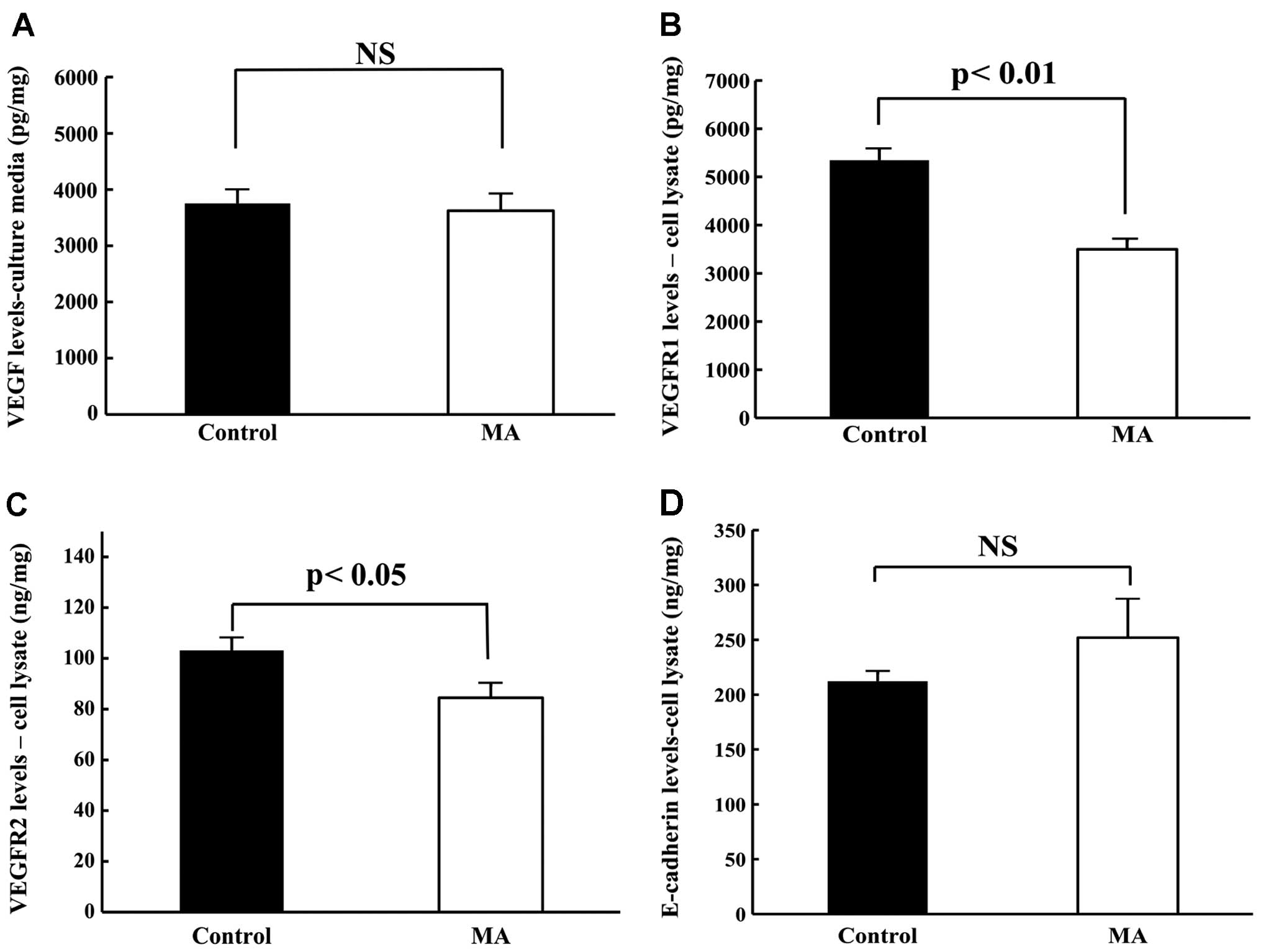
Levels of VEGF, VEGFR1, VEGFR2 and E-cadherin in
KPL-1 cells cultured with or without the IC50 dose of MA
for 72 h were compared. Although the VEGF levels in KPL-1 tumor
cells were similar (Fig. 11A), MA
treatment significantly decreased VEGFR1 and VEGFR2 levels and
tended to increase E-cadherin levels (Fig. 11B–D).
Discussion
In the present study, MA suppressed KPL-1 human
breast cancer cell growth in culture with an IC50 value
of 214.2 μM (65.7 μg/ml) for 72 h and significantly suppressed
KPL-1 tumor growth and regional (axillary) lymph node metastasis in
female athymic mice. Tumor suppression was due to decreased cell
proliferation. The body weight of the MA diet group, although it
was similar to the control diet-fed mice at the termination of the
present study, was significantly lighter during the experimental
period; the dose of food intake was less in the MA-fed mice. Thus,
the lighter body weight in the MA diet group may be due to
consuming a low amount of the MA diet. The serum MA level in MA
diet-fed mice was 237.2 μg/ml, which is higher than the
IC50 value. Thus, the IC50 dose of MA used in
the in vitro experiments is achievable and may not cause
serious side-effects in female mice. In contrast to n-3 PUFA, which
invariably exerts antiproliferation action on human tumor cell
lines, the n-9 series of MA causes different actions on different
human tumor cells, depending on their origin and cellular types
(21,22). MA may function in a cell-specific
manner. However, in agreement with MCF-7 human breast cancer cells,
the MA diet suppressed KPL-1 tumor growth and metastasis in female
athymic mice. Tumor growth is a balance between cell proliferation
and cell death. Mammary tumors of the VEGF knockout mice exhibit
cell-cycle arrest and induction of apoptosis (37). With the present dose of MA, the MA
diet suppressed KPL-1 tumor growth cytostatically by diminishing
cell proliferation; however, it was below the dose needed to cause
apoptosis.
Tumor angiogenesis is closely related to the growth
of breast cancer, and VEGF and its receptors are essential for
breast cancer growth (18–20). Cartilage is an avascular tissue and
contains high levels of MA; MA dose-dependently inhibits
VEGF-stimulated angiogenesis (17).
However, MA did not alter angiogenesis as evaluated by microvessel
density in KPL-1 tumors in athymic female mice. Although VEGF is
well known for its key roles in blood vessel growth, it also
promotes a range of other functions, such as cell adhesion,
survival, migration and invasion (40). VEGF, VEGFR1 and VEGFR2 were
immunohistochemically detected in KPL-1 cells. The presence of
VEGFR1 and VEGFR2 as well as VEGF on KPL-1 cells may raise the
possibility that VEGF may promote tumor growth not only by inducing
angiogenesis but directly through the activation of VEGFR1 and
VEGFR2 (41–43). In the present study, VEGF levels in
cultured KPL-1 cells treated with MA were comparable to the levels
in KPL-1 cells without MA treatment. However, VEGFR1 and VEGFR2
levels were significantly decreased in KPL-1 cells treated with MA.
As VEGF, VEGFR1 and VEGFR2 were co-localized in KPL-1 cells, the
present results suggest that VEGF signaling did not modulate
angiogenesis, however it directly modulated the growth of
VEGFR-positive tumor cells via an autocrine process, an endothelial
cell-independent pathway (44).
Expression of VEGF and its receptors (VEGF1 and VEGFR2) is
associated with poor outcome in breast cancer patients (45). VEGF knockout directly decreases the
proliferation of breast cancer cells (37).
Downregulation of E-cadherin is associated with loss
of cellular adhesiveness and initiates metastatic outgrowth
resulting in poor prognosis (46,47).
Downregulation of E-cadherin is required to initiate metastatic
outgrowth of breast cancer (47).
VEGF increases transcription factor Snail, which is associated with
breast cancer metastasis and reduces E-cadherin expression in
breast cancer cells (48). Although
MA tended to increase E-cadherin levels, the difference did not
reach statistical significance. VEGF increases the cellular
invasion of T-47D breast cancer cells on
Matrigel/fibronectin-coated transwell membranes (41). Therefore, VEGF, but not E-cadherin,
may contribute to the mechanisms of suppression of invasion and
metastasis in the KPL-1 tumor system.
In addition to affecting the VEGF pathway, MA may
function via other unknown mechanisms. In a human case-control
study of breast fat tissues, increased n-6/n-3 PUFA ratios and
decreased n-3 PUFA levels were found in breast cancer patients as
compared with controls (49,50).
Alteration in the n-6/n-3 PUFA ratio appears to be important
(51) and n-3 PUFA, namely EPA and
DHA (4) or n-6 PUFA, such as LA and
AA (11,12), is important. The MA diet-fed mice
had significantly decreased LA and AA levels, and the n-6/n-3 PUFA
ratio was significantly decreased in both the sera and the KPL-1
tumors, which may lead to growth suppression of KPL-1 cells. The
essential fatty acid LA cannot be synthesized in the body and must
be derived from the diet. Dietary LA shows a negative effect on the
incorporation of dietary MA into the plasma lipid fraction and
membrane phospholipids (52). LA is
converted via intermediates to dihomo-γ-linolenic acid and to AA.
Thus, an inverse relationship between n-9 PUFA MA and n-6 PUFA LA
and AA may exist. In the present MA diet, decreased expression of
n-6 PUFA, namely LA and AA, in the MA-fed mice, may suppress breast
cancer cell proliferation and invasion (4). However, n-6 content in the control
diet was considerably below 4%; n-6 PUFA (LA) at a dose of ~4% is
required for maximal acceleration of mammary tumorigenesis
(53).
In conclusion, MA effectively suppressed the growth
and metastasis of KPL-1 human breast cancer cells via VEGF
signaling.
Acknowledgements
The authors thank Ms. S Takebe (University of
Toyama) for her technical assistance and Ms. A Shudo (Kansai
Medical University) for manuscript preparation. This study was
supported in part by a Grant-in-Aid for Scientific Research (C)
(24592611) from the Ministry of Education, Culture, Sports, Science
and Technology of Japan, and by a grant from the Kiyoko Wada
Foundation of the Kansai Medical University Alumni Association
(2012).
References
|
1
|
Adami HO, Signorello LB and Trichopoulos
D: Towards an understanding of breast cancer etiology. Semin Cancer
Biol. 8:255–262. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tsubura A, Uehara N, Kiyozuka Y and
Shikata N: Dietary factors modifying breast cancer risk and
relation to time of intake. J Mammary Gland Biol Neoplasia.
10:87–100. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tsubura A, Yuri T, Yoshizawa K, Uehara N
and Takada H: Role of fatty acids in malignancy and visual
impairment: epidemiological evidence and experimental studies.
Histol Histopathol. 24:223–234. 2009.PubMed/NCBI
|
|
4
|
Chénais B and Blanckaert V: The Janus face
of lipids in human breast cancer: how polyunsaturated fatty acids
affect tumor cell hallmarks. Int J Breast Cancer.
2012:7125362012.PubMed/NCBI
|
|
5
|
de Lorgeril M and Salen P: New insights
into the health effects of dietary saturated and omega-6 and
omega-3 polyunsaturated fatty acids. BMC Med. 10:502012.PubMed/NCBI
|
|
6
|
Bartsch H, Nair J and Owen RW: Dietary
polyunsaturated fatty acids and cancers of the breast and
colorectum: emerging evidence for their role as risk modifiers.
Carcinogenesis. 20:2209–2218. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yuri T, Danbara N, Tsujita-Kyutoku M,
Fukunaga K, Takada H, Inoue Y, Hada T and Tsubura A: Dietary
docosahexaenoic acid suppresses N-methyl-N-nitrosourea-induced
mammary carcinogenesis in rats more effectively than
eicosapentaenoic acid. Nutr Cancer. 45:211–217. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wen ZH, Su YC, Lai PL, Zhang Y, Xu YF,
Zhao A, Yao GY, Jia CH, Lin J, Xu S, Wang L, Wang XK, Liu AL, Jiang
Y, Dai YF and Bai XC: Critical role of arachidonic acid-activated
mTOR signaling in breast carcinogenesis and angiogenesis. Oncogene.
32:160–170. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Patterson RE, Flatt SW, Newman VA,
Natarajan L, Rock CL, Thomson CA, Caan BJ, Parker BA and Pierce JP:
Marine fatty acid intake is associated with breast cancer
prognosis. J Nutr. 141:201–206. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Signori C, El-Bayoumy K, Russo J, Thompson
HJ, Richie JP, Hartman TJ and Manni A: Chemoprevention of breast
cancer by fish oil in preclinical models: trials and tribulations.
Cancer Res. 71:6091–6096. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Senzaki H, Iwamoto S, Ogura E, Kiyozuka Y,
Arita S, Kurebayashi J, Takada H, Hioki K and Tsubura A: Dietary
effects of fatty acids on growth and metastasis of KPL-1 human
breast cancer cells in vivo and in vitro. Anticancer Res.
18:1621–1627. 1998.PubMed/NCBI
|
|
12
|
Chang NW, Wu CT, Chen DR, Yeh CY and Lin
C: High levels of arachidonic acid and peroxisome
proliferator-activated receptor-alpha in breast cancer tissues are
associated with promoting cancer cell proliferation. J Nutr
Biochem. 24:274–281. 2013. View Article : Google Scholar
|
|
13
|
Taioli E, Nicolosi A and Wynder EL:
Dietary habits and breast cancer: a comparative study of United
States and Italian data. Nutr Cancer. 16:259–265. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cohen LA, Epstein M, Pittman B and
Rivenson A: The influence of different varieties of olive oil on
N-methylnitrosourea (NMU)-induced mammary tumorigenesis. Anticancer
Res. 20:2307–2312. 2000.PubMed/NCBI
|
|
15
|
Rose DP: Dietary fatty acids and cancer.
Am J Clin Nutr. 66(Suppl 4): 998S–1003S. 1997.PubMed/NCBI
|
|
16
|
Hamazaki T, Suzuki N, Widyowati R,
Miyahara T, Kadota S, Ochiai H and Hamazaki K: The depressive
effects of 5,8,11-eicosatrienoic acid (20:3n-9) on osteoblasts.
Lipids. 44:97–102. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hamazaki T, Nagasawa T, Hamazaki K and
Itomura M: Inhibitory effect of 5,8,11-eicosatrienoic acid on
angiogenesis. Prostaglandins Leukot Essent Fatty Acids. 86:221–224.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hicklin DJ and Ellis LM: Role of the
vascular endothelial growth factor pathway in tumor growth and
angiogenesis. J Clin Oncol. 23:1011–1027. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fox SB, Generali DG and Harris AL: Breast
tumour angiogenesis. Breast Cancer Res. 9:2162007. View Article : Google Scholar
|
|
20
|
Sharma PS, Sharma R and Tyagi T:
VEGF/VEGFR pathway inhibitors as anti-angiogenic agents: present
and future. Curr Cancer Drug Targets. 11:624–653. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Eynard AR, Jiang WG and Mansel RE:
Eicosatrienoic acid (20:3 n-9) inhibits the expression of
E-cadherin and desmoglein in human squamous cell carcinoma in
vitro. Prostaglandins Leukot Essent Fatty Acids. 59:371–377. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Heyd VL and Eynard AR: Effects of
eicosatrienoic acid (20:3 n-9, Mead’s acid) on some
promalignant-related properties of three human cancer cell lines.
Prostaglandins Other Lipid Mediat. 71:177–188. 2003.
|
|
23
|
Jones J and Walker R: Cell-cell and
cell-stromal interactions in breast cancer invasion and metastasis.
Int J Oncol. 11:609–616. 1997.PubMed/NCBI
|
|
24
|
Scully OJ, Bay BH, Yip G and Yu Y: Breast
cancer metastasis. Cancer Genomics Proteomics. 9:311–320. 2012.
|
|
25
|
Pidgeon GP, Lysaght J, Krishnamoorthy S,
Reynolds JV, O’Byrne K, Nie D and Honn KV: Lipoxygenase metabolism:
roles in tumor progression and survival. Cancer Metastasis Rev.
26:503–524. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hu N, Li Y, Zhao Y, Wang Q, You JC, Zhang
XD and Ye LH: A novel positive feedback loop involving
FASN/p-ERK1/2/5-LOX/LTB4/FASN sustains high growth of breast cancer
cells. Acta Pharmacol Sin. 32:921–929. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
James MJ, Gibson RA, Neumann MA and
Cleland LG: Effect of dietary supplementation with n-9
eicosatrienoic acid on leukotriene B4 synthesis in rats:
a novel approach to inhibition of eicosanoid synthesis. J Exp Med.
178:2261–2265. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cleland LG, Gibson RA, Neumann MA,
Hamazaki T, Akimoto K and James MJ: Dietary (n-9) eicosatrienoic
acid from a cultured fungus inhibits leukotriene B4
synthesis in rats and the effect is modified by dietary linoleic
acid. J Nutr. 126:1534–1540. 1996.PubMed/NCBI
|
|
29
|
Pouchieu C, Chajès V, Laporte F,
Kesse-Guyot E, Galan P, Hercberg S, Latino-Martel P and Touvier M:
Prospective associations between plasma saturated, monounsaturated
and polyunsaturated fatty acids and overall and breast cancer risk
- modulation by antioxidants: a nested case-control study. PLoS
One. 9:e904422014. View Article : Google Scholar
|
|
30
|
Kurebayashi J, Kurosumi M and Sonoo H: A
new human breast cancer cell line, KPL-1 secretes tumour-associated
antigens and grows rapidly in female athymic nude mice. Brit J
Cancer. 71:845–853. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kurebayashi J, Kanomata N, Moriya T,
Kozuka Y, Watanabe M and Sonoo H: Preferential antitumor effect of
the Src inhibitor dasatinib associated with a decreased proportion
of aldehyde dehydrogenase 1-positive cells in breast cancer cells
of the basal B subtype. BMC Cancer. 10:5682010. View Article : Google Scholar
|
|
32
|
Kanematsu S, Uehara N, Miki H, Yoshizawa
K, Kawanaka A, Yuri T and Tsubura A: Autophagy inhibition enhances
sulforaphane-induced apoptosis in human breast cancer cells.
Anticancer Res. 30:3381–3390. 2010.PubMed/NCBI
|
|
33
|
No authors listed. Report of the American
Institute of Nutrition Ad Hoc Committee on Standards for
Nutritional Studies. J Nutr. 107:1340–1348. 1977.PubMed/NCBI
|
|
34
|
Sakuradani E, Kamada N, Hirano Y,
Nishihara M, Kawashima H, Akimoto K, Higashiyama K, Ogawa J and
Shimizu S: Production of 5,8,11-eicosatrienoic acid by a Δ5 and Δ6
desaturation activity-enhanced mutant derived from a Δ12
desaturation activity-defective mutant of Mortierella alpina
1S-4. Appl Microbiol Biotechnol. 60:281–287. 2002.
|
|
35
|
Bligh EG and Dyer WJ: A rapid method of
total lipid extraction and purification. Can J Biochem Physiol.
3:911–917. 1959. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lai YC, Hamazaki K, Yoshizawa K, Kawanaka
A, Kuwata M, Kanematsu S, Hamazaki T, Takada H and Tsubura A:
Short-term pregnancy hormone treatment of
N-methyl-N-nitrosourea-induced mammary carcinogenesis
in relation to fatty acid composition of serum phospholipids in
female Lewis rats. In Vivo. 24:553–560. 2010.PubMed/NCBI
|
|
37
|
Schoeffner DJ, Matheny SL, Akahane T,
Factor V, Berry A, Merlino G and Thorgeirsson UP: VEGF contributes
to mammary tumor growth in transgenic mice through paracrine and
autocrine mechanisms. Lab Invest. 85:608–623. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kanematsu S, Yoshizawa K, Uehara N, Miki
H, Sasaki T, Kuro M, Lai YC, Kimura A, Yuri T and Tsubura A:
Sulforaphane inhibits the growth of KPL-1 human breast cancer cells
in vitro and suppresses the growth and metastasis of
orthotopically transplanted KPL-1 cells in female athymic mice.
Oncol Rep. 26:603–608. 2011.PubMed/NCBI
|
|
39
|
Redon CE, Weyemi U, Parekh PR, Huang D,
Burrell AS and Bonner WM: γ-H2AX and other histone
post-translational modifications in the clinic. Biochim Biophys
Acta. 1819:743–756. 2012.
|
|
40
|
Perrot-Applanat M and Di Benedetto M:
Autocrine functions of VEGF in breast tumor cells: adhesion,
survival, migration and invasion. Cell Adh Migr. 6:547–553. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Price DJ, Miralem T, Jiang S, Steinberg R
and Avraham H: Role of vascular endothelial growth factor in the
stimulation of cellular invasion and signaling of breast cancer
cells. Cell Growth Differ. 12:129–135. 2001.PubMed/NCBI
|
|
42
|
Weigand M, Hantel P, Kreienberg R and
Waltenberger J: Autocrine vascular endothelial growth factor
signalling in breast cancer. Evidence from cell lines and primary
breast cancer cultures in vitro. Angiogenesis. 8:197–204. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wu Y, Hooper AT, Zhong Z, Witte L, Bohlen
P, Rafii S and Hicklin DJ: The vascular endothelial growth factor
receptor (VEGFR-1) supports growth and survival of human breast
carcinoma. Int J Cancer. 119:1519–1529. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Guo S, Colbert LS, Fuller M, Zhang Y and
Gonzalez-Perez RR: Vascular endothelial growth factor receptor-2 in
breast cancer. Biochim Biophys Acta. 1806:108–121. 2010.PubMed/NCBI
|
|
45
|
Ghosh S, Sullivan CA, Zerkowski MP,
Molinaro AM, Rimm DL, Camp RL and Chung GG: High levels of vascular
endothelial growth factor and its receptors (VEGFR-1, VEGFR-2,
neuropilin-1) are associated with worse outcome in breast cancer.
Hum Pathol. 39:1835–1843. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Howard EM, Lau SK, Lyles RH, Birdsong GG,
Umbreit JN and Kochhar R: Expression of e-cadherin in high-risk
breast cancer. J Cancer Res Clin Oncol. 131:14–18. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wendt MK, Taylor MA, Schiemann BJ and
Schiemann WP: Downregulation of epithelial cadherin is required to
initiate metastatic outgrowth of breast cancer. Mol Biol Cell.
22:2423–2435. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wanami LS, Chen HY, Peiró S, García de
Herreros A and Bachelder RE: Vascular endothelial growth factor-A
stimulates Snail expression in breast tumor cells: implications for
tumor progression. Exp Cell Res. 314:2448–2453. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhu ZR, Agren J, Männistö S, Pietinen P,
Eskelinen M, Syrjänen K and Uusitupa M: Fatty acid composition of
breast adipose tissue in breast cancer patients and in patients
with benign breast disease. Nutr Cancer. 24:151–160. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Maillard V, Bougnoux P, Ferrari P, Jourdan
ML, Pinault M, Lavillonnière F, Body G, Le Floch O and Chajès V:
N-3 and N-6 fatty acids in breast adipose tissue and relative risk
of breast cancer in a case-control study in Tours, France. Int J
Cancer. 98:78–83. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Okuyama H, Kobayashi T and Watanabe S:
Dietary fatty acids - the n-6/n-3 balance and chronic elderly
diseases. Excess linoleic acid and relative n-3 deficiency syndrome
seen in Japan. Prog Lipid Res. 35:409–457. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Cleland LG, Neumann MA, Gibson RA,
Hamazaki T, Akimoto K and James MJ: Effect of dietary n-9
eicosatrienoic acid on the fatty acid composition of plasma lipid
fractions and tissue phospholipids. Lipids. 31:829–837. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Ip C, Carter CA and Ip MM: Requirement of
essential fatty acid for mammary tumorigenesis in the rat. Cancer
Res. 45:1997–2001. 1985.PubMed/NCBI
|