Introduction
Hepatocellular carcinoma (HCC), the predominant form
of primary liver cancer, is one of the most prevalent malignancies
and the third leading cause of cancer-related mortality worldwide
with ~700,000 deaths each year (1).
The incidence of HCC is dramatically increasing, and to date there
are no effective chemoprevention or systemic treatments available.
Therefore, it is of utmost importance to elucidate the molecular
mechanisms of HCC. An increasing number of studies have indicated
that chronic inflammation plays a crucial role in the occurrence
and development of HCC (2–4), yet the precise mechanisms are still
unclear.
Prostaglandin E2 (PGE2), a
bioactive lipid which is produced predominantly from arachidonic
acid by cyclooxygenases (COXs) and prostaglandin E synthases
(PGES), is generally considered to be a potent pro-inflammatory
mediator (5). Furthermore,
substantial evidence has shown that PGE2 is also
associated with several serious human diseases, including malignant
tumors (6,7). PGE2 may promote cancer cell
growth, adhesion, invasion, metastasis and angiogenesis (8), and thus participate in the
tumorigenesis and progression of numerous human cancers, such as
HCC (9–13), breast (14,15),
gastrointestinal (16) and prostate
cancer (17). PGE2
exerts diverse biological effects through four cognate E prostanoid
receptors (EP receptor) from EP1 to EP4 (5,18),
among which, the EP4 receptor is believed to be closely associated
with cancer cell proliferation and invasion in many types of human
cancer (19–23).
Moreover, the activation or overexpression of
protooncogenes is involved in HCC (24). The c-myc proto-oncogene is
the human cellular homologue of avian myelocytomatosis viral
oncogene (v-myc), encoding a transcription factor c-Myc
protein that upregulates the expression of many target genes and
thus promotes cell proliferation and tumorigenesis. In addition,
c-myc expression itself is regulated at multiple levels
including transcription, post-transcription and post-translation
(25,26). In HCC, the activation or
overexpression of the c-myc proto-oncogene has been well
documented (27,28). In addition, research supports a
central role for c-Myc in human hepatocarcinogenesis (29). Thus, it is vital to explore the
detailed molecular mechanisms of c-myc activation in
HCC.
Given our previous results showing that
PGE2 could notably enhance the cell growth and invasive
ability of HCC cells (10–13), and the involvement of c-myc
activation in hepatocarcinogenesis, the present study was designed
to evaluate our hypothesis that PGE2 may promote the
cell growth and invasion of HCC cells through upregulation of c-Myc
protein expression. Data from the present study revealed that
PGE2 significantly upregulated the expression of c-Myc
at the mRNA and protein levels both via the EP4 receptor and the
coupled GS/AC/cAMP/PKA/CREB signaling pathway, thus
promoting cell growth and invasiveness in HCC cells.
Materials and methods
Materials
The human HCC cell line Huh-7 was obtained from the
American Type Culture Collection (ATCC; Manassas, VA, USA).
Dulbecco’s modified Eagle’s medium (DMEM) and Lipofectamine 2000
were from Life Technologies (Grand Island, NY, USA).
PGE2, PGE1 alcohol, GW627368X and the cyclic
AMP EIA kit were from Cayman Chemical Co. (Ann Arbor, MI, USA).
SQ22536, forskolin, H89, actinomycin D (Act D) and cycloheximide
(CHX) were from Sigma-Aldrich (St. Louis, MO, USA). Cell Counting
Kit-8 (CCK-8) was from Dojindo Laboratories (Kumamoto, Japan). High
Pure RNA isolation kit was from Roche (Mannheim, Germany).
PrimeScript RT reagent kit was from Takara (Dalian, China).
Anti-c-Myc (no. 5605), anti-CREB (no. 9104), anti-phosphorylated
CREB (no. 9198, Ser133) antibodies were from Cell Signaling
Technology (Danvers, MA, USA); anti-β-actin mouse monoclonal
antibody (BM0627) was from Boster (Wuhan, China), and
anti-β-tubulin antibody (no. 21335) was from SAB (Signalway
Antibody; College Park, MD, USA). The protein assay dye reagent was
from Bio-Rad (Hercules, CA, USA). ECL Prime Western Blotting
Detection reagent was from GE Healthcare (Piscataway, NJ, USA). The
Transwell unit was from Corning (Cambridge, MA, USA). Matrigel was
from BD Biosciences (San Jose, CA, USA).
Cell line and culture
Human HCC Huh-7 cells were cultured in DMEM with 10%
fetal calf serum (FCS) at 37°C in a humidified 5% CO2
incubator. The experiments were performed when cells reached 80%
confluency and were conducted in serum-free medium.
Cell proliferation assay
Cell proliferation was determined using the CCK-8
kit from Dojindo Laboratories. This kit contains the WST-8 reagent,
a tetrazolium salt that can be reduced by mitochondrial
dehydrogenases in viable cells to produce an orange colored
formazan dye. Briefly, 600 μl of cell suspension (1×105
cells) was plated in each well of 24-well plates. After a 24-h
culture to allow reattachment, the cells were then incubated with
different treatments at the indicated concentrations and time
periods. Cell proliferation reagent, WST-8 (60 μl), was
subsequently added to each well. The incubation was continued from
30 min to 4 h at 37°C and absorbance at 450 nm was measured using
an automatic ELISA plate reader.
Cell invasion assays
Cell invasion assays were performed in
Matrigel-coated 24-well Transwell units. Cells (5×104)
were added to the upper Transwell chamber and media with 10% FCS
were added to the lower Transwell chamber. The serumfree media plus
pharmacological agents were added in the upper Transwell chamber.
After 24 h of incubation at 37°C, the cells were fixed with ethanol
and then stained by 0.1% crystal violet for 30 min at room
temperature. After washing the wells with PBS, the cells on the
upper surface of the filter were removed with a cotton swab. The
invaded cells on the lower surface of the membrane were solubilized
with 10% acetic acid for 10 min and quantified by measuring the
absorbance at 550 nm.
Western blotting
Cells were treated with different pharmacological
agents at 37°C for various times, as indicated in the experiments.
The cells were collected into lysis buffer (50 mM Tris-HCl pH 7.4,
150 mM NaCl, 0.5% sodium deoxycholate, 1% Nonidet P-40, 0.1% SDS, 1
mM PMSF, sodium orthovanadate, sodium fluoride and aprotinin) and
placed on ice for 30 min. Cell lysates were sonicated on ice for at
least 30 sec and then cleared by centrifugation at 15,000 × g for
30 min at 4°C. Protein concentrations of lysates were measured
using Bio-Rad protein assay dye reagent. Equal amounts of proteins
(40 μg) were separated by SDS-PAGE and transferred onto a
nitrocellulose membrane. Membranes were blocked with 5% non-fat
milk-TBST buffer for 1 h at room temperature and incubated with the
corresponding primary antibodies overnight at 4°C with gentle
shaking. Then, the membranes were washed by TBST and incubated for
2 h with the peroxidase-conjugated secondary anti-rabbit or
anti-mouse antibodies at room temperature. The signals were
detected using enhanced chemiluminescent reagent (ECL) and analyzed
using Image Lab 4.0 analysis software from Bio-Rad.
RNA isolation and real-time PCR
Total RNA from the cultured cells was isolated using
the High Pure RNA isolation kit according to the manufacturer’s
instructions. Reverse transcription was carried out with the
PrimeScript RT reagent kit according to the standard protocol. For
quantification of mRNA expression, real-time PCR was performed
using the following primer pairs: c-Myc, 5′-AGGCTATTCTGCCCATTT-3′
(forward) and 5′-TCGTAGTCGAGGTCATAGTTC-3′ (reverse); EP4,
5′-CATCTTACTCATTGCCACCT-3′ (forward) and 5′-TACTGAGCACTGTCTTTCTC-3′
(reverse); GAPDH, 5′-TTCCAGGAGCGAGATCCCT-3′ (forward), and
5′-CACCCATGACGAACATGGG-3′ (reverse). Real-time PCR analysis was
performed on Roche LightCycler Nano instrument using FastStart
Essential DNA Green Master Mix from Roche, and GAPDH was used as
the endogenous control. PCR conditions were pre-incubation at 95°C
for 10 min (1 cycle) followed by 40 cycles of 95°C for 20 sec, 60°C
for 20 sec and 72°C for 20 sec. All treatments and conditions were
performed in triplicate to calculate the statistical
significance.
siRNA interference
The siRNA targeting human EP4 receptor (siRNA ID:
s11455) was purchased from Life Technologies. The siRNA reagents
specific to c-Myc (no. 6341) and CREB (no. 6588) were from Cell
Signaling Technology. Huh-7 cells (2×105) were plated in
6-well plates for 24 h, resulting in a 30–50% confluent cell
monolayer. The cells were then transfected with the targeting siRNA
or the negative control siRNA (N.C. siRNA) from GenePharma
(Shanghai, China) using Lipofectamine 2000. After transfection,
depletion of target protein was confirmed by western blotting or
real-time PCR analysis, and the cells were subsequently used for
further experiments.
cAMP assay
Intracellular cAMP levels were measured using an
enzyme immunoassay kit. Briefly, Huh-7 cells were cultured in 35-mm
dishes until they reached 80% confluency and were then treated with
PGE1 alcohol or vehicle at 37°C for different times. The
cells were harvested in 0.1 M HCl solution, and incubated for 20
min at room temperature. After centrifugation at 1,000 × g for 10
min, the supernatant (50 μl) was analyzed for cAMP content
according to the manufacturer’s instructions.
Statistical analysis
Data are presented as means ± SD. P-values were
calculated using the Student’s t-test for unpaired samples with MS
Excel software. The results were considered significantly different
at P<0.05.
Results
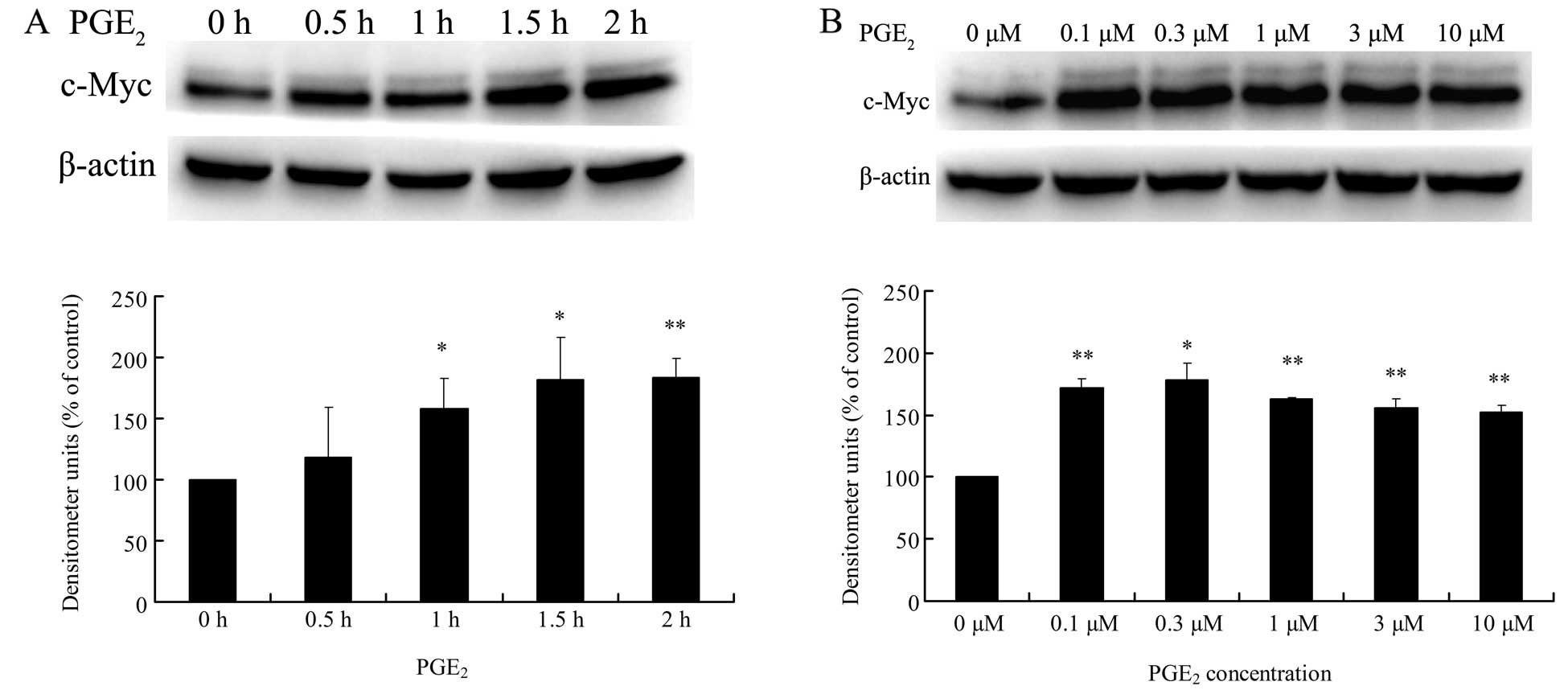
PGE2 upregulates c-Myc mRNA
and protein expression in HCC cells
To determine the direct effect of PGE2 on
c-Myc expression in HCC cells, Huh-7 cells were treated with
PGE2 at various doses or for different times, and then
the levels of mRNA and protein expression of c-Myc were analyzed by
real-time PCR and western blotting, respectively. Results from the
western blotting experiments showed that 3 μM PGE2
treatment significantly increased c-Myc protein expression from 0.5
to 2 h, and at 2 h it reached a maximum value which was 183% fold
of the value at 0 h (Fig. 1A).
Moreover, the expression levels of c-Myc protein were all
upregulated by 2 h PGE2 treatment with a dose from 0.1
to 10 μM (Fig. 1B). On the other
hand, the mRNA level of c-Myc was obviously elevated by 3 μM
PGE2 from 0.5 h, and it peaked at 1 h which was
5.16-fold of that at 0 h and then declined to the basal level at 2
h (Fig. 1C). In addition, treatment
of cells with 0.1 to 10 μM PGE2 for 1 h markedly
increased the mRNA level of c-Myc in the Huh-7 cells (Fig. 1D). These data indicate that
PGE2 upregulates c-Myc expression at the mRNA and
protein levels in the HCC cells.
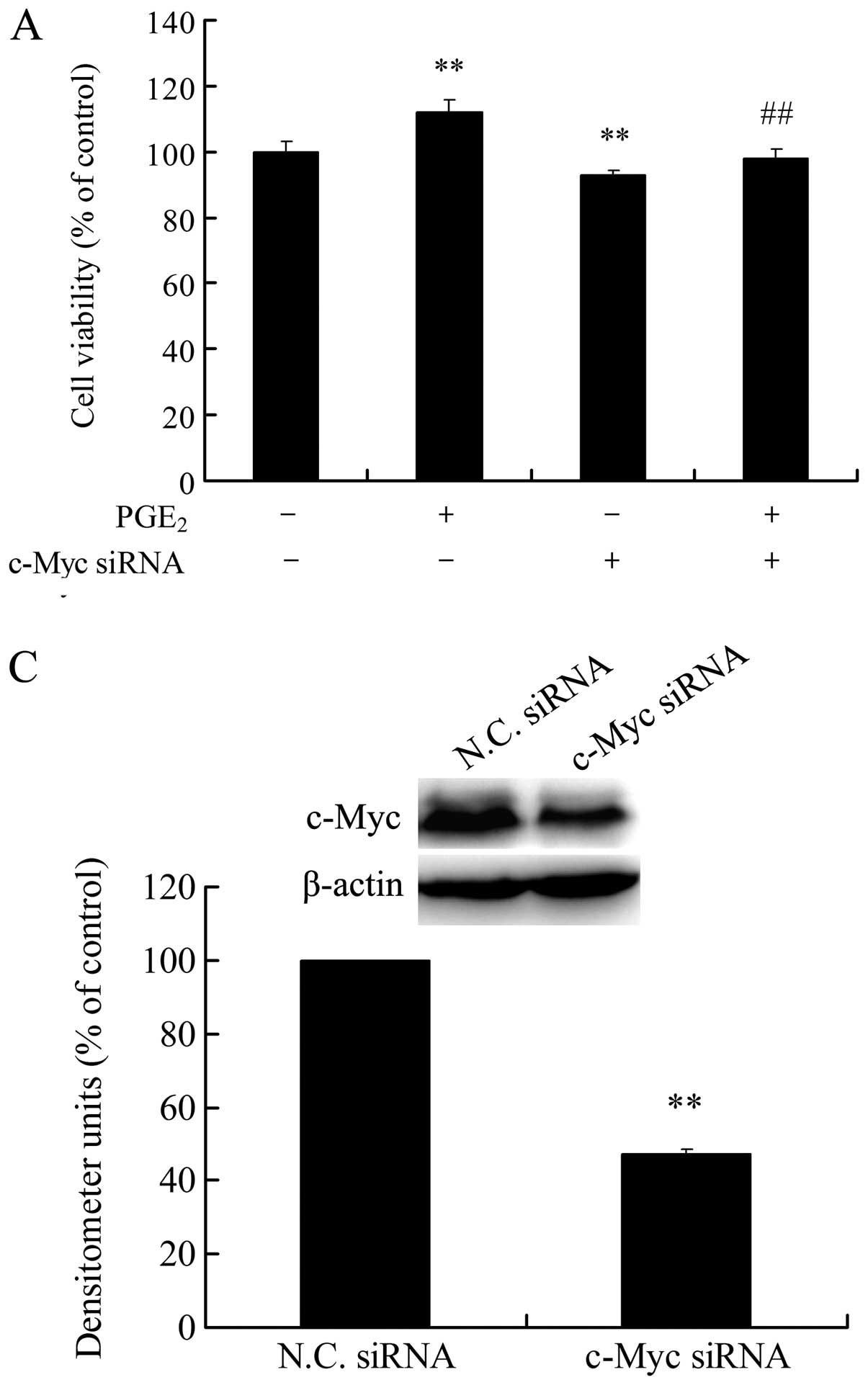
Knockdown of c-Myc blocks
PGE2-induced HCC cell growth and invasion
Our previous results demonstrated that
PGE2 promotes cell growth and invasion in HCC cells
(10–13). Now, we revealed that PGE2
directly upregulated c-Myc protein expression in the HCC cells, a
key transcription factor with cancer-promoting effects. Thus, the
role of c-Myc upregulation in PGE2-induced HCC cell
growth and invasion need to be further investigated. As shown in
Fig. 2A, c-Myc siRNA significantly
lowered the basal proliferation and blocked PGE2-induced
proliferation in the HCC Huh-7 cells. In addition, c-Myc siRNA
showed a similar inhibitory effect on PGE2-induced HCC
cell invasion (Fig. 2B). Depletion
of c-Myc protein by siRNA transfection was confirmed by western
blot analysis (Fig. 2C). These
results showed that the upregulation of c-Myc protein plays an
important role in PGE2-induced HCC cell growth and
invasion.
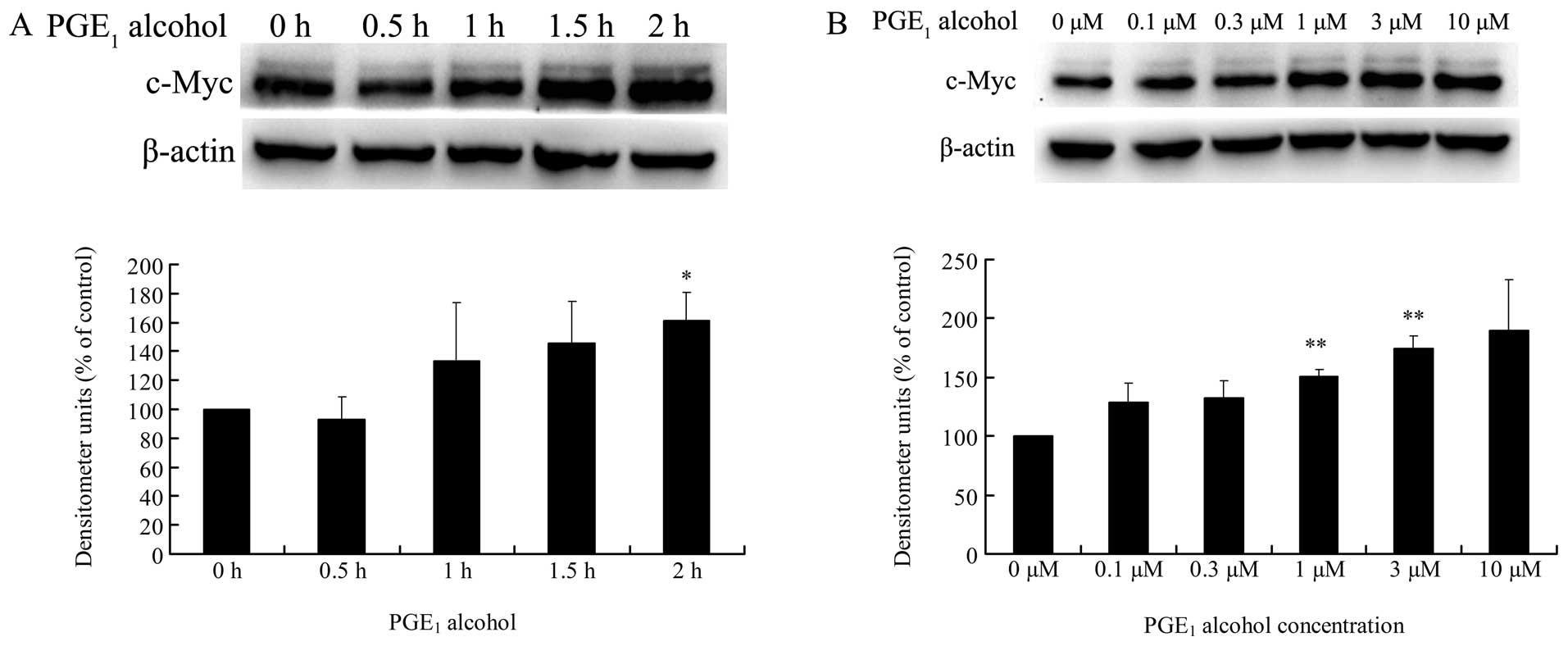
EP4 receptor is involved in
PGE2-induced c-Myc expression in HCC cells
EP4 receptor is closely associated with cancer cell
growth and invasion in various human cancers (19–23).
Thus, we investigated whether the EP4 receptor is also involved in
PGE2-induced c-Myc expression in HCC cells. As shown in
Fig. 3A and B, treatment of Huh-7
cells with PGE1 alcohol, the EP4 receptor selective
agonist, significantly increased the expression level of c-Myc
protein in a time- and dose-dependent manner. In addition, the mRNA
expression level of c-Myc was also upregulated by PGE1
alcohol in Huh-7 cells in a dose-dependent manner (Fig. 3C and D). These results suggest a key
role of the EP4 receptor in the regulation of c-Myc expression in
HCC cells.
To further confirm the involvement of the EP4
receptor in PGE2-induced c-Myc expression, we examined
the effects of an EP4 receptor selective antagonist or EP4 receptor
siRNA on PGE2-induced c-Myc expression in HCC cells. As
shown in Fig. 4A and B,
pretreatment of Huh-7 cells with GW627368X, the EP4 receptor
selective antagonist, markedly reduced PGE2-or
PGE1 alcohol-induced c-Myc expression at the protein and
mRNA levels. In addition, EP4 receptor siRNA also blocked the
upregulation of c-Myc protein expression by PGE2 or
PGE1 alcohol in the Huh-7 cells (Fig. 4C). The interference efficacy of EP4
receptor siRNA was verified by real-time PCR analysis, showing that
EP4 siRNA significantly lowered the mRNA expression of the EP4
receptor in the Huh-7 cells (Fig.
4D). These observations indicate that the EP4 receptor is
involved in PGE2-induced c-Myc expression in HCC
cells.
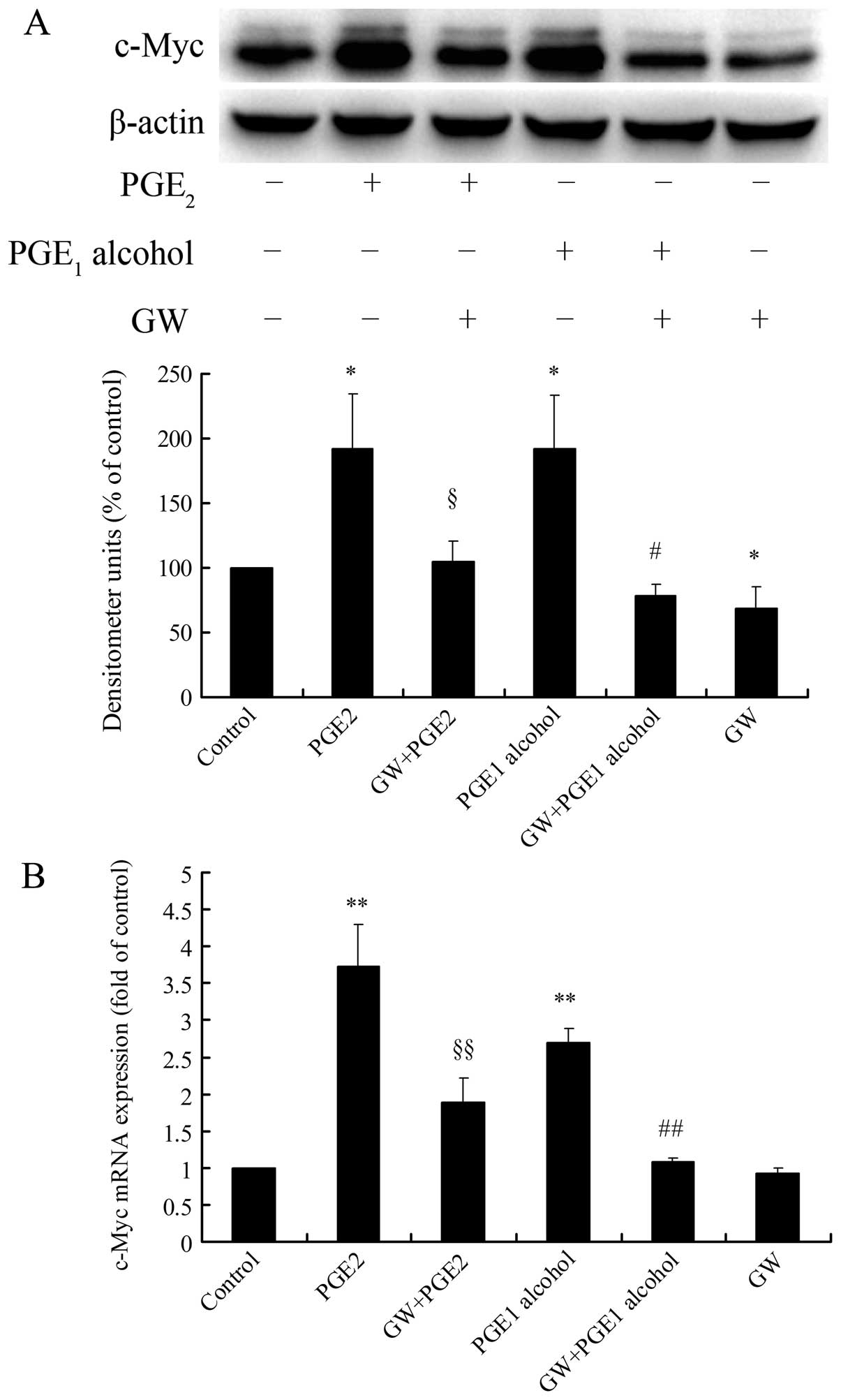 | Figure 4EP4 receptor antagonism or siRNA
interference attenuates PGE2-induced c-Myc expression in
Huh-7 cells. (A and B) Effect of the EP4 receptor selective
antagonist GW627368X on PGE2-induced c-Myc expression.
Huh-7 cells were pretreated for 1 h with GW627368X (4 μM) followed
by stimulation with PGE2 (3 μM) or PGE1
alcohol (3 μM), and protein expression of c-Myc was determined by
western blotting after 2 h (A) and mRNA expression of c-Myc was
examined by real-time PCR after 1 h (B). (C) Effect of EP4 receptor
siRNA on PGE2-induced c-Myc protein expression. Huh-7
cells were transfected with EP4 siRNA or N.C. siRNA for 72 h and
then stimulated with PGE2 (3 μM) or PGE1
alcohol (3 μM) in serum-free medium for 2 h, and protein expression
of c-Myc was determined by western blotting. Quantitative analysis
of c-Myc protein expression was carried out by calculating the
ratio between c-Myc protein and β-actin expression levels from
three different experiments. (D) RNAi efficiency of EP4 siRNA in
Huh-7 cells. After transfection of Huh-7 cells with EP4 siRNA or
N.C. siRNA for 72 h, the mRNA expression of the EP4 receptor was
examined by real-time PCR. Results are presented as the means ± SD
of three independent experiments. *P<0.05,
**P<0.01 compared with the control;
§P<0.05, §§P<0.01 compared with the
PGE2 treatment; #P<0.05,
##P<0.01 compared with the PGE1 alcohol
treatment. GW, GW627368X. PGE2, prostaglandin
E2; PGE1, prostaglandin E1; N.C.,
negative control. |
EP4 receptor-mediated c-Myc protein
upregulation depends on de novo biosynthesis of c-Myc mRNA and its
protein in HCC cells
As a key protein in cells, c-Myc expression is
tightly regulated at multiple levels, including transcription,
post-transcription and post-translation (25,26).
To elucidate the mechanisms of EP4 receptor-mediated c-Myc protein
upregulation, Act D and CHX, inhibitors of de novo RNA
synthesis and de novo protein synthesis, respectively, were
used. As shown in Fig. 5A,
pretreatment with Act D or CHX significantly reduced EP4
receptor-mediated c-Myc protein upregulation in the Huh-7 cells,
suggesting a crucial role of de novo biosynthesis of c-Myc
mRNA and its protein in EP4 receptor-mediated c-Myc protein
upregulation. The pharmacological effects of Act D or CHX on the
mRNA level of c-Myc were further examined by real-time PCR
experiment, and the results showed that Act D markedly lowered the
basal and EP4 receptor-mediated c-Myc mRNA expression, while CHX,
the inhibitor of de novo protein synthesis, had no such
effect (Fig. 5B). These findings
indicate that EP4 receptor-mediated c-Myc protein upregulation
greatly depends on the de novo biosynthesis of c-Myc mRNA
and its protein in HCC cells.
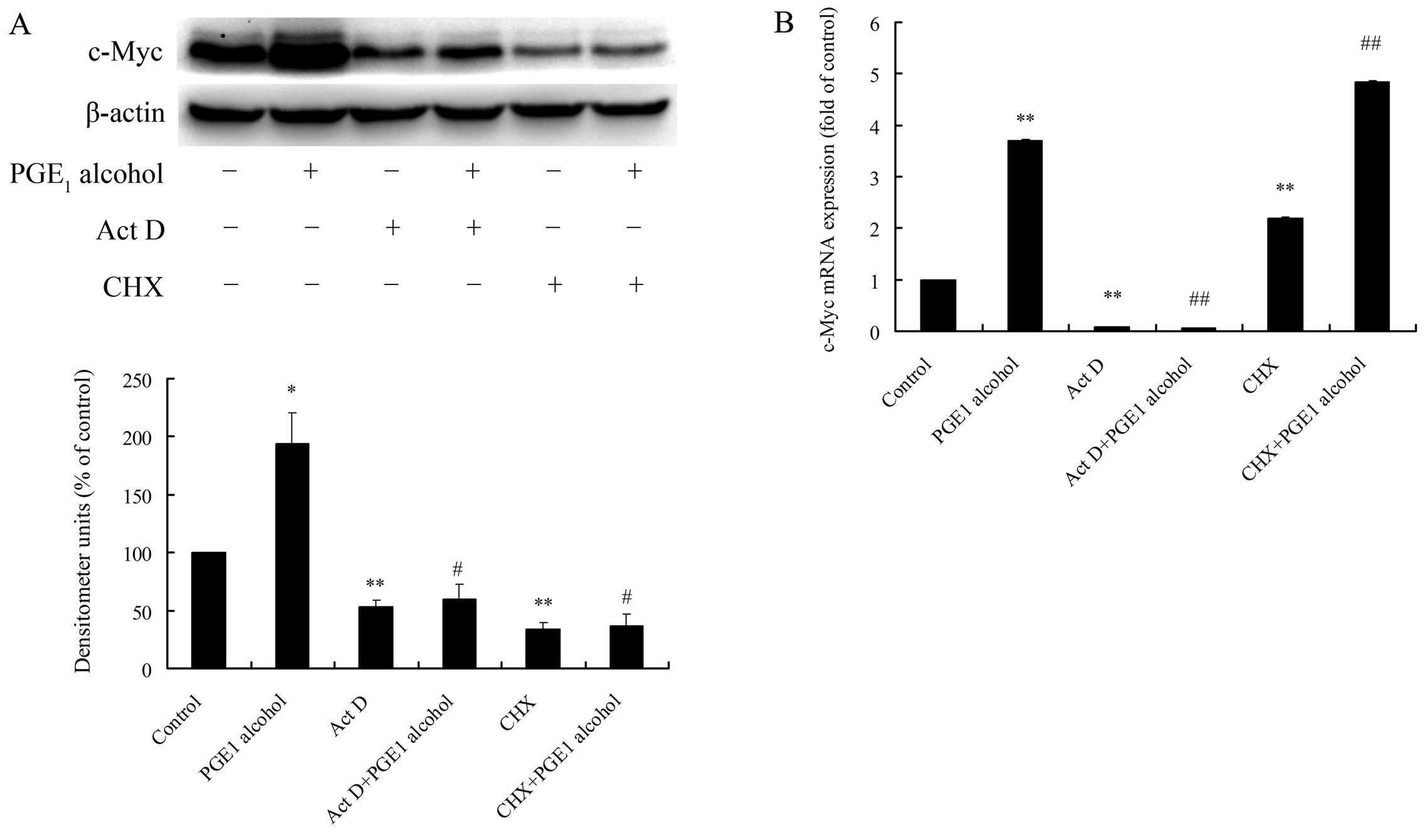 | Figure 5Effects of Act D and CHX, de
novo biosynthesis inhibitors of RNA and protein, respectively,
on EP4 receptor-mediated c-Myc protein upregulation in Huh-7 cells.
Huh-7 cells were pretreated for 1 h with Act D (5 μg/ml) or CHX (50
μg/ml) followed by stimulation with 3 μM PGE1 alcohol,
and protein expression of c-Myc was determined by western blotting
after 2 h (A) and mRNA expression of c-Myc was examined by
real-time PCR after 1 h (B). Quantitative analysis of c-Myc protein
expression was carried out by calculating the ratio between c-Myc
protein and β-actin expression levels from three different
experiments. Results are presented as the means ± SD of three
independent experiments. *P<0.05,
**P<0.01 compared with the control;
#P<0.05, ##P<0.01 compared with the
PGE1 alcohol treatment. PGE2, prostaglandin
E2. Act D, actinomycin D; CHX, cycloheximide;
PGE1, prostaglandin E1. |
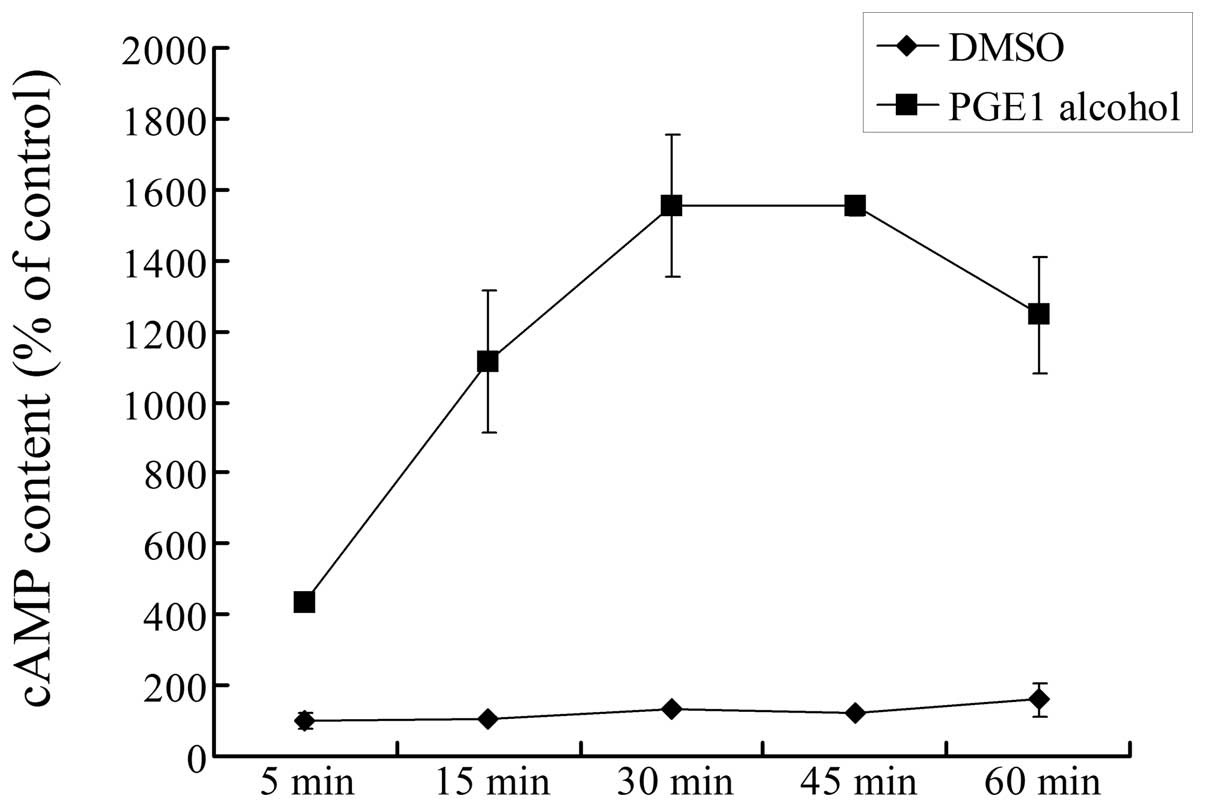
The GS/AC/cAMP signaling
pathway is involved in EP4 receptor-mediated c-Myc upregulation in
HCC cells
The EP4 receptor, a G protein coupled receptor, is
usually coupled with GS protein to activate adenylate
cyclase (AC) and elevate intracellular cAMP levels (5,18). To
confirm the downstream signaling pathway of the EP4 receptor in HCC
cells, we measured the direct effect of the EP4 receptor agonist on
the intracellular cAMP level in Huh-7 cells. As shown in Fig. 6, stimulation of cells with
PGE1 alcohol notably increased the intracellular cAMP
level in a time-dependent manner, and at around 30–45 min the cAMP
level reached a maximum value which was ~15-fold of the control. By
contrast, the vehicle DMSO had no effect on the cAMP level. These
data demonstrated that the EP4 receptor is coupled to the
GS/AC/cAMP signaling pathway in HCC cells.
To determine the role of the GS/AC/cAMP
signaling pathway in EP4 receptor-mediated c-Myc upregulation,
Huh-7 cells were treated with a specific activator or inhibitor of
AC, and then their effects on c-Myc expression in HCC cells were
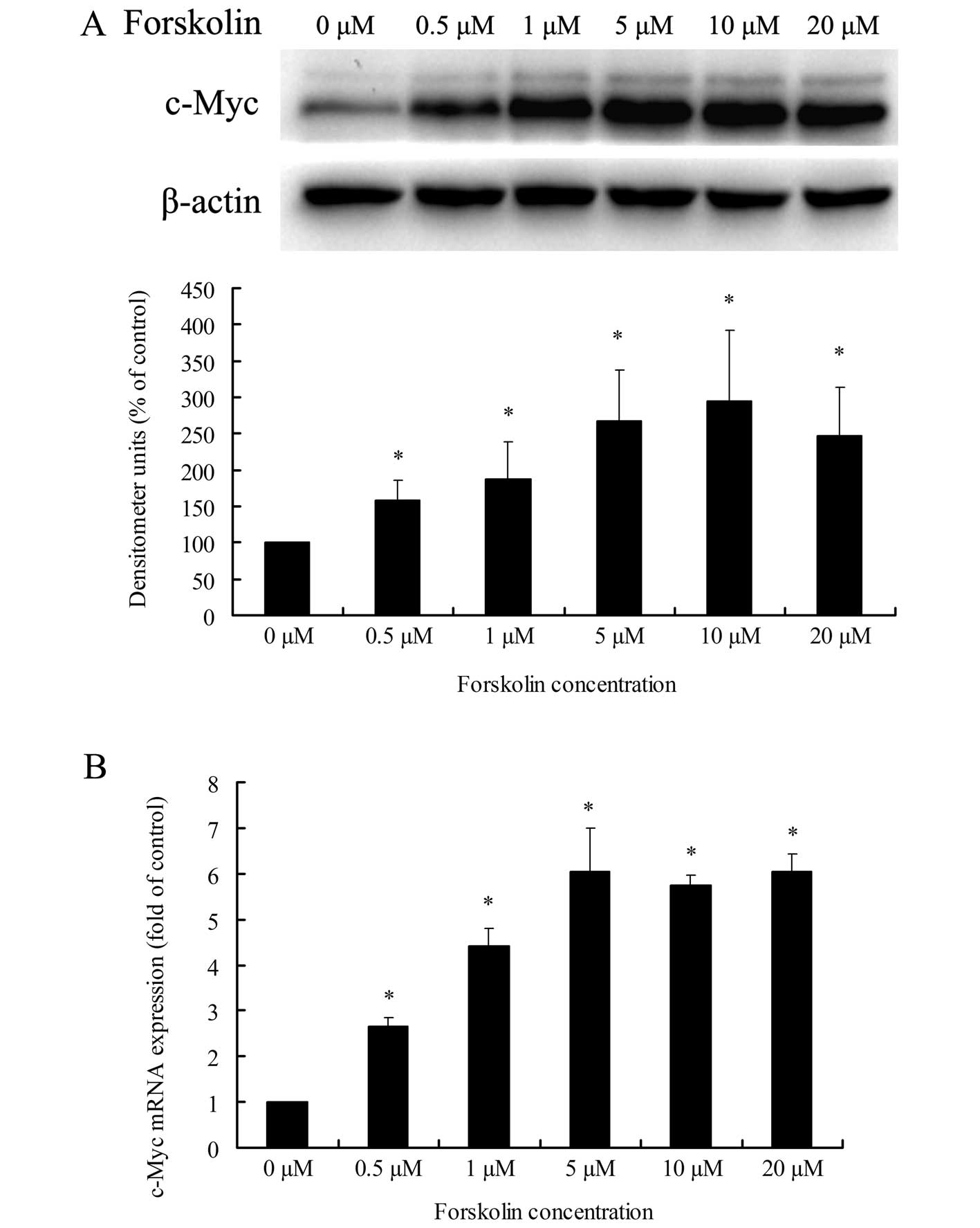
examined. As shown in Fig. 7A and
B, treatment of Huh-7 cells with forskolin, the specific
activator of AC, resulted in a dose-dependent increase in c-Myc
expression at the protein and mRNA levels. Pretreatment of cells
with SQ22536, the specific inhibitor of AC, markedly reduced EP4
receptor-mediated c-Myc upregulation at the protein level and
partly at the mRNA level (Fig. 7C and
D). These results revealed that the GS/AC/cAMP
signaling pathway is involved in EP4 receptor-mediated c-Myc
upregulation in HCC cells.
Involvement of the PKA/CREB pathway in
EP4 receptor-mediated c-Myc upregulation in HCC cells
An elevated intracellular cAMP level leads to
protein kinase A (PKA) activation, while activated PKA could
transfer into the cell nucleus and phosphorylate transcription
factor CREB protein, thus regulating gene expression (30). Firstly, we investigated the
involvement of PKA activation in EP4 receptor-mediated c-Myc
upregulation in HCC cells by using the PKA-specific inhibitor H89.
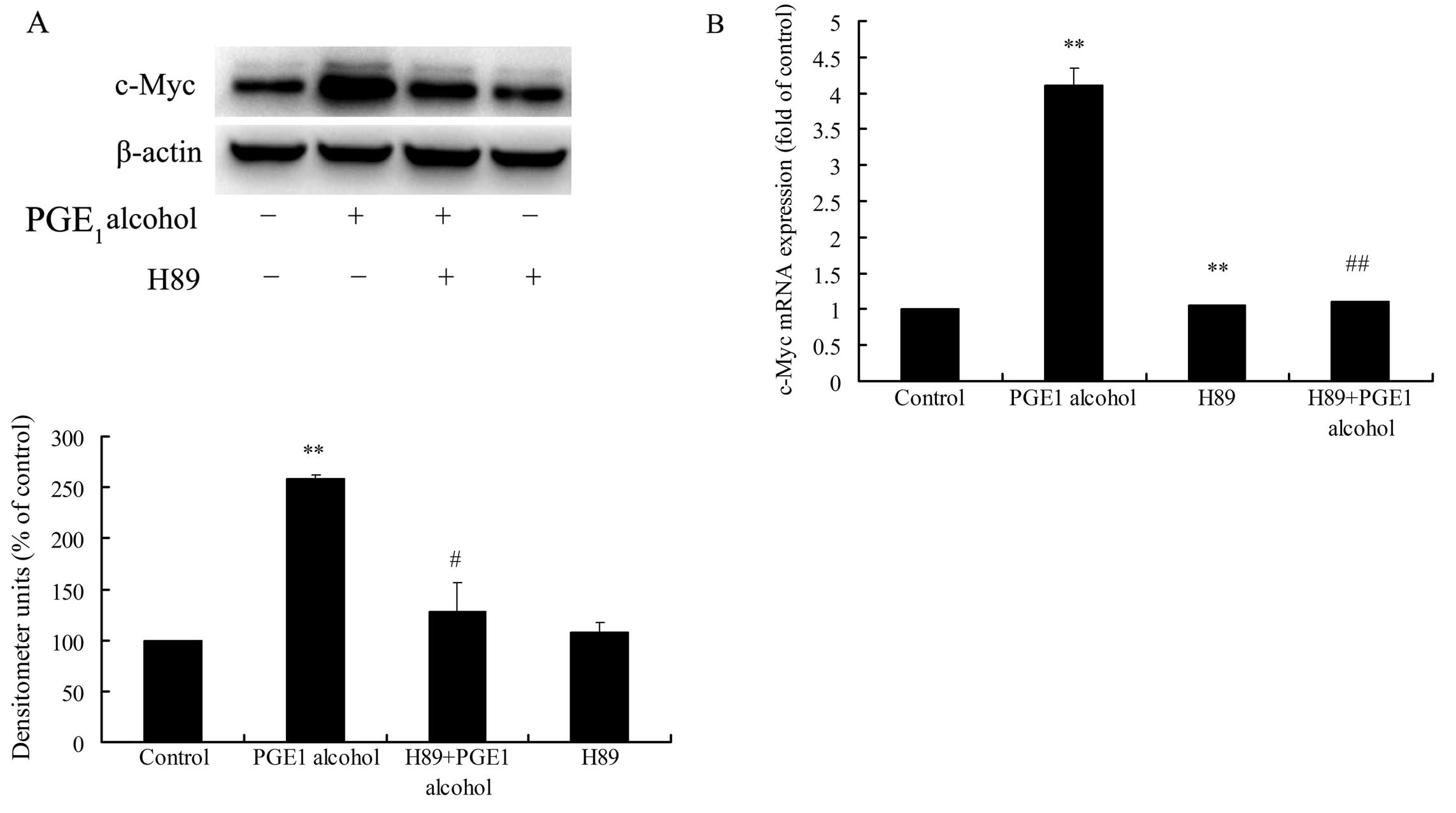
As shown in Fig. 8A and B,
pretreatment of Huh-7 cells with H89 significantly reduced EP4
receptor-mediated c-Myc upregulation at the protein and mRNA
levels, suggesting an important role of PKA in EP4
receptor-mediated c-Myc upregulation. Secondly, the role of CREB in
EP4 receptor-mediated c-Myc upregulation was also examined.
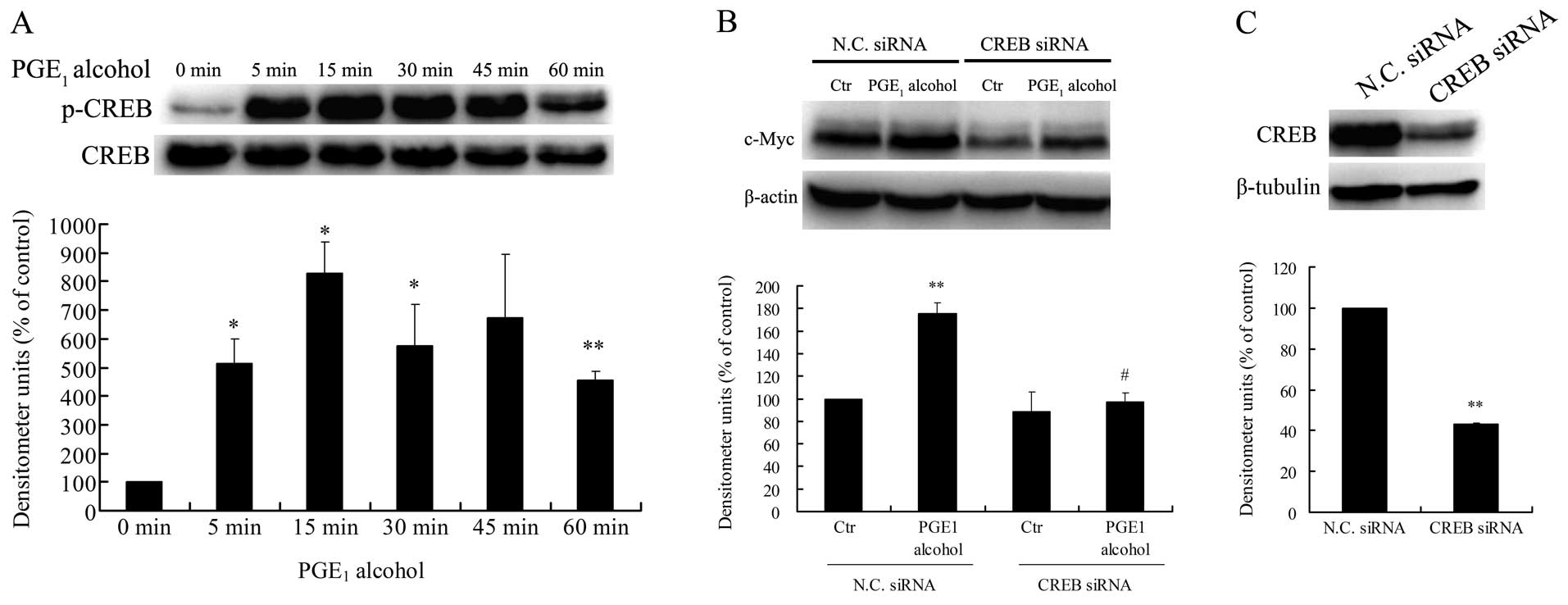
Stimulation of Huh-7 cells with the EP4 receptor agonist
PGE1 alcohol led to a significant increase in the
phosphorylation of CREB at Ser133 (Fig.
9A), a crucial event for transcriptional activation by CREB.
Meanwhile, the CREB siRNA markedly reduced EP4 receptor-mediated
c-Myc protein upregulation in Huh-7 cells (Fig. 9B). The downregulation of CREB
protein by siRNA transfection in the Huh-7 cells was confirmed by
western blotting (Fig. 9C). Based
on these findings, the PKA/CREB pathway is also involved in EP4
receptor-mediated c-Myc upregulation in HCC cells.
Discussion
Hepatocellular carcinoma (HCC) represents a major
health problem worldwide. To date, our understanding of the
molecular mechanisms of this disease remains rudimentary. A large
body of studies support a pivotal role of chronic inflammation in
the pathogenesis of HCC (2–4). Yet, the underlying mechanisms are not
well understood.
PGE2, an inflammatory mediator and the
predominant product of COX-2, has been shown to be involved in
various human cancers, including HCC. Our previous results revealed
that PGE2 significantly enhanced the cell growth,
migration and invasion in HCC cells (10–13).
Although several signaling pathways have been identified such as
transactivation of EGFR receptor (31), phosphorylation of FAK kinase
(12,13), the detailed mechanisms of
PGE2 in HCC remain to be further studied.
c-Myc protein encoded by the proto-oncogene
c-myc functions as a transcription factor. After dimerizing
with its partner protein Max, c-Myc binds to E box sequence
elements (5′-CACGTG-3′) to activate the transcription of many
target genes, and thus promotes cell proliferation and
tumorigenesis (26). In addition,
substantial studies confirmed that c-myc is involved in
human hepatocarcinogenesis. Approximately 30% of human HCC samples
exhibit gene amplification of c-myc (27,28),
and the major etiological factors of HCC including hepatitis C or B
virus infection, and aflatoxin could induce overexpression of c-Myc
protein (32–34). However, the exact mechanisms of
c-myc activation in HCC are largely unknown.
Since PGE2 and c-Myc are both involved in
HCC, and have the potential for causing tumorigenesis, the internal
relationship between PGE2 and c-Myc in HCC is of
particular interest to us. In the present study, we found that
PGE2 directly upregulated c-Myc expression at the mRNA
and protein levels, and knockdown of c-Myc protein greatly
suppressed PGE2-induced HCC cell growth and invasion in
Huh-7 cells. These findings firmly confirm that c-Myc is a critical
regulator in PGE2-induced HCC cell growth and invasion,
suggesting a probable molecular mechanism for inflammation-induced
hepatocarcinogenesis.
PGE2 exerts biological effects through
four types of EP receptors on the cell surface membrane, designated
as EP1, EP2, EP3 and EP4 (5,18).
Studies indicate that the EP4 receptor plays a crucial role in
PGE2-mediated tumorigenesis in many types of cancer. For
example, PGE2 promotes renal cancer cell invasion via
the EP4 receptor and small GTPase Rap signaling (19). In colon cancer cells,
PGE2 induces S100p expression to enhance cancer cell
growth and migration via the EP4 receptor signaling (20). In lung cancer cells, PGE2
promotes cell migration via EP4-β Arrestin1-c-Src signal-some
(21). Likewise, we found that
PGE2 induced c-Myc expression to promote cell growth and
invasion in HCC cells mainly through the EP4 receptor.
Usually, c-Myc expression is tightly controlled by a
number of mechanisms at the transcriptional, post-transcriptional
and post-translational levels. Many transcription factors such as
TCF, FBP, CNBP, NF-κB and AP-1 can bind to the promoter of the
human c-myc gene, and upregulate its expression at the
transcriptional level (25,41). In addition, the c-Myc protein itself
can be post-translational modified at multiple sites by
phosphorylation or ubiquitination. Two key phosphorylation sites of
Ser62 and Thr58 located within the N terminus are important for the
regulation of c-Myc protein stabilization, yet have opposite
function. Ser62 phosphorylation stabilizes c-Myc protein, while T58
phosphorylation leads to its protein degradation (35). In human esophageal squamous cell
carcinoma cells, PGE2 was shown to upregulate c-Myc
protein expression by stimulating Ser62 phosphorylation and then
stabilizing its protein at the post-translational level (36). However, the present study revealed
that in HCC cells the EP4 receptor-mediated c-Myc protein
upregulation largely depended on de novo biosynthesis of
c-Myc mRNA and its protein.
As a G protein coupled receptor, the EP4 receptor is
believed to be coupled with GS protein to activate AC
and elevate intracellular cAMP levels (5,18). Our
results confirmed that the EP4 receptor activated the
GS/AC/cAMP signaling pathway in HCC cells. In addition,
we demonstrated that this canonical pathway is involved in EP4
receptor-mediated c-Myc upregulation in HCC cells. This finding is
similar to a study in human umbilical cord blood-derived
mesenchymal stem cells (hUCB-MSCs), which showed that
PGE2 upregulated the expression of c-Myc and VEGF via
EP2/cAMP signaling and thus promoted cell proliferation of
hUCB-MSCs (37).
In cells, an elevated cAMP level subsequently
activates three main targets including protein kinase A (PKA), the
exchange protein activated by cAMP (Epac) and the cyclicnucleotide-
gated ion channels (CNGCs) (30).
Among them, PKA has been shown to regulate many aspects of cell
functions, such as metabolism, signal transduction and gene
expression (38). The effect of PKA
on gene transcription is mainly achieved by direct phosphorylation
of the transcription factor CREB at the site of Ser133 following
stimulation of gene transcription by activated CREB (39,40).
By using a specific inhibitor and siRNA, we demonstrated that the
signaling pathway of PKA/CREB is also involved in the EP4
receptor-mediated c-Myc upregulation in HCC cells.
CREB, one member of the bZIP (basic domain/leucine
zipper) transcription factor family, can activate numerous target
genes including c-fos and JunD through cAMP response elements
(CREs). Full CRE is an 8 bp palindrome (5′-TGACGTCA-3′), and the
half motif (CGTCA) is also active for CREB binding and cAMP
responsiveness. It is reported that of 105 genes with functional
CREs identified in the literature, approximately half contain a
full palindrome, with the other half containing a single CGTCA
motif (40). In the present study,
we found that CREB is involved in the upregulation of c-Myc
expression in HCC cells. Yet, there are no data or study which
shows that the promoter of the human c-myc gene contains a
or more full CRE (41). Based on
sequence analysis, we found that two half CGTCA motifs locate in
the upstream of transcription start site (TSS) of the human
c-myc gene, at −3,005 and −3,759 bp, respectively.
Therefore, we speculated that CREB upregulates the expression of
c-myc by following possible mechanisms: by binding to these
two half CGTCA motifs; indirect action, by stimulation of the
expression of other transcription factors such as AP-1 and then
activation of c-Myc expression or other unknown mechanisms. Thus,
the exact mechanisms involved in c-Myc upregulation by CREB need to
be further investigated.
In summary, the present study revealed that
PGE2 directly upregulated c-Myc expression via the
EP4R/GS/AC/cAMP/PKA/CREB signaling pathway to promote
cell growth and invasion in human HCC cells. This finding provides
a further insight into the mechanisms by which PGE2
enhances HCC cell growth and invasion. Targeting of the
PGE2/EP4R/c-Myc pathway may be a new therapeutic
strategy to prevent and cure human HCC.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (nos. 30871015 and 81172003),
and by a project funded by the Priority Academic Program
Development (PAPD) of Jiangsu Higher Education Institutions.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
2
|
Nakagawa H and Maeda S: Inflammation- and
stress-related signaling pathways in hepatocarcinogenesis. World J
Gastroenterol. 18:4071–4081. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ramakrishna G, Rastogi A, Trehanpati N,
Sen B, Khosla R and Sarin SK: From cirrhosis to hepatocellular
carcinoma: new molecular insights on inflammation and cellular
senescence. Liver Cancer. 2:367–383. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Berasain C, Castillo J, Perugorria MJ,
Latasa MU, Prieto J and Avila MA: Inflammation and liver cancer:
new molecular links. Ann NY Acad Sci. 1155:206–221. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hata AN and Breyer RM: Pharmacology and
signaling of prostaglandin receptors: multiple roles in
inflammation and immune modulation. Pharmacol Ther. 103:147–166.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang D and Dubois RN: Prostaglandins and
cancer. Gut. 55:115–122. 2006. View Article : Google Scholar
|
|
7
|
Legler DF, Bruckner M, Uetz-von Allmen E
and Krause P: Prostaglandin E2 at new glance: novel
insights in functional diversity offer therapeutic chances. Int J
Biochem Cell Biol. 42:198–201. 2010.
|
|
8
|
Rizzo MT: Cyclooxygenase-2 in oncogenesis.
Clin Chim Acta. 412:671–687. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mayoral R, Fernández-Martínez A, Boscá L
and Martín-Sanz P: Prostaglandin E2 promotes migration
and adhesion in hepatocellular carcinoma cells. Carcinogenesis.
26:753–761. 2005.PubMed/NCBI
|
|
10
|
Leng J, Han C, Demetris AJ, Michalopoulos
GK and Wu T: Cyclooxygenase-2 promotes hepatocellular carcinoma
cell growth through Akt activation: evidence for Akt inhibition in
celecoxib-induced apoptosis. Hepatology. 38:756–768. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ma J, Chen M, Xia SK, et al: Prostaglandin
E2 promotes liver cancer cell growth by the upregulation
of FUSE-binding protein 1 expression. Int J Oncol. 42:1093–1104.
2013.
|
|
12
|
Bai XM, Zhang W, Liu NB, et al: Focal
adhesion kinase: Important to prostaglandin E2-mediated
adhesion, migration and invasion in hepatocellular carcinoma cells.
Oncol Rep. 21:129–136. 2009.PubMed/NCBI
|
|
13
|
Bai X, Wang J, Zhang L, et al:
Prostaglandin E2 receptor EP1-mediated phosphorylation
of focal adhesion kinase enhances cell adhesion and migration in
hepatocellular carcinoma cells. Int J Oncol. 42:1833–1841.
2013.
|
|
14
|
Reader J, Holt D and Fulton A:
Prostaglandin E2 EP receptors as therapeutic targets in
breast cancer. Cancer Metastasis Rev. 30:449–463. 2011.
|
|
15
|
Timoshenko AV, Xu G, Chakrabarti S, Lala
PK and Chakraborty C: Role of prostaglandin E2 receptors
in migration of murine and human breast cancer cells. Exp Cell Res.
289:265–274. 2003.PubMed/NCBI
|
|
16
|
Wu WK, Sung JJ, Lee CW, Yu J and Cho CH:
Cyclooxygenase-2 in tumorigenesis of gastrointestinal cancers: an
update on the molecular mechanisms. Cancer Lett. 295:7–16. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jain S, Chakraborty G, Raja R, Kale S and
Kundu GC: Prostaglandin E2 regulates tumor angiogenesis
in prostate cancer. Cancer Res. 68:7750–7759. 2008.
|
|
18
|
Sugimoto Y and Narumiya S: Prostaglandin E
receptors. J Biol Chem. 282:11613–11617. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wu J, Zhang Y, Frilot N, Kim JI, Kim WJ
and Daaka Y: Prostaglandin E2 regulates renal cell
carcinoma invasion through the EP4 receptor-Rap GTPase signal
transduction pathway. J Biol Chem. 286:33954–33962. 2011.
|
|
20
|
Chandramouli A, Mercado-Pimentel ME,
Hutchinson A, et al: The induction of S100p expression by the
Prostaglandin E2 (PGE2)/EP4 receptor
signaling pathway in colon cancer cells. Cancer Biol Ther.
10:1056–1066. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kim JI, Lakshmikanthan V, Frilot N and
Daaka Y: Prostaglandin E2 promotes lung cancer cell
migration via EP4-βArrestin1-c-Src signalsome. Mol Cancer Res.
8:569–577. 2010.
|
|
22
|
Oshima H, Popivanova BK, Oguma K, Kong D,
Ishikawa TO and Oshima M: Activation of epidermal growth factor
receptor signaling by the prostaglandin E2 receptor EP4
pathway during gastric tumorigenesis. Cancer Sci. 102:713–719.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Robertson FM, Simeone AM, Mazumdar A, et
al: Molecular and pharmacological blockade of the EP4 receptor
selectively inhibits both proliferation and invasion of human
inflammatory breast cancer cells. J Exp Ther Oncol. 7:299–312.
2008.PubMed/NCBI
|
|
24
|
Zender L, Villanueva A, Tovar V, Sia D,
Chiang DY and Llovet JM: Cancer gene discovery in hepatocellular
carcinoma. J Hepatol. 52:921–929. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Levens D: You don’t muck with MYC. Genes
Cancer. 1:547–554. 2010.
|
|
26
|
Dang CV: MYC on the path to cancer.
Cell. 149:22–35. 2012. View Article : Google Scholar
|
|
27
|
Kawate S, Fukusato T, Ohwada S, Watanuki A
and Morishita Y: Amplification of c-myc in hepatocellular
carcinoma: correlation with clinicopathologic features,
proliferative activity and p53 overexpression. Oncology.
57:157–163. 1999.
|
|
28
|
Chan KL, Guan XY and Ng IO:
High-throughput tissue microarray analysis of c-myc
activation in chronic liver diseases and hepatocellular carcinoma.
Hum Pathol. 35:1324–1331. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kaposi-Novak P, Libbrecht L, Woo HG, et
al: Central role of c-Myc during malignant conversion in human
hepatocarcinogenesis. Cancer Res. 69:2775–2782. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fimia GM and Sassone-Corsi P: Cyclic AMP
signalling. J Cell Sci. 114:1971–1972. 2001.PubMed/NCBI
|
|
31
|
Han C, Michalopoulos GK and Wu T:
Prostaglandin E2 receptor EP1 transactivates
EGFR/MET receptor tyrosine kinases and enhances invasiveness in
human hepatocellular carcinoma cells. J Cell Physiol. 207:261–270.
2006.
|
|
32
|
Higgs MR, Lerat H and Pawlotsky JM:
Hepatitis C virus-induced activation of β-catenin promotes c-Myc
expression and a cascade of pro-carcinogenetic events. Oncogene.
32:4683–4693. 2013.
|
|
33
|
Iizuka N, Tsunedomi R, Tamesa T, et al:
Involvement of c-myc-regulated genes in hepatocellular carcinoma
related to genotype-C hepatitis B virus. J Cancer Res Clin Oncol.
132:473–481. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tashiro F, Morimura S, Hayashi K, et al:
Expression of the c-Ha-ras and c-myc genes in aflatoxin B1-induced
hepatocellular carcinomas. Biochem Biophys Res Commun. 138:858–864.
1986. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Junttila MR and Westermarck J: Mechanisms
of MYC stabilization in human malignancies. Cell Cycle. 7:592–596.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yu L, Wu WK, Li ZJ, Li HT, Wu YC and Cho
CH: Prostaglandin E2 promotes cell proliferation
via protein kinase C/extracellular signal regulated kinase
pathway-dependent induction of c-Myc expression in human esophageal
squamous cell carcinoma cells. Int J Cancer. 125:2540–2546.
2009.PubMed/NCBI
|
|
37
|
Jang MW, Yun SP, Park JH, Ryu JM, Lee JH
and Han HJ: Cooperation of Epac1/Rap1/Akt and PKA in prostaglandin
E2-induced proliferation of human umbilical cord blood
derived mesenchymal stem cells: involvement of c-Myc and VEGF
expression. J Cell Physiol. 227:3756–3767. 2012.PubMed/NCBI
|
|
38
|
Cheng X, Ji Z, Tsalkova T and Mei F: Epac
and PKA: a tale of two intracellular cAMP receptors. Acta Biochim
Biophys Sin. 40:651–662. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sands WA and Palmer TM: Regulating gene
transcription in response to cyclic AMP elevation. Cell Signal.
20:460–466. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Mayr B and Montminy M: Transcriptional
regulation by the phosphorylation-dependent factor CREB. Nat Rev
Mol Cell Biol. 2:599–609. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wierstra I and Alves J: The c-myc
promoter: still MysterY and challenge. Adv Cancer Res. 99:113–333.
2008.
|