Introduction
The death rate of patients with ovarian carcinoma is
one of the highest for gynecologic malignancies (1). Paclitaxel plus platinum therapy is
gaining acceptance as the standard clinical chemotherapy regimen
for ovarian cancer. Paclitaxel is an important new agent for
ovarian cancer treatment and is highly effective as the first-line
therapy for advanced ovarian cancer. However, the emergence of a
paclitaxel-resistant tumor sub-population ultimately leads to
treatment failure. Therefore, determining the mechanism underlying
paclitaxel resistance is important.
Comparative genomic hybridization (CGH) detects the
chromosomal loci DNA copy-number changes in a single experiment.
CGH analyses of ovarian cancer cell lines resistant to paclitaxel
and platinum revealed that they harbor drug resistance-related
chromosomal abnormalities (2,3). For
example, the increased DNA copy-number at 6q21–25 and decreased DNA
copy-number at 7q21–36 and 10q12–15, respectively, are associated
with platinum resistance, and the increased DNA copy-number at
7q11.2–21 is related to paclitaxel resistance (3). In the present study, we conducted CGH
analyses to compare the genomic alteration present in
paclitaxel-resistant ovarian carcinoma cell lines with those
present in their parental cell lines. These analyses revealed high
amplification of chromosome 2p21 in paclitaxel-resistant OC3/TAX300
cells. Certain drug resistance-related genes are located on
chromosome 2p21, including hMSH2, a member of the mismatch
repair (MMR) gene family. MMR, which corrects base mismatch,
ensures DNA replication fidelity, and maintains genomic
stability.
In human cells, the mechanism of MMR mainly includes
three processes: mismatch identification, mismatch excision, and
DNA resynthesis. MMR involves hMSH1, hMSH2, hMSH6, PMS1, and PMS2
proteins (4,5). The dysfunction of MMR, which causes
the failure of mismatch base repair, will induce the genomic
instability caused by the increased frequency of spontaneous
mutations, leading to tumorigenesis. Studies have shown that MMR
dysfunction (most frequently hMSH1 or hMSH2) is an
important genetic risk factor for hereditary nonpolyposis
colorectal cancer, which is also called Lynch syndrome (6). Lynch syndrome is a hereditary syndrome
related to some familiar cancers, such as colorectal cancer, as
well as extracolonic cancers such as endometrial, gastric, ovarian,
pancreatic, ureteral, and brain cancers, and sebaceous adenomas
(6–8). Deficiency of MMR proteins leads to an
accumulation of DNA replication errors and allows the persistence
of mismatch mutations, particularly in areas of the genome with
short repetitive DNA known as ‘microsatellites’; this phenomenon is
known as microsatellite instability (MSI) (9). More than 90% of colorectal cancers
related to Lynch syndrome manifest MSI. MSI is also associated with
15–20% of sporadic colorectal cancers (10–12).
However, if hMSH2 is overexpressed in tumor
cells, the DNA damage in these cells can be repaired rapidly,
leading to tumor progression, deterioration and chemotherapy
resistance, ultimately resulting in poor prognosis (4,13). For
decades, studies have confirmed that an increase in the ability of
cells to repair damaged DNA is a crucial factor in determining the
resistance elicited against DNA-damaging agents such as alkylating
agents and platinum-based compounds (13–16).
Paclitaxel, the most common chemotherapeutic drug in
ovarian cancer treatment, can generate free radicals leading to
irreversible oxidative DNA damage (17). Therefore, we designed the present
study to investigate the relationship between hMSH2 and
paclitaxel resistance. In previous studies, we established the
paclitaxel-resistant ovarian carcinoma cell lines OC3/TAX300 and
OC3/TAX50 by exposing OC3 ovarian carcinoma cells to various doses
of paclitaxel (18). The resistance
index (RI) of OC3/TAX300 and OC3/TAX50 cell lines is 6.70 and 2.52,
respectively. In the present study, we analyzed hMSH2
expression among different ovarian cancer cell lines and tissues.
We employed siRNA techniques to assess the morphological features,
proliferation, and apoptosis susceptibility in the
paclitaxel-resistant OC3/TAX300 cell line.
Materials and methods
Ethics statement
The study was approved by the Ethics Committee of
the Beijing Shijitan Hospital of Capital Medical University.
Written informed consent was obtained from all the patients and
families before surgery. All procedures were performed in
accordance with the Declaration of Helsinki.
Cell lines and culture conditions
The OC3 human ovarian carcinoma cell line was
provided by the Basic Medical Research Institute (Beijing, China)
and was used in several studies (19). The paclitaxel-resistant ovarian
carcinoma cell lines OC3/TAX300 and OC3/TAX50 were established in
previous studies (18). Cells were
cultured in RPMI-1640 medium (Gibco, Carlsbad, CA, USA)
supplemented with 10% bovine calf serum (DingGuo Co. Ltd., Beijing,
China), and 0.1% each of penicillin and streptomycin at 37°C in an
incubator with a 5% CO2 atmosphere.
Reagents and antibodies
Rabbit anti-human hMSH2 monoclonal antibody
was purchased from Cell Signaling Technology Co. (Boston, MA, USA).
TRIzol® reagent kit and Lipofectamine® were
supplied by Invitrogen (Life Technologies Co., Carlsbad, CA, USA).
Dimethyl sulfoxide (DMSO) was purchased from Sigma-Aldrich (St.
Louis, MO, USA). The Annexin V-PE/7-AAD (7-amino-actinomycin D)
apoptosis test kit was from Kaiji Technology Co. Ltd. (Nanjing,
China). Paclitaxel was purchased from ChenXin Medicine Co. Ltd.
(Jining, China). Phosphate-buffered saline, propidium iodide,
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide
(MTT), and microplates were purchased from DingGuo Co. Ltd.
Clinical samples
We collected 54 ovarian cancer tissue samples
(preserved in liquid nitrogen) from the specimen repository of
Beijing Shijitan Hospital from March 2008 to May 2013. The tissue
samples were divided into 2 sets as follows: 33 tissue samples with
paclitaxel chemotherapy before surgery and 21 without paclitaxel
chemotherapy before surgery; 30 samples with low-differentiated
ovarian carcinoma tissue and 24 with moderate-to-highly
differentiated ovarian carcinoma tissue.
Comparative genomic hybridization
(CGH)
Genomic DNA was extracted from the
paclitaxel-resistant cell lines, OC3/TAX300 and OC3/TAX50, the
paclitaxel-sensitive cell line OC3, and the peripheral blood of
healthy women volunteers using the standard phenol/chloroform
method. OC3/TAX300, OC3/TAX50 and OC3 DNA were labeled with
fluorescein-dUTP (green fluorescence). DNA from normal peripheral
blood was labeled with rhodamine-dUTP (red fluorescence) using a
nick-translation method. Metaphase chromosome spreads prepared from
the lymphocytes of the healthy women were hybridized with a sample
DNA probe or a matched normal peripheral-blood DNA probe,
respectively. The differential fluorescence of chromosomes was
observed using a fluorescence microscope. The fluorescence
intensity, fluorescence ratio, and analysis diagrams were compared
using a fluorescence image analysis system.
Reverse transcription-polymerase chain
reaction (RT-PCR) assay
The hMSH2 primers, designed and synthesized
by Shenggong Co. Ltd., (Shanghai, China) were: forward, 5′-CAA TTG
AAA GGA GTC TCC ACG-3′ [21 base pairs (bp)]; and reverse, 5′-AAA
CTC CTC AAG TTC CAG GG-3′ (20 bp). The length of the amplified DNA
fragment was 411 bp. RNA was extracted using the TRIzol reagent
kit. The PCR reaction mixture (20 μl total volume) composition was
as follows: 2 μl 10X PCR buffer, 1.5 μl 10 mmol/l deoxynucleoside
triphosphate (dNTP), 2.4 μl MgCl2 (25 mmol/l), 2 μl each
forward and reverse primer, 1 μl cDNA, 0.1 μl Taq DNA
polymerase, and 9 μl distilled water. The PCR reaction conditions
were as follows: denaturation at 94°C for 5 min; 35 cycles each of
denaturation at 95°C for 50 sec, annealing at 55°C for 1 min, and
extension at 72°C for 2 min; followed by a final extension at 72°C
for 7 min. A 10-μl sample of each reaction was subjected to
electrophoresis on a 2% agarose gel, and the gel was stained with
ethidium bromide after electrophoresis for DNA band
visualization.
Transfection with siRNA
The small interfering RNA (siRNA) oligonucleotides
were synthesized by Jikai Gene Chemical Technology Co. Ltd.
(Shanghai, China). According to the principles of siRNA design
using the RNA online tools of Invitrogen and the mRNA sequences of
hMSH2 in GenBank (Gene ID: 4436), 3 siRNA targeting
different parts of hMSH2 mRNA and an siRNA with a random
sequence were designed (hMSH2-siRNA-#1,
hMSH2-siRNA-#2, hMSH2-siRNA-#3 and negative-siRNA;
Table I). The BLAST sequence
alignment algorithm was used to exclude homology (http://blast.ncbi.nlm.nih.gov/Blast.cgi)
(20).
 | Table IsiRNA sequences. |
Table I
siRNA sequences.
| Genes | Sequence |
|---|
|
hMSH2-siRNA-#1 |
CTTGCTGAATAAGTGTAAA |
|
hMSH2-siRNA-#2 |
TGGCAATCTCTCTCAGTTT |
|
hMSH2-siRNA-#3 |
AGTAATGGAATGAAGGTAA |
| Negative-siRNA |
TTCTCCGAACGTGTCACGT |
We constructed pGCSIL-GFP-hMSH2 and
pGCSIL-GFP-negative lentivirus vectors. 293T cells were
cotransfected with plasmid pGCSIL-GFP, pHelper1.0, and pHelper2.0
to produce a virus stock (Genechem Co. Ltd., Shanghai, China). The
OC3/TAX300 ovarian carcinoma cell line was subjected to 3 types of
treatments (groups): i) experimental (EX) groups, in which cells
were transfected with the hMSH2-siRNA-1#-plasmid,
hMSH2-siRNA-2#-plasmid, and hMSH2-siRNA-3#-plasmid;
the titer of the virus was 2×109, 4×108 and
3×108 TU/ml, respectively; ii) negative control (NC)
group, in which cells were transfected using a
negative-siRNA-plasmid that has been used as the negative control
in a number of studies (21,22),
and has only 16 consecutive bases that are identical to 2 genes in
zebrafish among all sequences in the GenBank database; iii) blank
control (BC) group, in which cells were not transfected.
The OC3/TAX300 ovarian carcinoma cell line was
cultured in RPMI-1640 medium supplemented with 10% bovine calf
serum at 37°C in an incubator with a 5% CO2 atmosphere
and passaged every 2 days. When the cells reached the logarithmic
growth phase, they were added to the wells of a 96-well plate at a
concentration of 3.0–5.0×104 cells/ml (90 μl/well). When
the cells were 70–80% confluent, they were transfected in the
presence of Lipofectamine 2000 (LF2000, Invitrogen), according to
the manufacturer’s protocol. Transfection efficiency was determined
72 h after transfection, by measuring the green fluorescence
intensity of the cells in each well by fluorescence microscopy.
Transfection efficiency was determined using the formula:
Transfection efficiency (%) = (the number of fluorescent cells/the
total number of cells) × 100%. A cell line with better transfection
efficiency was used for subsequent experiments. All experiments
were performed in triplicate.
Real time-PCR analysis
Total RNA was extracted from the cultured cells
using TRIzol reagent. hMSH2 primers were designed according
to sequence data obtained from GenBank, and ACTB (β-actin)
was used as an internal control. The primer sequences were as
follows: 5′-AAG AAG CCC AGG ATG CCA TT-3′ (sense) and 5′-AGC ATC
TAG CTG AGC TAA CAC ATC A-3′ (antisense) for hMSH2; 5′-AGG
TCA TCA CCA TTG GCA ATG-3′ (sense) and 5′-GGT AGT TTC GTG GAT GCC
ACA-3′ (antisense) for ACTB. Real time-PCR cycling
parameters were as follows: 50°C for 2 min; 95°C for 10 min; and 40
cycles each of 95°C for 15 sec and 60°C for 1 min for the
amplification curve and 95°C for 15 sec, 60°C for 15 sec, and 95°C
for 15 sec for the dissociation curve. The data were analyzed using
SDS 2.2 software and exported to an Excel spreadsheet. Target gene
expression was normalized to that of ACTB. The mRNA expression
ratio of hMSH2/ACTB was calculated using
2−ΔΔCt analysis, which represents the relative
expression values of hMSH2 mRNA. Ct is the number of cycles
when the DNA concentration reached the threshold. The formula is as
follows: ΔCt = Ct (hMSH2) - Ct (ACTB). ΔΔCt = ΔCt
(experimental group) - ΔCt (blank control).
Western blot analysis
The transfected cells were centrifuged at 4°C (5,000
rpm, 5 min) and the supernatant was saved. Total cell lysates were
prepared using cell-lysis buffer. After the cell lysates were
incubated for 30 min on ice, they were centrifuged at 4°C (12,000
rpm, 10 min) and heated at 100°C for 5 min. The extracted proteins
(30–40 μl) were separated using 10% sodium
dodecylsulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and
were electrophoretically transferred onto an Immobilon-P membrane.
The membranes were incubated in a blocking solution for 1–3 h at
room temperature and then incubated with rabbit anti-human
hMSH2 monoclonal antibodies (diluted 1:1,000) at 4°C
overnight. After the membranes were washed three times with
Tris-buffered saline with Tween-20, they were incubated for 1–2 h
with a fluorescent secondary antibody (diluted 1:5,000) at room
temperature. The membranes were washed as described above and
analyzed using a two-color infrared imaging system (Odyssey;
Li-COR, Lincoln, NE, USA). The gray level of each band was
calculated using the image processing software Image J (National
Institutes of Health, Bethesda, MD, USA).
MTT assay
The IC50 value of paclitaxel was
determined in each of the 3 groups. Cells in the logarithmic growth
phase were seeded into 96-well plates at a concentration of
1×104 cells/well. On reaching confluency, the cells were
treated with paclitaxel at various concentrations: 0 (control), 1,
2, 4, 8, 16 and 32 μg/ml for 48 h. IC50 of paclitaxel
was determined in triplicate for each group. The cells were
incubated at 37°C in an incubator with a 5% CO2
atmosphere. At the end of the treatment, the medium was removed by
vacuum and replaced with 200 μl of fresh RPMI-1640 medium. Twenty
microliters of MTT stock solution (5 mg/ml) was added to each well
after which the cells were incubated for an additional 4 h. The
supernatant was then removed and cell pellets were resuspended in
150 μl DMSO by using 5 min of constant agitation. Cell viability
was determined by measuring the absorbance at 492 nm using a
microplate reader. The following equation was used to calculate
cell growth inhibition rate: Cell growth inhibition rate = [1 -
(absorbance value of each well treated with paclitaxel/absorbance
value of the control well without paclitaxel)] × 100%.
IC50 of paclitaxel was measured by chartography.
Flow cytometric analysis of apoptosis and
the cell cycle
Cells in logarithmic phase were treated with 2 μg/ml
of paclitaxel for 24 and 48 h. After the culture media was removed,
the cells were trypsinized (0.25% trypsin without EDTA), washed
twice with phosphate-buffered saline, and made into a single-cell
suspension at a concentration of 1.5×106 cells/ml. Next,
1 ml of the cell suspension was centrifuged at 200 rcf for 10 min
at 4°C, and the pellet was resuspended in 500 μl flow cytometry
binding buffer. In accordance with the instructions of the Annexin
V-PE/7-AAD apoptosis test kit, 20 μl propidium iodide was added to
the suspension, and the tube was incubated at room temperature for
1 h in the dark. Flow cytometric analysis was then performed to
determine cell viability and to analyze the progression of the cell
cycle after the treatment.
Electron microscopic observations of
apoptosis-induced morphological changes
Cells in each of the 3 groups were treated with 2
μg/ml paclitaxel for 48 h and fixed in 4% gluteraldehyde at 4°C for
1.5 h. The cells were rinsed with rinse solution (0.18 M sucrose in
0.1 MPB, 4°C) twice and further incubated with osmium tetroxide for
1 h. Cells were then washed in distilled water, dehydrated with
graded ethanol, and embedded in solidifying medium for ultra-thin
sectioning. The sections were dyed with uranyl acetate and lead
citrate. Typical ultrastructural changes in cell apoptosis were
visualized under transmission electron microscopy.
Statistical analysis
Statistical analysis was performed using SPSS
software (version 17.0 for Windows). Data are presented as mean
values ± standard deviation (SD) and comparisons were performed
using a t-test. P≤0.05 was considered to indicate a statistically
significant difference.
Results
CGH and RT-PCR analysis show hMSH2
overexpression in paclitaxel-resistant ovarian carcinoma cells
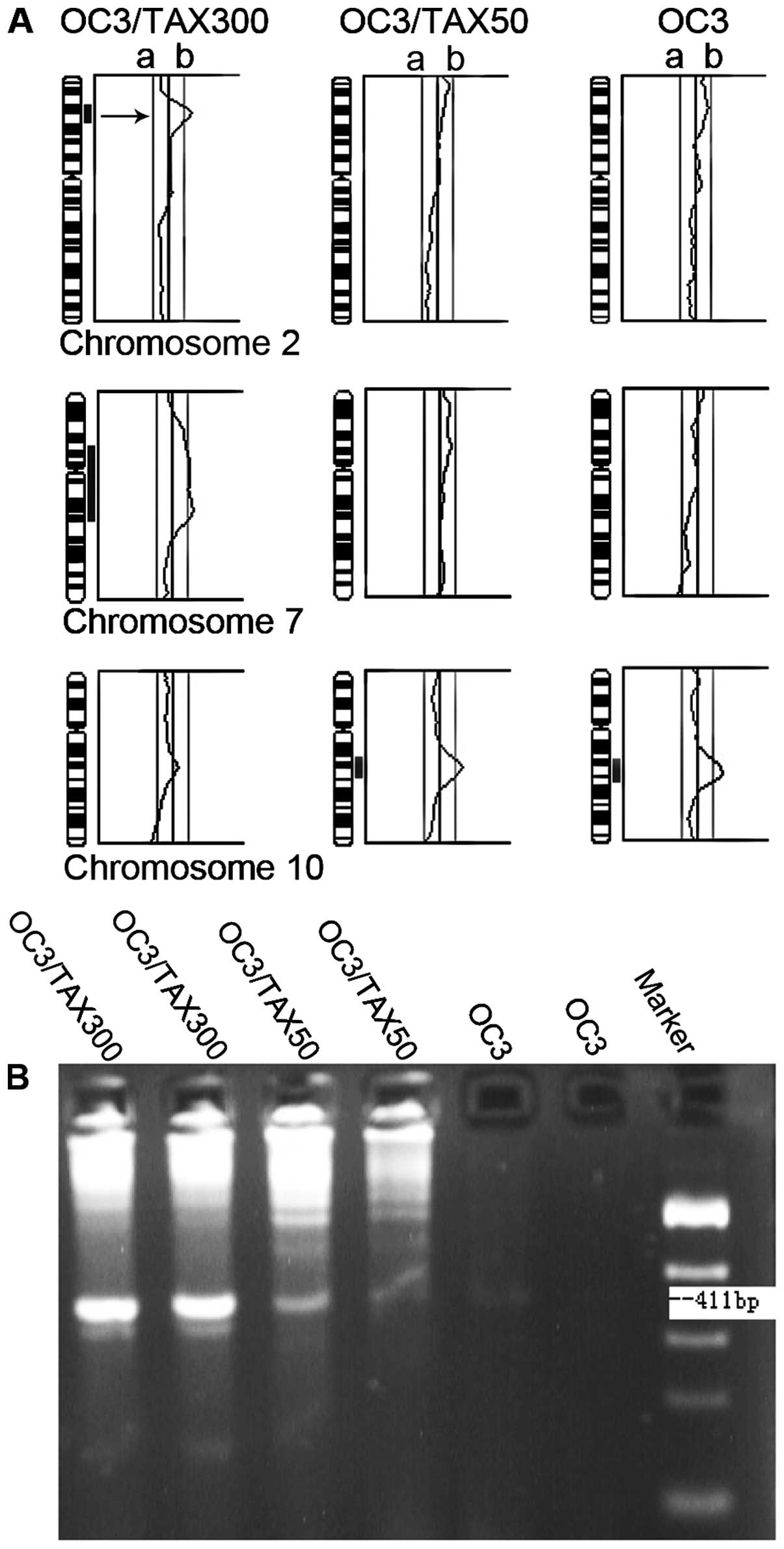
CGH analysis revealed extensive genomic alterations
in all 3 cell lines (Fig. 1A).
Chromosome 2p21 amplification was detected only in the OC3/TAX300
cells. The PCR results correlated closely with the CGH results,
which revealed chromosome 2p21 (gene locus of hMSH2)
amplification (Fig. 1B). The
expression rates of paclitaxel-treated and paclitaxel-untreated
tissue samples were 93.9 (31/33) and 47.6% (10/21), respectively
(P=0.0001). The expression rate was 93.3 and 54.2% in
low-differentiated ovarian carcinoma tissue samples and
moderate-to-highly differentiated tissue samples, respectively
(P=0.0008).
hMSH2-siRNA inhibits hMSH2 mRNA and
protein expression in OC3/TAX300 cells
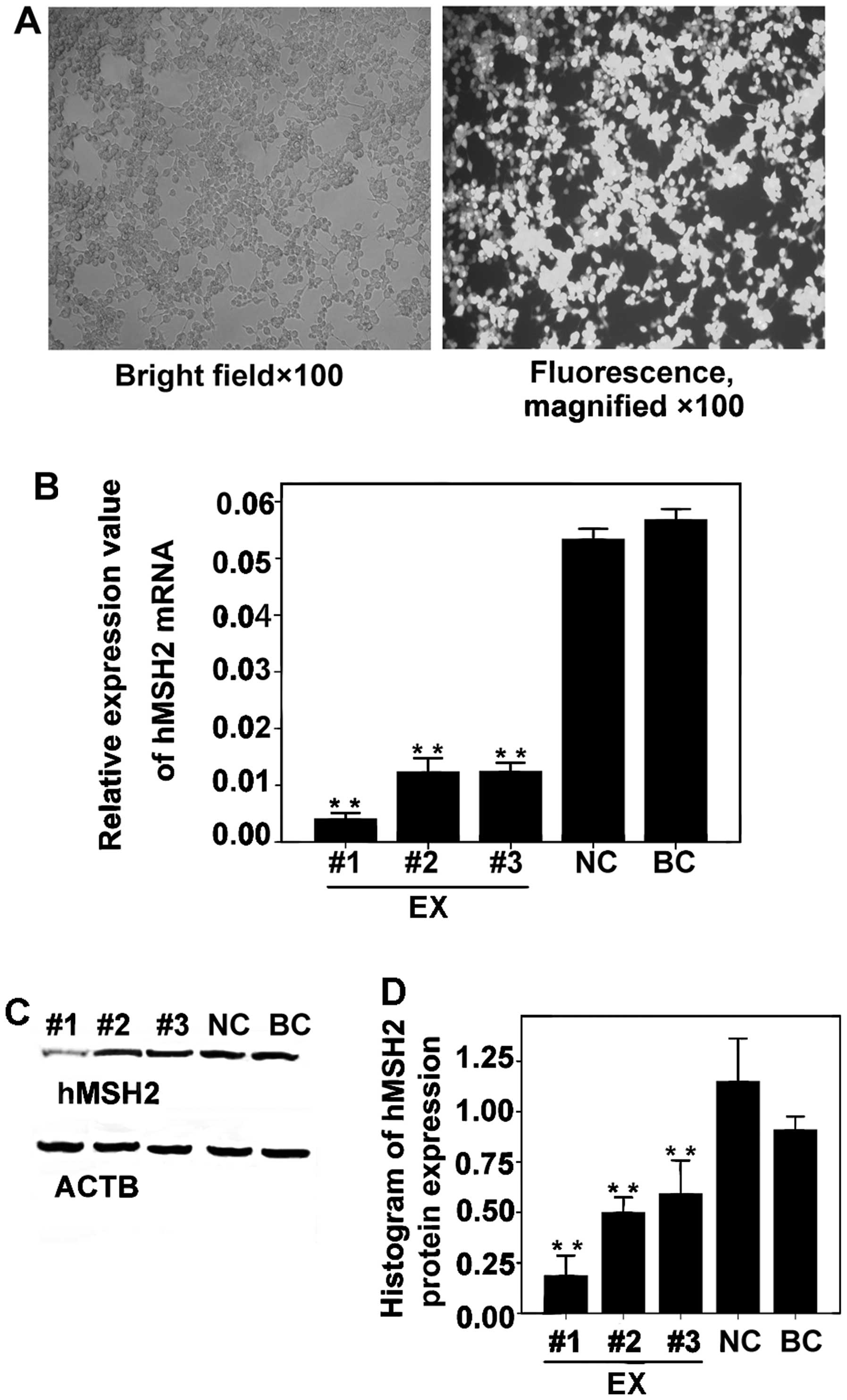
We found that the cells were transfected at the
highest efficiency and that almost all cells emitted green
fluorescence (Fig. 2A). The
relative expression levels of hMSH2 mRNA in the EX groups
were significantly reduced, particularly by hMSH2-siRNA-#1
in comparison with the results of the NC (P<0.001) and BC groups
(P<0.001; Fig. 2B). hMSH2
protein expression in the EX group was significantly inhibited,
particularly by hMSH2-siRNA-#1, in comparison with the
results for the NC (P<0.001) and BC (P<0.001) groups
(Fig. 2C and D). No significant
difference was observed between the inhibition of hMSH2
expression in the NC and BC groups (P>0.05). Therefore,
hMSH2-siRNA-1# was used in the subsequent experiments.
Paclitaxel resistance is reversed by
inhibition of hMSH2 expression
Cell growth inhibition rate curves were constructed
for paclitaxel using the following concentration gradient: 0, 1, 2,
4, 8, 16 and 32 μg/ml (Fig. 3A).
The inhibitory effect of paclitaxel on cell growth in the EX group
was the most obvious. The IC50 for paclitaxel in the EX
group (2.078 μg/ml) was considerably lower than that in the NC
group (13.778 μg/ml; P<0.001) and BC group (14.056 μg/ml;
P<0.001; Fig. 3B). No
significant difference was observed between the IC50
values of the NC and BC groups (P>0.05).
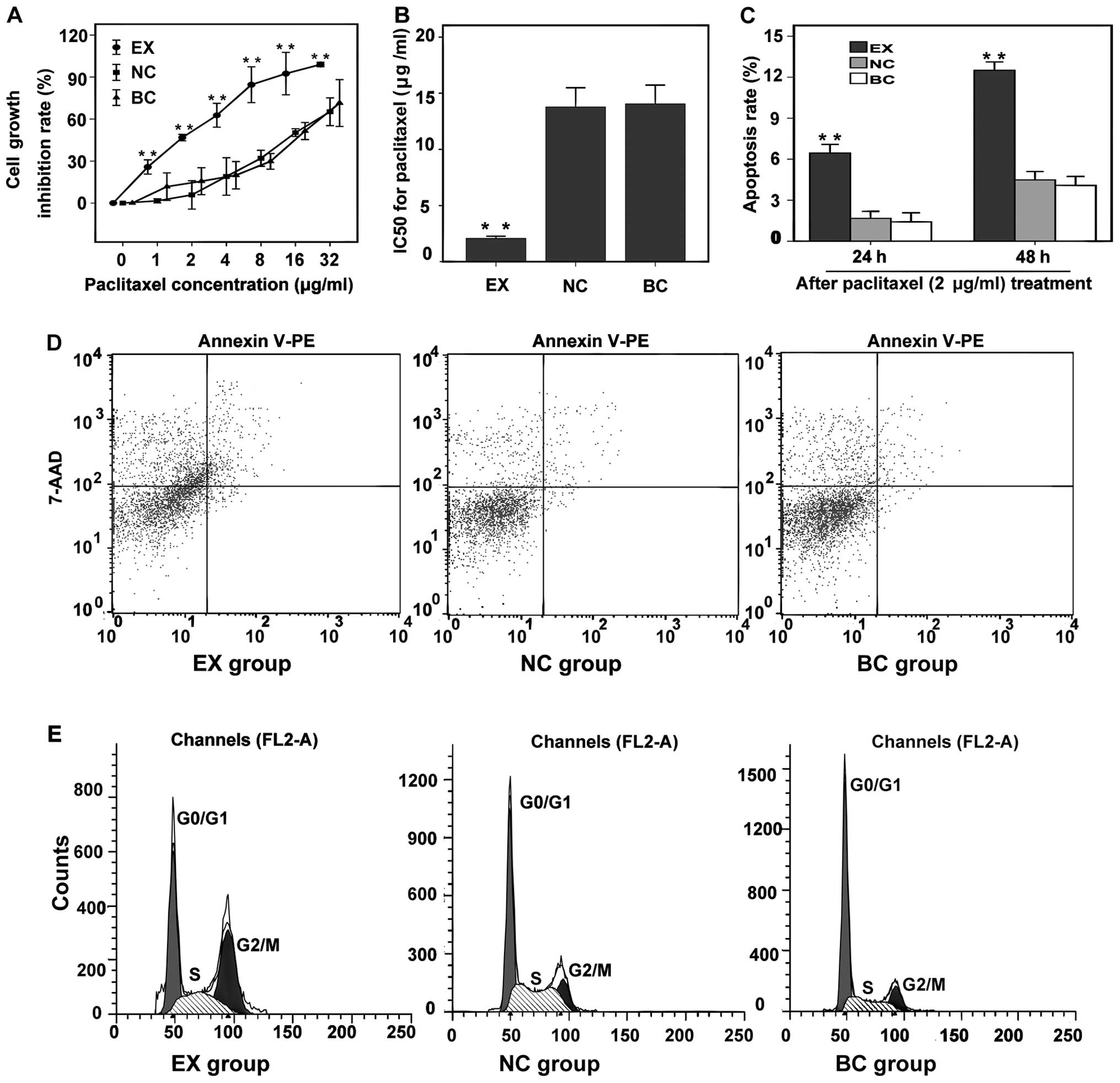 | Figure 3Effect of the inhibition of
hMSH2 expression on paclitaxel resistance. (A) Cell growth
inhibition rate curves of cells in all groups after paclitaxel
treatment in the following concentration gradient: 0, 1, 2, 4, 8,
16 and 32 μg/ml. (B) The IC50 for paclitaxel in the 3
groups. (C) Apoptosis rate at 24 and 48 h after paclitaxel (2
μg/ml) treatment (%). (D) Apoptosis rate at 48 h after paclitaxel
treatment (%). Lower left quadrant, living cells; lower right
quadrant, early apoptotic cells; upper right quadrant, late
apoptotic cells; upper left quadrant, necrotic cells. Apoptosis
rate = number of apoptotic cells/total cells × 100%. (E) Cell cycle
analysis at 48 h after paclitaxel (2 μg/ml) treatment. EX group,
experiment group; NC group, negative control group; BC group, blank
control group. |
Apoptosis rate and arrest of the
G2/M phase of the cell cycle are significantly increased
in paclitaxel-treated transfected cells
The cell cycle progression and apoptosis rate were
analyzed after paclitaxel treatment at a concentration of 2 μg/ml.
This concentration was selected because it was approximately
equivalent to the IC50 value in the EX group.
The apoptosis rates for the EX group increased with
time and were significantly higher at 24 and 48 h (6.45 and 12.46%,
respectively) than those for the NC (1.67 and 4.46%, respectively;
P<0.001) and BC groups (1.42 and 4.09%, respectively;
P<0.001) (Fig. 3C and D). No
significant difference was observed between the apoptosis rates for
the NC and BC groups (P>0.05). The G2/M ratios for
the EX group after paclitaxel treatment for 24 and 48 h were
significantly higher (20.61 and 38.02%, respectively) than those
for the NC (10.84 and 13%, respectively; P<0.001) and BC groups
(10.45 and 13.17%, respectively; P<0.001) (Figs. 3E, 4A
and B). The S-phase ratios for the EX group after paclitaxel
treatment for 24 were significantly higher (25.70%) than that for
the NC (21.85%, P<0.001) and BC groups (22.29%, P<0.001)
(Fig. 4A). The G0/G1 ratios for the
EX group after paclitaxel treatment for 24 and 48 h were
significantly lower (53.69 and 38.23%, respectively) than those for
the NC (67.32 and 64.43%, respectively; P<0.001) and BC groups
(67.25 and 63.97%, respectively; P<0.001) (Figs. 3E, 4A
and B). No significant difference was observed between the cell
cycle progression (P>0.05) in the NC and BC groups.
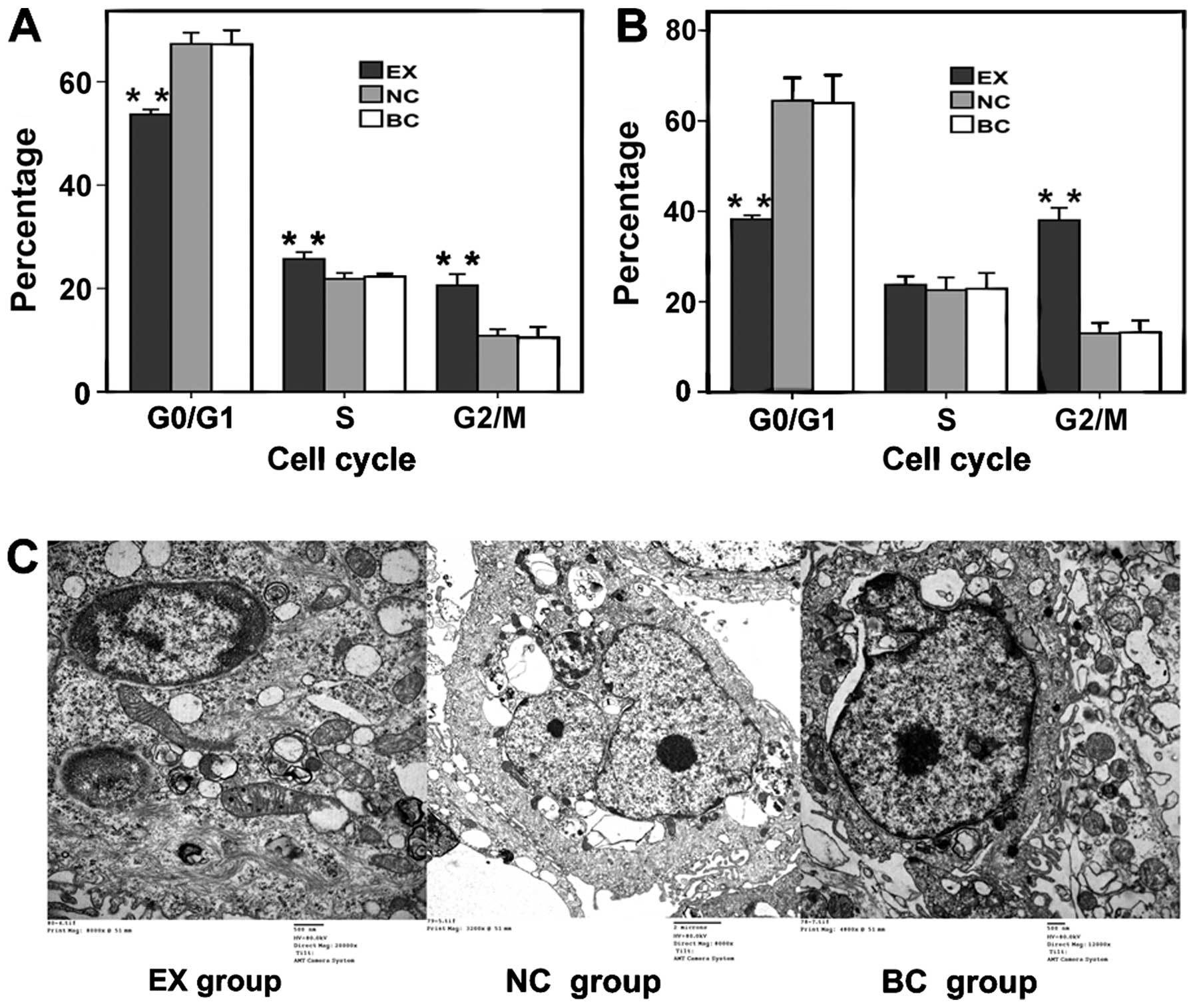 | Figure 4(A) Cell cycle analysis 24 h after
paclitaxel (2 μg/ml) treatment in the 3 groups. (B) Cell cycle
analysis 48 h after paclitaxel (2 μg/ml) treatment in the 3 groups.
(C) Ultrastructural changes in the cells undergoing apoptosis. EX
group, chromatin margination, nuclear condensation, and
intracytoplasmic vacuoles (scale bar, 500 nm). NC group, cell with
nearly normal morphological features, central nuclei and clear
nucleoli (scale bar, 2 μm). BC group, mild chromatin pyknosis and
marginalized mild swelling of mitochondria (scale bar, 500 nm). EX
group, experiment group; NC group, negative control group; BC
group, blank control group. |
Ultrastructural changes in cells undergoing
apoptosis were observed using electron microscopy. Compared with
the NC and BC group cells, EX group cells exhibited more visible
cell shrinkage, severe chromatin margination, nuclear condensation,
fragmentation, and apoptotic body formation than the control cells.
Moreover, the nucleolus disappeared, the mitochondria swelled
markedly, mitochondrial cristae disappeared, and other signs of
apoptosis appeared. In the NC and BC groups, the nucleus was
centrally located in the cells and the nucleolus was clear; the
chromatin was pyknotic and marginalized with mild mitochondrial
swelling (Fig. 4C).
Discussion
Our study investigated the relationship between
hMSH2 and paclitaxel resistance in ovarian cancer cell lines
for the first time. We found hMSH2 overexpression in the
paclitaxel-resistant cell lines, which was established by exposing
OC3 ovarian carcinoma cells to different paclitaxel doses, as well
as in the paclitaxel-treated ovarian carcinoma tissues. These
findings indicate that hMSH2 upregulation is related to
paclitaxel treatment. We propose the following mechanism to explain
this phenomenon: DNA damage to carcinoma cells treated with
chemotherapeutic drugs activates hMSH2 expression.
hMSH2 repairs the drug-induced damage to DNA and prevents
tumor cell death.
Subsequent experiments suggested that the poor
prognosis of patients with low-differentiated ovarian carcinomas is
related to the development of drug resistance. There are many
reports concerning the upregulation of hMSH2 expression in
tumors and its correlation with prognosis. For example, Ciavattini
et al (23) reported that
the frequency of hMSH2 expression in patients with invasive
and pre-invasive cervical cancer was higher than that of normal
cervical epithelium. Materna et al (24) analyzed 73 ovarian carcinoma samples
and found that patients with undetectable hMSH2 expression
experienced higher overall survival rates than those with
detectable hMSH2 expression (81 and 42%, respectively;
P=0.013). Vageli et al (25)
reported that patients with lung adenocarcinoma showing increased
hMSH2 expression had poor outcomes compared with those with
low hMSH2 expression. However, contradicting results were
obtained for patients with squamous cell carcinoma of the lung.
Nadin et al (26) found
that, at 24 h after chemotherapy, hMSH1 and hMSH2
expression in approximately 83% of cisplatin-treated cancer
patients with complete responses was higher than the mean value,
indicating that the MMR pathway is important for correcting
cisplatin-induced DNA damage. Similarly, higher hMSH2
expression was detected in urinary tract and prostate cancers
(27–29).
We further investigated whether we could reverse
paclitaxel resistance by inhibiting hMSH2 expression through
RNAi technology. We found that the IC50 of paclitaxel
was significantly decreased, and the apoptosis rate and
G2/M ratios of the cell cycle were significantly
increased in paclitaxel treated-transfected cells. The results
further confirm that low expression of hMSH2 could reduce drug
resistance, sensitivity to paclitaxel can be restored by knocking
down the hMSH2 gene, and the failure of chemotherapy is
associated with the elevated DNA damage repair capacity (13–16,30).
DNA repair systems eliminate the damaged or
mismatched nucleotides of DNA and have been confirmed as being
closely associated with resistance to chemotherapeutic drugs. DNA
repair mechanisms are classified into the following 5 categories
according to their basic chemical role in repairing damage
(13,31–35).
First, direct repair (DR) is a simple way to repair DNA damage.
O6-methylguanine-DNA methyltransferase (MGMT) is the
most important immediate repair enzyme. Wiewrodt et al
(31) found that MGMT activity of
recurrent glioblastomas significantly increased after radiotherapy
or a combined treatment with alkylating agents (e.g. temozolomide
and chloroethylnitrosoureas). Second, MMR has a mechanism similar
to that of DR. Third, base-excision repair, which is the most
active DNA repair pathway in mammals, is primarily responsible for
the repair of DNA base damage such as base loss and DNA single
strand breaks caused by spontaneous hydrolysis, reactive oxygen
species, or alkylating agents (32,33).
Fourth, nucleotide-excision repair is the major repair process
active against larger damages in the DNA of normal cells and is
composed of a multi-enzyme DNA repair pathway. One of the most
crucial enzymes is the excision repair cross complementing 1
(ERCC1). A high level of ERCC1 expression has been reported in
cisplatin-resistant tumors (34–36).
Fifth, double-strand break repair (DSBR)comprises 2 sub-pathways:
homologous recombination (HR), and non-homologous end joining
(NHEJ). Both are mediated by complex pathways that involve a series
of components and multi-step reaction processes. The key enzyme in
DSBR is Rad51. Studies have found that Rad51 overexpression is an
important cause of gefitinib and cisplatin resistance in lung
cancer. Knockdown of Rad51 significantly enhanced cell death after
cisplatin treatment (37).
DNA repair system provides a potential target for
reversing resistance to chemotherapy (38,39).
In recent years, many researchers have synthesized agents that
target special DNA repair proteins and have reported that
inhibiting DNA damage repair systems may restore the response to
chemotherapy in some resistant tumors; combining DNA repair
inhibitors with chemotherapeutic drugs can also significantly
improve therapy effectiveness. Several clinical trials have been
carried out using different DNA damage repair inhibitors that
target several enzymes such as PARP, DNA-PK and MGMT (16,36,37,40,41).
Therefore, blocking hMSH2 expression may be
an important new way to treat cancer. However, there are still
several barriers to the application of this strategy, since drug
resistance is not the result of hMSH2 expression alone and
depends on many other complex pathways and the multiple molecular
structure of DNA (8,42). Studies involving the mechanisms
underlying the relationship between hMSH2 and drug
resistance should continue in the future.
Our experiments are the first to indicate that the
expression of hMSH2 is upregulated in ovarian carcinoma cell
lines and tissues after paclitaxel treatment, and hMSH2
overexpression is related to paclitaxel resistance and poor
prognosis. Inhibition of hMSH2 expression indicates a new
direction in adjuvant therapy for ovarian carcinoma.
Acknowledgements
The present study was supported by the National High
Technology Research and Development Program of China (grant no.
2006AA02A245).
References
|
1
|
Jemal A, Siegel R, Xu J and Ward E: Cancer
statistics, 2010. CA Cancer J Clin. 60:277–300. 2010. View Article : Google Scholar
|
|
2
|
Osterberg L, Levan K, Partheen K, Delle U,
Olsson B, Sundfeldt K, et al: Specific copy number alterations
associated with docetaxel/carboplatin response in ovarian
carcinomas. Anticancer Res. 30:4451–4458. 2010.PubMed/NCBI
|
|
3
|
Takano M, Kudo K, Goto T, Yamamoto K, Kita
T and Kikuchi Y: Analyses by comparative genomic hybridization of
genes relating with cisplatin-resistance in ovarian cancer. Hum
Cell. 14:267–271. 2001.PubMed/NCBI
|
|
4
|
Marcelis CL, van der Putten HW, Tops C,
Lutgens LC and Moog U: Chemotherapy resistant ovarian cancer in
carriers of an hMSH2 mutation? Fam Cancer. 1:107–109. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Honda M, Okuno Y, Hengel SR, et al:
Mismatch repair protein hMSH2-hMSH6 recognizes mismatches and forms
sliding clamps within a D-loop recombination intermediate. Proc
Natl Acad Sci USA. 111:E316–E325. 2014. View Article : Google Scholar
|
|
6
|
Lynch HT, Lynch PM, Lanspa SJ, Snyder CL,
Lynch JF and Boland CR: Review of the Lynch syndrome: history,
molecular genetics, screening, differential diagnosis, and
medicolegal ramifications. Clin Genet. 76:1–18. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Masuda K, Banno K, Hirasawa A, et al:
Relationship of lower uterine segment cancer with Lynch syndrome: A
novel case with an hMLH1 germline mutation. Oncol Rep.
28:1537–1543. 2012.PubMed/NCBI
|
|
8
|
Yasin SL and Rainbow AJ: A combination of
MSH2 DNA mismatch repair deficiency and expression of the SV40
large T antigen results in cisplatin resistance of mouse embryonic
fibroblasts. Int J Oncol. 39:719–726. 2011.
|
|
9
|
Kheirelseid EA, Miller N, Chang KH, Curran
C, Henessey E, Sheehan M and Kerin MJ: Mismatch repair protein
expression in colorectal cancer. J Gastrointest Oncol. 4:397–408.
2013.PubMed/NCBI
|
|
10
|
Shah SN, Hile SE and Eckert KA: Defective
mismatch repair, microsatellite mutation bias, and variability in
clinical cancer phenotypes. Cancer Res. 70:431–435. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Poulogiannis G, Frayling IM and Arends MJ:
DNA mismatch repair deficiency in sporadic colorectal cancer and
Lynch syndrome. Histopathology. 56:167–179. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kamat N, Khidhir MA, Alashari MM and
Rannug U: Microsatellite instability and loss of heterozygosity
detected in middle-aged patients with sporadic colon cancer: A
retrospective study. Oncol Lett. 6:1413–1420. 2013.PubMed/NCBI
|
|
13
|
Chaney SG and Sancar A: DNA repair:
enzymatic mechanisms and relevance to drug response. J Natl Cancer
Inst. 88:1346–1360. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ding J, Miao ZH, Meng LH and Geng MY:
Emerging cancer therapeutic opportunities target DNA-repair
systems. Trends Pharmacol Sci. 27:338–344. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fox M and Roberts JJ: Drug resistance and
DNA repair. Cancer Metastasis Rev. 6:261–281. 1987. View Article : Google Scholar
|
|
16
|
Sánchez-Pérez I: DNA repair inhibitors in
cancer treatment. Clin Transl Oncol. 8:642–646. 2006.
|
|
17
|
Alzoubi K, Khabour O, Khader M, Mhaidat N
and Al-Azzam S: Evaluation of vitamin B12 effects on DNA damage
induced by paclitaxel. Drug Chem Toxicol. Nov 11–2013.(Epub ahead
of print). View Article : Google Scholar
|
|
18
|
Zhang J, Zhao J, Zhang W, et al:
Establishment of paclitaxel-resistant cell line and the underlying
mechanism on drug resistance. Int J Gynecol Cancer. 22:1450–1456.
2012.PubMed/NCBI
|
|
19
|
Zhang L, Liu P, Li H and Xue F: Effect of
histone deacetylase inhibitors on cell apoptosis and expression of
the tumor suppressor genes RUNX3 and ARHI in ovarian tumors. Mol
Med Rep. 7:1705–1709. 2013.PubMed/NCBI
|
|
20
|
Newell PD, Fricker AD, Roco CA,
Chandrangsu P and Merkel SM: A small-group activity introducing the
use and interpretation of BLAST. J Microbiol Biol Educ. 14:238–243.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lin X, Yu Y, Zhao H, Zhang Y, Manela J and
Tonetti DA: Overexpression of PKCα is required to impart estradiol
inhibition and tamoxifen-resistance in a T47D human breast cancer
tumor model. Carcinogenesis. 27:1538–1546. 2006.
|
|
22
|
Mi J, Zhang X, Liu Y, Reddy SK, Rabbani
ZN, Sullenger BA and Clary BM: NF-kappaB inhibition by an
adenovirus expressed aptamer sensitizes TNF-alpha-induced
apoptosis. Biochem Biophys Res Commun. 359:475–480. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ciavattini A, Piccioni M, Tranquilli AL,
Filosa A, Pieramici T and Goteri G: Immunohistochemical expression
of DNA mismatch repair (MMR) system proteins (hMLH1, hMSH2) in
cervical preinvasive and invasive lesions. Pathol Res Pract.
201:21–25. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Materna V, Surowiak P, Markwitz E,
Spaczynski M, Drag-Zalesinska M, Zabel M and Lage H: Expression of
factors involved in regulation of DNA mismatch repair- and
apoptosis pathways in ovarian cancer patients. Oncol Rep.
17:505–516. 2007.PubMed/NCBI
|
|
25
|
Vageli DP, Zaravinos A, Daniil Z, et al:
hMSH2 and hMLH1 gene expression patterns differ between lung
adenocarcinoma and squamous cell carcinoma: correlation with
patient survival and response to adjuvant chemotherapy treatment.
Int J Biol Markers. 27:e400–e404. 2013. View Article : Google Scholar
|
|
26
|
Nadin SB, Vargas-Roig LM, Drago G, Ibarra
J and Ciocca DR: DNA damage and repair in peripheral blood
lymphocytes from healthy individuals and cancer patients: a pilot
study on the implications in the clinical response to chemotherapy.
Cancer Lett. 239:84–97. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Leach FS, Hsieh JT, Molberg K, Saboorian
MH, McConnell JD and Sagalowsky AI: Expression of the human
mismatch repair gene hMSH2: a potential marker for
urothelial malignancy. Cancer. 88:2333–2341. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Prtilo A, Leach FS, Markwalder R, et al:
Tissue microarray analysis of hMSH2 expression predicts
outcome in men with prostate cancer. J Urol. 174:1814–1818.
2005.PubMed/NCBI
|
|
29
|
Li M, Zhang Q, Liu L, Lu W, Wei H, Li RW
and Lu S: Expression of the mismatch repair gene hMLH1 is enhanced
in non-small cell lung cancer with EGFR mutations. PLoS One.
8:e785002013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Aebi S, Fink D, Gordon R, Kim HK, Zheng H,
Fink JL and Howell SB: Resistance to cytotoxic drugs in DNA
mismatch repair-deficient cells. Clin Cancer Res. 3:1763–1777.
1997.PubMed/NCBI
|
|
31
|
Wiewrodt D, Nagel G, Dreimüller N,
Hundsberger T, Perneczky A and Kaina B: MGMT in primary and
recurrent human glioblastomas after radiation and chemotherapy and
comparison with p53 status and clinical outcome. Int J Cancer.
122:1391–1399. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cadet J, Douki T, Gasparutto D and Ravanat
JL: Oxidative damage to DNA: formation, measurement and biochemical
features. Mutat Res. 531:5–23. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gurubhagavatula S, Liu G, Park S, et al:
XPD and XRCC1 genetic polymorphisms are prognostic factors in
advanced non-small-cell lung cancer patients treated with platinum
chemotherapy. J Clin Oncol. 22:2594–2601. 2004. View Article : Google Scholar
|
|
34
|
Chen HY, Shao CJ, Chen FR, Kwan AL and
Chen ZP: Role of ERCC1 promoter hypermethylation in drug resistance
to cisplatin in human gliomas. Int J Cancer. 126:1944–1954.
2010.PubMed/NCBI
|
|
35
|
Steffensen KD, Smoter M, Waldstrøm M, et
al: Resistance to first line platinum paclitaxel chemotherapy in
serous epithelial ovarian cancer: The prediction value of ERCC1 and
Tau expression. Int J Oncol. 44:1736–1744. 2014.PubMed/NCBI
|
|
36
|
Li QQ, Lee RX, Liang H, Wang G, Li JM,
Zhong Y and Reed E: β-Elemene enhances susceptibility to cisplatin
in resistant ovarian carcinoma cells via downregulation of ERCC-1
and XIAP and inactivation of JNK. Int J Oncol. 43:721–728.
2013.
|
|
37
|
Ko JC, Ciou SC, Cheng CM, et al:
Involvement of Rad51 in cytotoxicity induced by epidermal growth
factor receptor inhibitor (gefitinib, Iressa®) and
chemotherapeutic agents in human lung cancer cells. Carcinogenesis.
29:1448–1458. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhu Y, Hu J, Hu Y and Liu W: Targeting DNA
repair pathways: a novel approach to reduce cancer therapeutic
resistance. Cancer Treat Rev. 35:590–596. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Curtin N: Therapeutic potential of drugs
to modulate DNA repair in cancer. Expert Opin Ther Targets.
11:783–799. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Mohammed MZ, Vyjayanti VN, Laughton CA, et
al: Development and evaluation of human AP endonuclease inhibitors
in melanoma and glioma cell lines. Br J Cancer. 104:653–663. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Rodon J, Iniesta MD and Papadopoulos K:
Development of PARP inhibitors in oncology. Expert Opin Investig
Drugs. 18:31–43. 2009. View Article : Google Scholar
|
|
42
|
Lenglet G and David-Cordonnier MH:
DNA-destabilizing agents as an alternative approach for targeting
DNA: mechanisms of action and cellular consequences. J Nucleic
Acids. 2010:2909352010. View Article : Google Scholar : PubMed/NCBI
|