Introduction
Glioblastoma is the most common and lethal type of
primary brain tumor, accounting for 82% of all malignant glioma
cases (1). Despite multimodal
treatment including surgical resection, adjuvant TMZ-based
chemotherapy and radio therapy, the prognosis of glioblastoma
remains poor, with a median overall survival time of only 15 months
from the time of diagnosis (2). It
is now commonly recognized that the rapid proliferation of
glioblastoma cells, coupled with its widely invasive growth and
persistently aggressive biological behavior, lead to treatment
failure and tumor recurrence (3).
Therefore, the development of novel agents with promising
therapeutic results and minimized toxicities for the treatment of
glioblastoma is urgently needed.
Honokiol
[(3,5-di-(2-propenyl)-1,1-biphenyl-2,2-diol; HNK] is a natural
bioactive molecular compound isolated from the Magnolia
officinalis, a well-known Chinese traditional medicine that has
been used for thousands of years in the treatment of anxiety,
thrombotic stroke and gastrointestinal complaints. Accumulating
studies have demonstrated that HNK exerts multiple pharmacological
activities including anti-inflammatory, anti-bacterial,
anti-angiogenic, anti-thrombocytic and anti-oxidative activity
(4). Moreover, extensive
mechanistic and preclinical studies have indicated that HNK
exhibits potent antitumor activity against a variety of human
cancer cell lines (5–8), without appreciable toxicity (9). The antitumor properties of HNK in
relation to cell cycle arrest (7,8),
apoptosis (5,6) and paraptosis (10), or autophagy (11) have been well characterized. However,
few studies have reported the antineoplastic effects of HNK on
glioblastoma cells and it is not known how apoptosis is induced by
HNK on glioblastoma cells and through which associated pathway this
compound acts. Wang et al (12) reported that HNK could effectively
cross blood-brain barrier (BBB) and blood-cerebrospinal fluid
barrier (BCSFB) and inhibit brain tumor growth in rat 9L
intracerebral gliosarcoma model and human U251 xenograft glioma
model. Another study verified that HNK exerts an anticancer effect
on T98G human glioblastoma cells through the induction of apoptosis
and inhibition of adhesion molecules ICAM-1 and VCAM-1 expression
(13).
In the present study, we investigated the
therapeutic potential of HNK against U87 cells in vitro.
This study demonstrated that HNK inhibits growth of glioblastoma
cells by causing a slight G0/G1 phase cell
cycle arrest and inducing apoptosis via both caspase-dependent and
-independent pathways. We also demonstrated that HNK exerts
antiproliferative activity on glioblastoma cells through the
inhibition of signal transducers and activator of transcription 3
(STAT3) signaling, and ERK1/2 signaling pathway, as well as through
activation of the p38 MAPK signaling pathway. Our findings suggest
that HNK treatment could be a promising therapeutic strategy in
human glioblastoma.
Materials and methods
Cell lines and reagents
The human glioma cell lines U87, U251 and T98G were
grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented
with 10% fetal calf serum (FCS), 100 IU/ml penicillin and 100 IU/ml
streptomycin in a 5% CO2 atmosphere at 37°C. HNK with a
purity of up to 99.5% was kindly provided by Professor Xiao Wang
(Shandong Analysis and Test Center, Shandong Academy of Sciences),
and stock solutions were prepared in dimethyl sulfoxide (DMSO) at a
concentration of 100 mg/ml and dissolved in DMEM medium prior to
use. Cell Counting Kit-8 (CCK8) was purchased from Dojindo
Laboratories (Kumamoto, Japan). Propidium iodide (PI), Rhodamine
123 (Rh123) were purchased from Sigma. Hoechst 33342, p38 inhibitor
(SB203580), JNK1/2 inhibitor (SP600125), MEK-1/2 inhibitor (U0126)
and caspase inhibitor (Z-VAD-fmk) were obtained from Beyotime
Biotechnology Inc. (Nantong, China). Annexin V (FITC) apoptosis
detection kit was obtained from BD Biosciences (San Diego, CA,
USA). Antibodies against Bax, Bcl-2, cleaved caspase-3, −9 and −8,
Fas, FasL, survivin, phospho-ERK1/2, ERK1/2, phospho-p38, p38,
phospho-JNK were purchased from Cell Signaling Technology (Beverly,
MA, USA). JNK antibody was purchased from Santa Cruz. Horseradish
peroxidase-labeled IgG anti-mouse and anti-rabbit antibodies were
supplied by Proteintech Group Inc. (Chicago, IL, USA). GAPDH-HRP
antibody and anti-LC3αβ antibody were obtained from Epitomics.
Cell proliferation assay
Cell proliferation was analyzed by Cell Counting
Kit-8 (CCK-8; Dojindo Laboratories), according to the
manufacturer’s protocol, as previously described (14). Cell viability was measured using
trypan blue exclusion and live cells were enumerated. Cell counts
were expressed as mean ± standard deviation (SD).
Cell cycle distribution
After exposure to 10 μg/ml HNK for different
exposure durations, cells (3×105) were washed twice with
ice-cold phosphate-buffered saline (PBS), and fixed in cold 75%
ethanol at 4°C for at least 24 h. Then, the cells were rinsed with
PBS, resuspended in 1 ml of cell cycle buffer (0.38 mm Na-citrate,
0.5 mg/ml RNase A, and 20 μg/ml propidium iodide) at room
temperature for 40 min and the cells were analyzed using an EPICS
XL flow cytometer with EXPO32™ ADC software (Beckman Coulter,
Miami, FL, USA).
Reverse transcription-PCR assays
Cells were incubated with 10 μg/ml HNK for 24 and 48
h. Then, total RNA was extracted using TRIzol (Invitrogen,
Carlsbad, CA, USA) according to the manufacturer’s instructions.
RNA purity determination, cDNA synthesis, and reverse
transcription-PCR (RT-PCR) were performed as previously described
(16). The primers used were all
synthesized by Sangon Co., Ltd. (Shanghai, China) and the sequences
were as follows: cyclin E (301 bp) sense,
5′-ATACAGACCCACAGAGACAG-3′ and antisense,
5′-TGCCATCCACAGAAATACTT-3′; p27 (270 bp) sense,
5′-ATGTCAAACGTGCGAGTGTC-3′ and antisense, 5′-TCT
GTAGTAGTCGGGCAA-3′; P21 (341 bp) sense, 5′-CCCTCC
TGGCTCTTGATACCC-3′ and antisense, 5′-TGCCCTTCTT
CTTGTGTGTCCC-3′.
Detection of apoptosis
After HNK treatment for 12 or 24 h, cells were
observed under inverted microscope and photographed. Then, the
cells were collected and washed with PBS, fixed with 4%
paraformaldehyde for 15 min, and stained with Hoechst 33342 (10
μg/ml) for 5 min at room temperature in the dark. After washing
three times, cells were resuspended by PBS. Stained nuclei were
observed by a fluorescence microscope and photographed.
HNK-induced U87 cells undergoing apoptosis were
further determined using an Annexin V/FITC apoptosis kit. Briefly,
after exposure to 10, 20 μg/ml of HNK for different time points,
the cells were harvested, washed twice with cold PBS and
resuspended in 100 μl 1X binding buffer containing 5 μl of Annexin
V incubation reagent and 10 μl of PI. After incubation at room
temperature in the dark for 15 min, analysis was carried out using
a Beckman Coulter EPICS XL cytometer.
Caspase inhibition
To evaluate the involvement of caspases in
HNK-induced apoptosis, cells were preincubated with the pan-caspase
inhibitor Z-VAD-fmk (25 μM) for 1 h prior to incubation with HNK.
Annexin V/FITC analysis was performed as previously described.
Analysis of mitochondrial membrane
potential (ΔΨm)
ΔΨm was assessed using fluorescent dye Rh123 and
flow cytometric analysis. Briefly, after HNK treatment for 6 h,
non-adherent cells were collected, and attached cells were
trypsinized and washed twice with PBS. Cells (1×106) in
different groups were incubated with 10 mg/ml Rh123 for 30 min at
37°C. Then, cells were washed twice and resuspended in PBS followed
by flow cytometric analysis. The change in the mean fluorescence
intensity reflects the modification of ΔΨm, which drives the uptake
and accumulation of Rh123 in the mitochondria.
Western blot analysis
Cells were lysed with lysis buffer (50 mM Tris-HCl
pH 7.4, 150 mM NaCl, 1 mM PMSF, 1 mM EDTA, 1% Triton X-100, 0.5%
deoxycholate, 1 μg/ml leupeptin, 2 μg/ml aprotinin, 1 mM
Na3VO4 and 0.1% SDS). Protein concentration
of each sample was determined by the Bradford protein assay. Equal
quantities of protein were subjected to SDS-PAGE and were then
transferred to PVDF membranes. The membranes were blocked with 5%
milk for 1 h and incubated with primary rabbit monoclonal
antibodies against cleaved caspase-3 (1:1,000), −8 and −9, Fas,
FasL, Bcl-2 (1:1,000) and Bax (1:1,000), survivin, phospho-ERK1/2,
ERK1/2, phospho-p38, p38, phospho-JNK, JNK, phospho-STAT3 and STAT3
overnight at 4°C followed by incubation with HRP-conjugated
secondary antibodies for 1 h. The protein bands were visualized
using Immobilon Western Chemiluminescent HRP substrate (Millipore,
Billerica, MA, USA) and pictured by LAS-4000 mini luminescent image
analyzer (Fujifilm, Tokyo, Japan).
Statistical analysis
All experiments were performed in triplicate. Data
are represented as means ± standard deviation (SD). Statistical
analyses were performed using Student’s t-test or one-way ANOVA. In
all analyses, p<0.05 was considered to indicate a statistically
significant difference.
Results
HNK inhibits growth in glioblastoma
cells
To determine the effect of HNK on the cellular
proliferation in glioblastomas, we performed dose-response and
time-course studies in three established cell lines, U251, U87 and
T98G. DMSO, which was used to dissolve HNK, served as the control.
As shown in Fig. 1A, HNK markedly
inhibited growth of all three cell lines in a dose- and
time-dependent manner from 5 to 40 μg/ml HNK at 12–72 h. Based on
the results obtained in terms of cell growth inhibition, we found
that U87 cells were most sensitive to HNK and were selected for the
following steps of our study. Treatment of U87 cells with 40 μg/ml
HNK for 12 h resulted in a great loss of viability (as shown by
trypan blue staining), suggesting that treatment with HNK caused
cell necrosis at this concentration. In comparison, the viability
of cells treated with 20 μg/ml remained at 92% (data not shown).
Therefore, the cells treated with 10 and 20 μg/ml HNK were adopted
to perform the subsequent experiments.
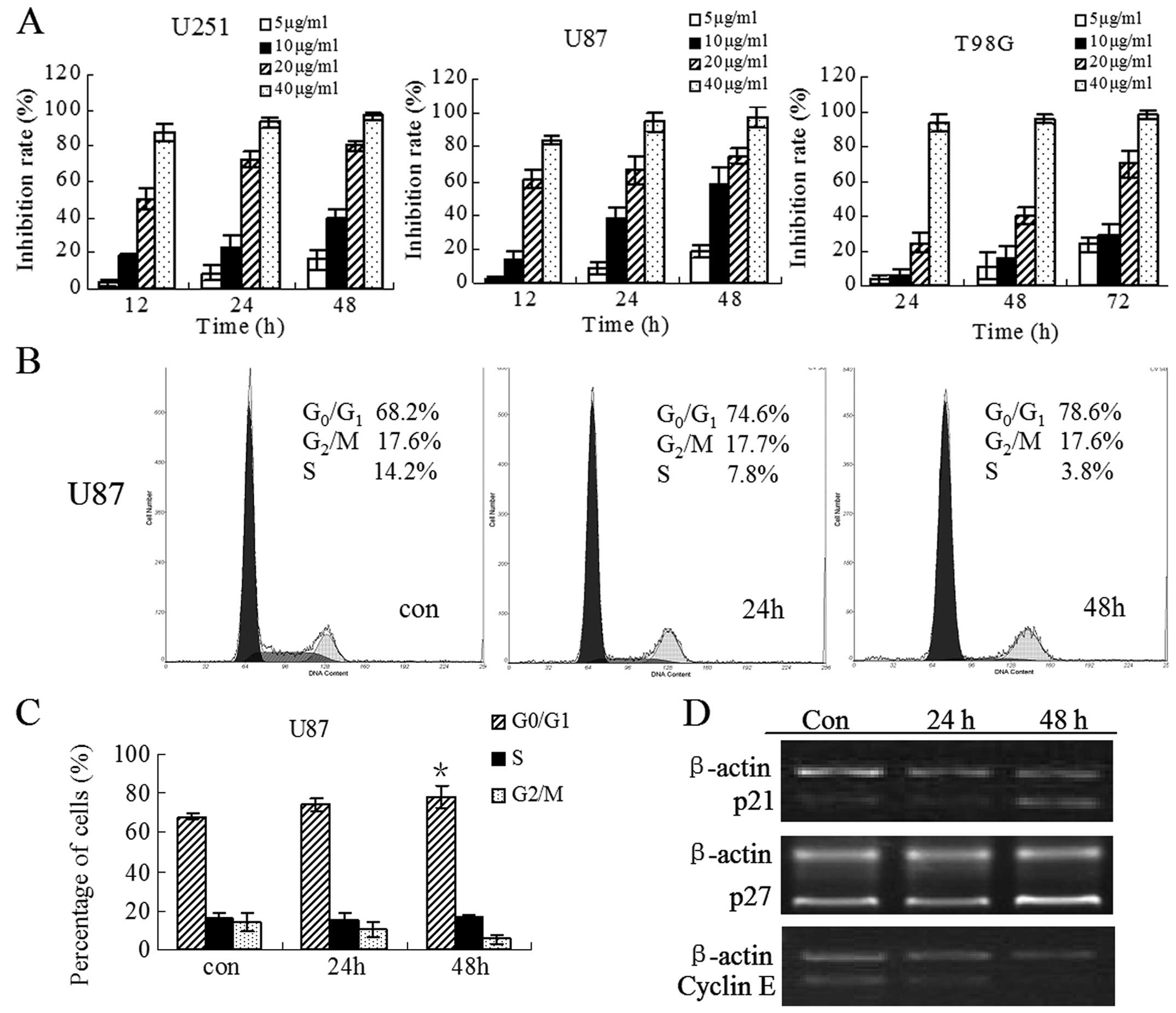 | Figure 1Evaluation of cell growth and cell
cycle distribution of glioblastoma cells after treatment with HNK.
(A) U251, U87 and T98G cells were incubated with HNK at a
concentration of 5, 10, 20, 40 μg/ml for the indicated time points
and the number of surviving cells was determined using CCK8 assay
as described in Materials and methods. (B) The cells were treated
with 10 μg/ml HNK for 0, 24 and 48 h, they were then centrifuged,
incubated in 70% ethanol, stained with PI, and then the DNA content
was analyzed by flow cytometry as described in Materials and
methods. One representative experiment of three is shown. (C) The
histograms show that there was a slight increase of
G0/G1 population with concomitant decrease of
S and G2/M population after treatment with HNK. All
results are described as means ± SD of three independent
experiments. *P<0.05 compared with control. (D) U87
cells were treated with HNK for 24 and 48 h, then the mRNA
expression of p21, p27 and cyclin E was detected by RT-PCR. Each
amplification was performed at least three times. Semi-quantitative
analysis was performed using the ImageJ software program. |
HNK induces a slight
G0/G1 arrest in U87 cells
To determine whether the growth inhibitory effect of
HNK is due to the arrest of cell cycle, the distribution of
cellular DNA content in U87 cells was examined in the presence of
HNK at 10 μg/ml for 24 and 48 h. Representative flow histograms and
quantitative data depicting cell cycle distribution in U87 cultures
are shown in Fig. 1B and C. HNK
caused a slight accumulation in G0/G1 phase
cell population (68.2 in control vs. 74.6 and 78.6% in cells
treated for 24 and 48 h, respectively), accompanied by a
concomitant reduction in S-phase cell population (14.2 in control
vs. 7.8 and 3.8% in HNK-treated cells for 24 and 48 h,
respectively). However, the slight cell cycle block is not
completely in parallel to the marked inhibition of cell
proliferation at the same time points, indicating that a
G0/G1 phase cell cycle arrest may be partly
associated with cell growth inhibition by HNK.
In order to further explore the cell cycle arrest in
U87 cells under the influence of HNK, the mRNA expression of cell
cycle regulatory genes involved in G1/S transition and S
phase was detected by RT-PCR (Fig.
1D). The results indicated that cyclin E was significantly
reduced after HNK treatment for 48 h. In contrast, the expression
of p21 and p27 was pronouncedly upregulated compared to the control
at this time point, while there was no detectable change at 24 h.
Altered expression of cyclin E, p21 and p27 may account for an
arrest in G0/G1 phase, in line with the
change of cell cycle distribution at the same time points.
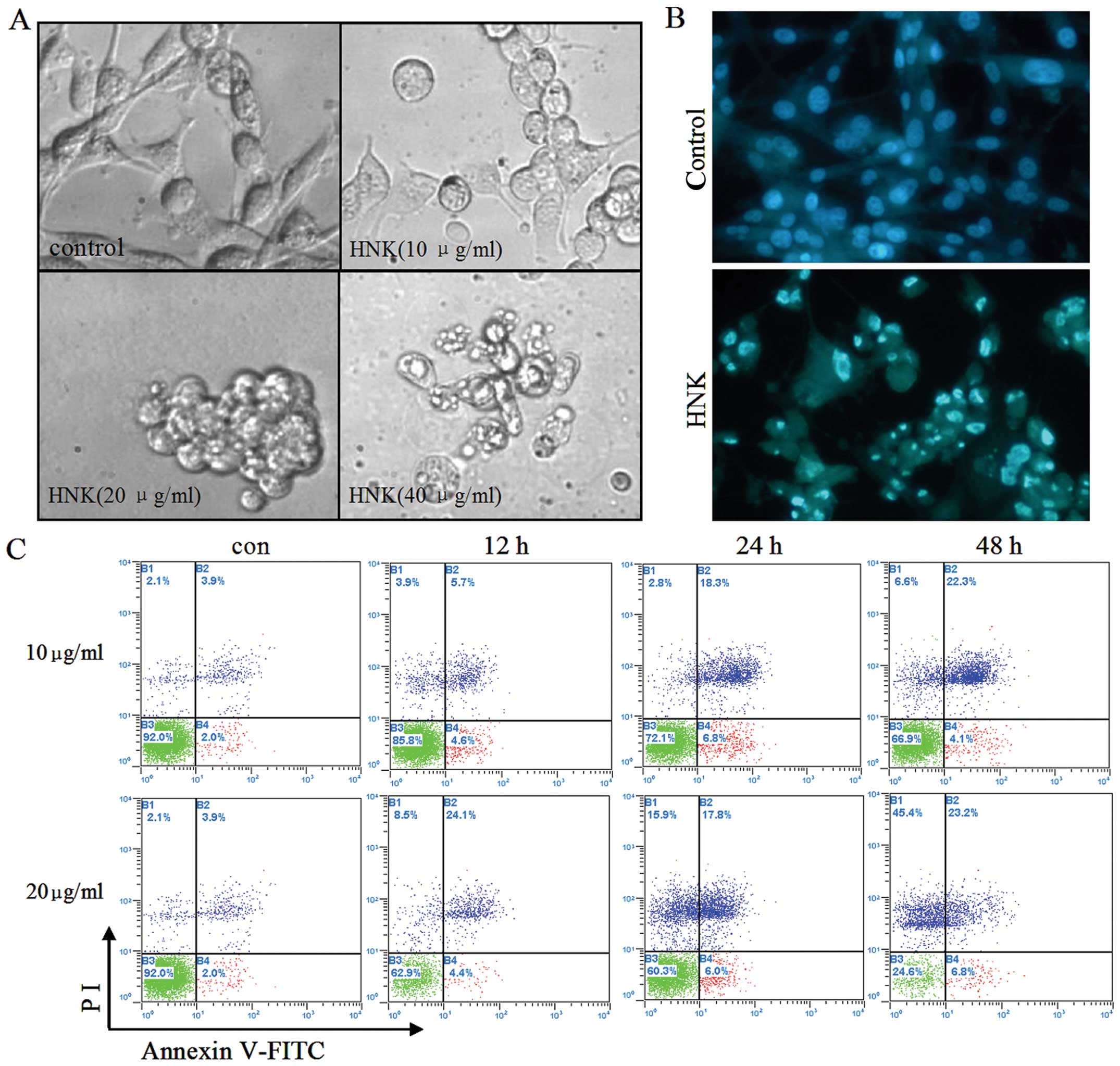
HNK induces apoptosis in U87 cells
To further clarify whether the growth inhibitory
effect of HNK was associated with apoptosis, in the first step,
morphological changes of U87 cells were observed under an inverted
light microscope and an inverted fluorescence microscope after
Hoechst 33342 staining. It was noted that after 10 μg/ml HNK
treatment for 12 h, the number of U87 cells moderately decreased
and a proportion of the cells shrank, detached from the culture
plate and became round. Furthermore, cells treated with 20 μg/ml
HNK displayed greater morphological changes. The cells were
completely detached from the culture plate and apoptotic bodies
were formed. Even at the concentration of 40 μg/ml, most of the
cells underwent necrosis characterized by structural properties
such as a loss of membrane integrity, cellular swelling and
organelle degradation (Fig. 2A).
Trypan blue staining further confirmed the result (data not shown).
Accordingly, Hoechst 33342 staining results showed that the nuclei
of control cells were round and large in size, exhibiting
homogeneous blue fluorescence. By contrast, parts of cells treated
with 10 μg/ml HNK for 24 h exhibited condensed or fragmented nuclei
which is characteristic of cell apoptosis (Fig. 2B). In order to confirm apoptosis
induced by HNK, U87 cells treated with 10, 20 μg/ml HNK for
different durations were analyzed for Annexin V/PI double staining
by flow cytometry. A time-dependent study indicated that HNK at 10
μg/ml induced 10.3, 25.1 and 26.4% of total apoptotic U87 cells
following 12, 24 and 48 h treatment. While at 20 μg/mg, apoptotic
cells reached 28.5, 23.3 and 30% after 12, 24 and 48 h treatment,
accompanied by elevated necrotic cells for 8.5, 15.3 and 45.4%
(Fig. 2C).
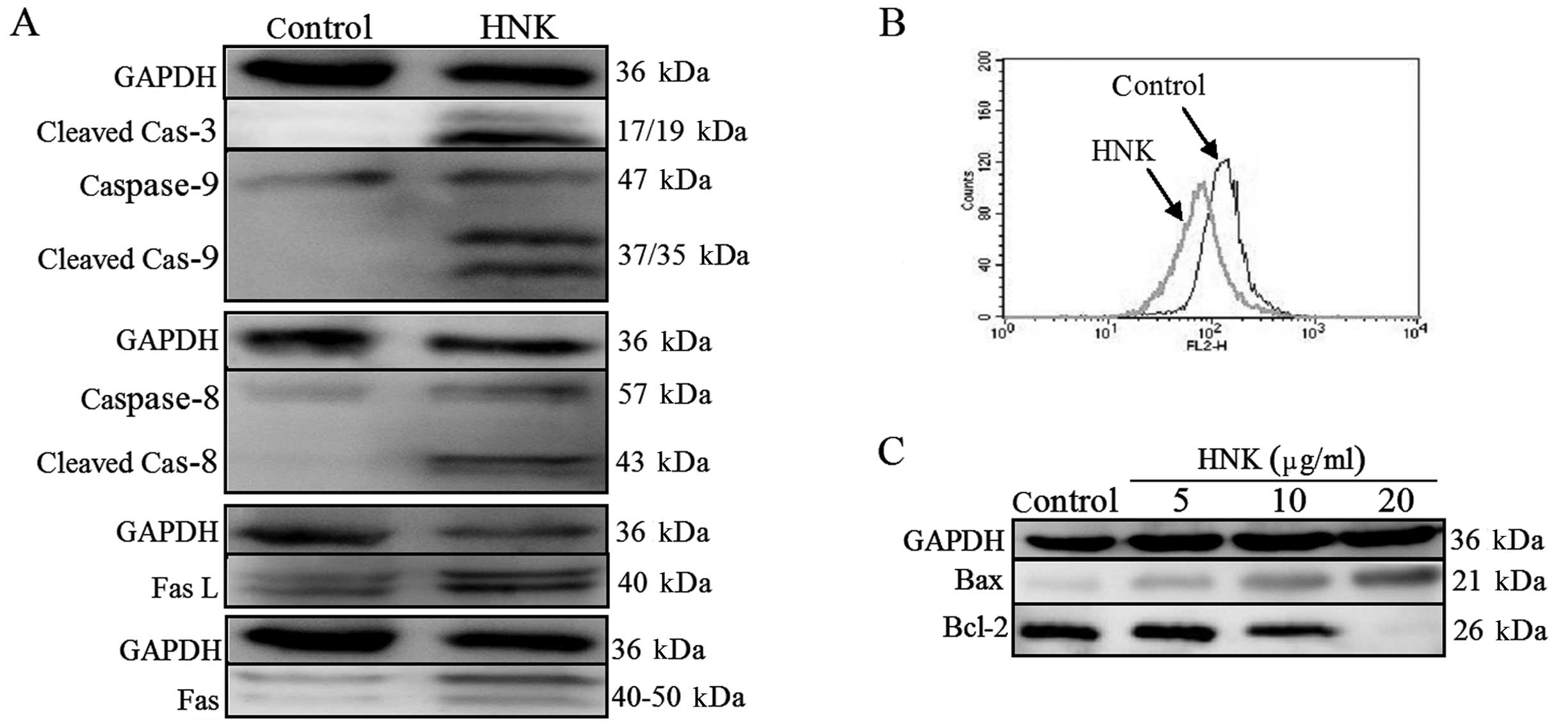
HNK results in the activation of
caspase-3, −8 and −9
Caspase family of cysteinyl-proteases plays the key
role in the initiation and execution of programmed cell death
(16). Thus, the activities of
effector caspase (caspase-3) and initiator caspase (caspase-8 and
−9) were investigated by western blotting. Fig. 3A shows that the cleaved fragments of
caspase-3, −8 and −9, which indicate the activation of caspase-3,
−8 and −9, were observed to be more evident after treatment of U87
cells with 10 μg/ml HNK for 24 h in comparison with those in
control cells. The results suggest that both the extrinsic pathway
(death receptor pathway) and intrinsic pathway (mitochondrial
pathway) were involved in HNK-induced apoptosis.
HNK-induced apoptosis is mediated through
intrinsic pathways
To further address the intrinsic pathway,
mitochondrial membrane potential (ΔΨm) was monitored by flow
cytometry using Rh123. As shown in Fig.
3B, control cells exhibited strong staining for Rh123, whereas
treatment with HNK for 6 h led to the cell population shifting to
the left, indicating that HNK induced a marked loss of ΔΨm. Such
findings were consistent with the activation of caspase-9, which is
often the result of disruption of ΔΨm.
It is well known that the Bcl-2 family of proteins
represents key regulators of the mitochondrial apoptotic pathway
(17). The influence of HNK on the
expression of Bax and Bcl-2 was examined. As shown in Fig. 3C, treatment of U87 cells with HNK
for 24 h led to an upregulation in the expression of Bax and a
concurrent downregulation in the expression of Bcl-2 in a
dose-dependent manner, accordingly leading to an increase in the
Bax/Bcl-2 ratio (data not shown). Taken together, the data suggest
that the mitochondrial pathway is likely to be involved in the
apoptotic process induced by HNK in glioblastoma cells.
HNK-induced apoptosis is mediated through
Fas-mediated extrinsic pathways
To characterize the role of the extrinsic pathway in
HNK-induced apoptosis, we detected protein expression of Fas and
FasL by western blotting. The results indicated that HNK induced a
significant elevation in Fas and FasL (Fig. 3A). The data presented herein suggest
that activation of the extrinsic Fas-related pathway may also play
a role in HNK-induced apoptosis.
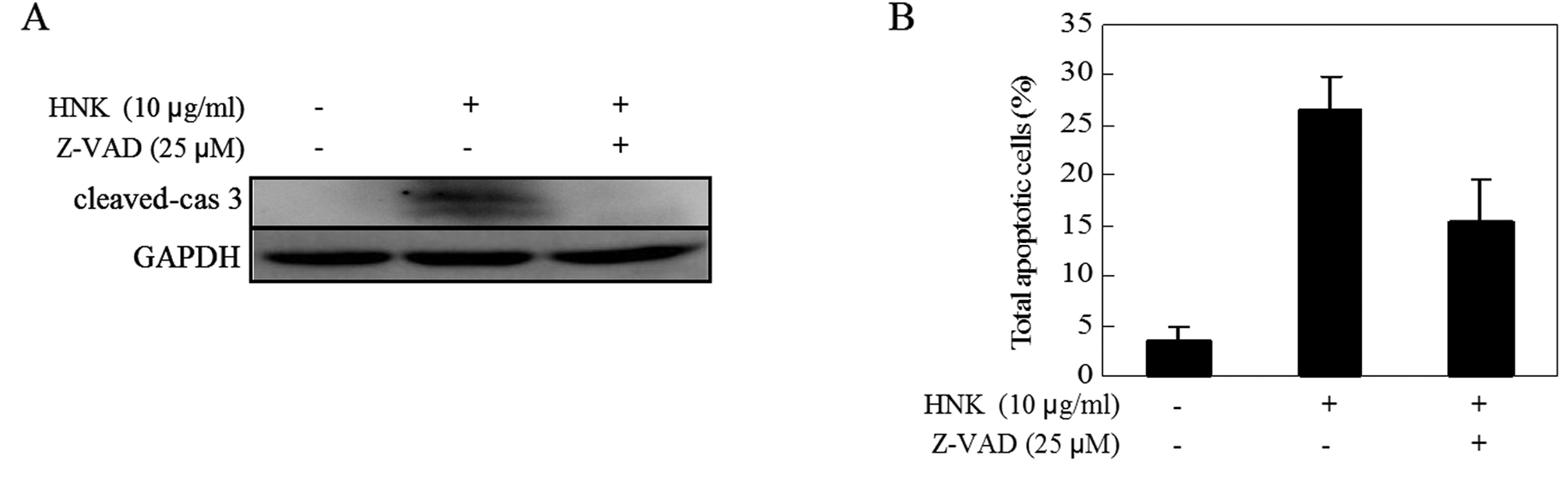
The caspase-independent pathway is also
involved in HNK-induced apoptosis
A previous study revealed that both
caspase-dependent and -independent mechanisms are involved in
HNK-induced apoptosis in human multiple myeloma (18). To verify whether caspases are
necessary in apoptosis induced by HNK in U87 cells, we pretreated
cells with pan-caspase inhibitor Z-VAD-fmk (25 μM) for 1 h prior to
HNK exposure, and checked for signs of apoptosis. As shown in
Fig. 4, we found that although
caspase-3 activity was almost completely blocked by the pan-caspase
inhibitor Z-VAD-FMK, the inhibitor did not block HNK-induced
apoptosis. These results suggest that caspase-independent pathways
may be involved in HNK-induced apoptosis.
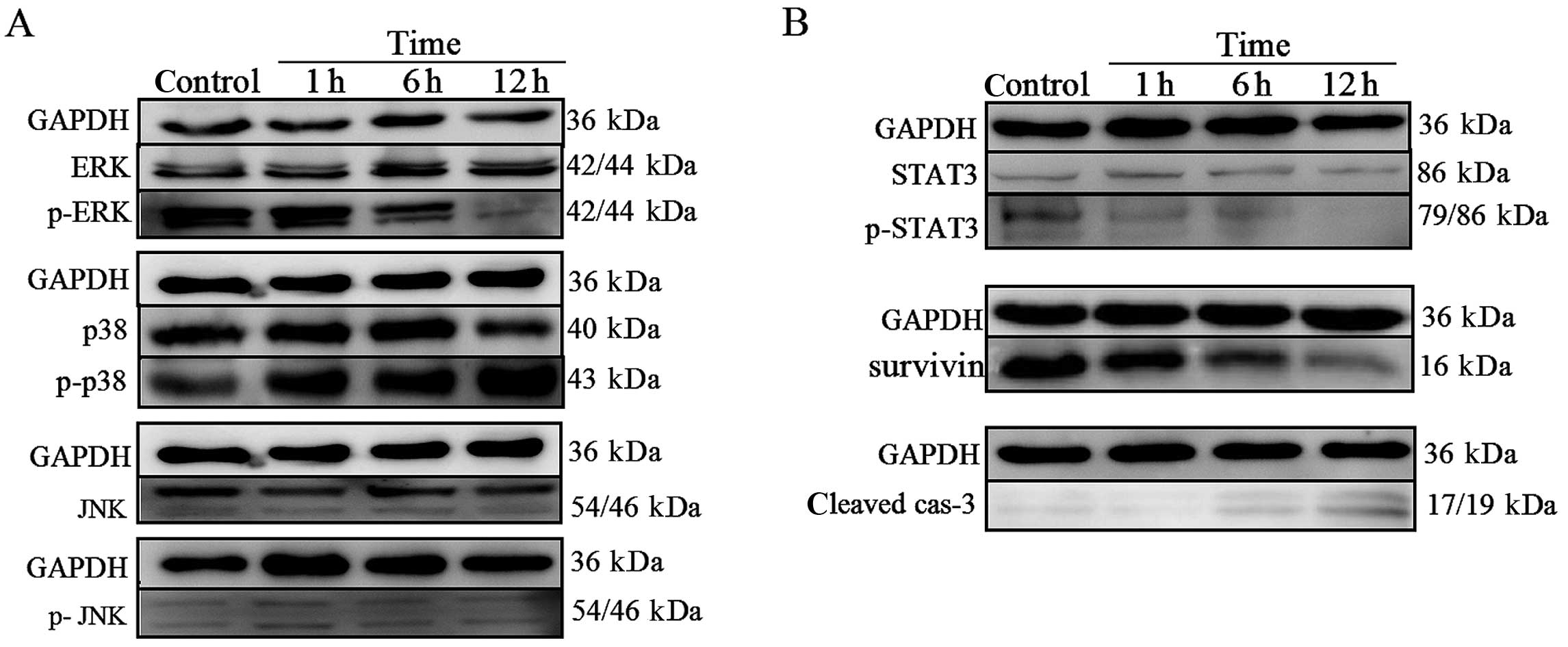
The MAPK signaling pathway is involved in
HNK-induced cell death in U87 cells
To define the intracellular mechanisms by which HNK
affects human glioblastoma cell survival in vitro, we
analyzed the activation of intracellular pathways related to
glioblastoma development MAPK pathways. We performed western
blotting to detect the protein phosphorylation state of ERK, p38
and JNK in control cells and in cells treated for 1, 6 and 12 h
with HNK. There were no detectable changes in expression of total
ERK, p38, JNK and phosphorylated-JNK protein level (Fig. 5A). In contrast, phosphorylation of
ERK was reduced within 12 h from the onset of HNK treatment. Also,
the treatment caused a significant increase in the activation of
p38. These findings strongly indicated that activated p38 and
abrogated ERK1/2 may both be involved in the HNK-induced apoptosis
of U87 cells (Fig. 5A).
The STAT3 signaling pathway is also
involved in HNK-induced apoptosis
STAT3, a signal transducer and activator of
transcription 3, is overexpressed in gliomas (19). Its involvement in tumor genesis can
be attributed to its ability to induce cell proliferation and
inhibit apoptosis (20). Therefore,
to investigate whether the STAT3 signaling pathway was involved in
HNK-induced U87 cells death, we performed western blotting to
assess the protein phosphorylation state and the protein expression
of survivin (its target gene). The results indicated that HNK
suppressed the phosphorylation of STAT3 on Tyr705 for activation,
without affecting the total abundance of STAT3 (Fig. 5B). In parallel, survivin, as one of
the downstream targets of STAT3 (21), was downregulated significantly, in
line with the upregulation of cleaved caspase-3 expression
(Fig. 5B). Hence, STAT3 signaling
pathway may contribute to apoptosis induced by HNK.
Discussion
HNK has been shown to have a broad spectrum of
biological activities, including anti-inflammatory,
anti-angiogenic, anti-thrombocytic and anticancer activity, based
on recent discoveries in basic and translational research (4). Despite numerous studies being
conducted to investigate the effect of HNK on a variety of human
cancer cell lines, specific information on the glioblastoma lineage
remains scant (12,13). In the present study, we confirmed
that HNK exhibits strong dose- and time-dependent anticancer
activity in all three glioblastoma cell lines: U87, U251 and T98G
cells. According to the inhibition rate curve, we found that the
U87 cell line was the most sensitive to HNK. Thus, the U87 cell
line was selected to perform the following experiments.
To gain insight into the molecular mechanism
involved in the growth arrest of glioblastoma by HNK, we first
detected the cell cycle distribution and found that HNK
particularly enhanced the amount of G0/G1
phase cells and diminished the amount of S-phase cells, as
demonstrated previously in other cell lines (8,9). In
parallel to the alteration of cell cycle distribution, we observed
the downregulation of cyclin E mRNA and upregulation of p21 and p27
mRNA. Collectively, these may account for HNK-mediated
G0/G1 phase cell cycle arrest in U87 cells
since cyclin E is essential for the G1-to-S phase
transition (22), while the Cdk
inhibitor p21 and p27 regulates G1-S transition by binding to and
inhibiting kinase activity of Cdk/cyclin complexes (23). It is interesting to note, however,
that a slight G0/G1 arrest does not concur
with marked growth inhibition by HNK at the same concentration and
time point, indicating that there are at least one other mechanism
responsible for the proliferation inhibition of U87 cells.
In the present study, the cell morphological changes
and Annexin V/PI staining results, together with activation of
caspase-3, confirmed pronounced apoptosis induced by HNK in
accordance with earlier studies on the effect of HNK in human
glioma cells (12,13), also in parallel to marked growth
inhibition by HNK at the same time point. It suggests that the
apoptosis may primarily contribute to the antiproliferative
activity of HNK on U87 cells.
It is well known that classical apoptosis is
mediated by two major pathways, known as the intrinsic
(mitochondrial-mediated) and the extrinsic (receptor-mediated)
pathway (24). The extrinsic
pathway is initiated by the binding of a death ligand [e.g., FasL,
tumor necrosis factor (TNF)-related apoptosis-inducing ligand
(TRAIL)] to its corresponding death receptor (e.g. Fas and TRAIL
receptors DR4 and DR5), which lead to a sequential activation of
caspase-8 and −3. The intrinsic pathway is characterized by the
collapse of ΔΨm, causing the release of cytochrome c or
Smac/DIABLO from the mitochondrial intermembrane space to the
cytosol and the formation of the apoptosome complex consisting of
cytochrome c, Apaf-1 and caspase-9. Then, caspase-9 is
activated at the apoptosome and in turn activates caspase-3,
committing the cell to apoptosis. It has been well documented that
HNK-induced apoptosis is characterized by the activation of
caspase-3, −8 and −9 in human multiple myeloma cells and B-cell
chronic lymphocytic leukemia cells (18,25).
However, whether the same phenomenon as that of the above mentioned
cell lines occurs in glioblastoma cells remains to be elucidated.
In the present study, we noted that treatment of U87 with HNK did
result in pronounced increase in caspase-8 activation, as well as a
significant elevation in Fas and FasL expression, which highlighted
the involvement of the extrinsic pathway. Furthermore, we observed
increased cleaved fragments of caspase-9 and loss of ΔΨ after HNK
treatment in U87 cells, similar to previous reports in other cell
types (18,25). The Bcl-2 family of proteins plays an
important role in the regulation of mitochondrial-mediated
apoptosis (26). The family
consists of anti-apoptotic proteins, such as Bcl-2 and
Bcl-xL, as well as pro-apoptotic members, such as Bax,
Bid, Bax and Bak. Accumulating evidence suggests it is the relative
ratio of anti-apoptotic and pro-apoptotic Bcl-2 family proteins
rather than the levels of individual proteins that plays a major
role in determining the survival or death of cells (27). Consistent with a previous report on
human colorectal cells (6), our
data indicated that HNK increased expression of Bax and decreased
expression of Bcl-2, finally resulting in downregulation of
Bcl-2/Bax ratio and further confirmed that the intrinsic apoptotic
pathway is also involved in HNK-induced apoptosis in U87 cells.
A previous study revealed that both
caspase-dependent and -independent mechanisms are involved in
HNK-induced apoptosis in human multiple myeloma (18). Similarly, we found that pretreatment
with the pan-caspase inhibitor Z-VAD-FMK completely abrogated the
cleaved caspase-3 fragments, indicating that caspase activation may
be fully blocked by pan-caspase inhibitor. The apoptosis induced by
HNK was not accordingly abrogated by addition of the pan-caspase
inhibitor Z-VAD-FMK, which suggests that the caspase-independent
pathway may also be involved in apoptosis induced by HNK in U87
cells except for caspase-dependent pathway.
Numerous studies have indicated that HNK can
stimulate apoptosis by modulating multiple signaling pathways
including nuclear factor-κB (NF-κB), signal transducers and
activator of transcription 3 (STAT3), mammalian target of rapamycin
(m-TOR) which have considerable relevance during cancer initiation
and progression (4). However, the
role of MAPK and STAT3 signaling pathway in HNK-induced apoptosis
in U87 glioblastoma cells has never been examined. We then focused
on the status of STAT3 and MAPK activation in HNK-treated
glioblastoma cells.
MAPK families play an important role in
proliferation, differentiation, development, transformation and
apoptosis (28). At least three
MAPK families have been studied in detail: extracellular
signal-regulated kinases (ERKs), c-Jun NH2-terminal kinases (JNKs)
and p38 MAPKs (29). It has been
reported that activated mitogen-activated protein kinase MAPK
cascade leading to ERK1/2 phosphorylation is expressed in the
majority of glial neoplasms and pharmacological targeting of the
activated MEK/ERK1/2 module with the MEK inhibitor U0126 attenuates
cell proliferation (30). Contrary
to ERK1/2 MAPK, in some cases the stress-activated protein kinase
(SAPK, also known as JNK; c-Jun amino-terminal kinase) was revealed
to be activated in response to stress stimuli such as drug
treatment (31). It has been
reported that mimosine (32) and
scriptaid (33) could induce
apoptosis and inhibit glioma cell proliferation by elevating JNK
activation. In the present study, we found the levels of
phosphorylated ERK1/2 decreased in a time-dependent manner
following HNK treatment, suggesting that HNK-induced apoptosis in
U87 cells may be mediated by ERK1/2 downregulation. In contrast to
previous studies, we detected no changes in phosphorylated JNK,
which means SAPK/JNK may not contribute to apoptosis induced by HNK
in U87 cells. Regarding another MAPK family-p38, the downstream
effect of ROS accumulation, several studies revealed that
activation of p38 may be required for apoptosis in human leukemic
cells (34,35). The present study showed the same
phenomenon occurred in U87 cells after treatment with HNK. We
speculate a quick burst of ROS (9,36) may
account for the activation of p38 in other cells treated with HNK,
although we did not determine the modulation of ROS level in this
study.
It has been confirmed that high STAT3 activity is
frequently present in human tumors, including glioblastoma
(19). Activation of STAT3 and its
target genes, such as c-myc (37),
survivin (21), bcl-2, can lead to
cell-cycle progression, pro-angiogenesis, anti- apoptotic effects,
tumor invasion and metastasis (20). Thus, STAT3 has emerged as a
promising molecular target for glioblastoma therapy. A previous
study revealed that blockage of the STAT3 signaling pathway with a
decoy oligonucleotide suppresses growth of human malignant glioma
cells (38). HNK was reported to
inhibit the STAT3 signaling pathway in hepatocellular carcinoma
cells (39). Here, we observed
significant downregulation of constitutive STAT3 activation is in
line with apoptosis exhibited through modulation of cleaved
caspase-3 protein. In addition, survivin, a downstream target of
STAT3, accordingly decreased. Collectively, STAT3 signaling may be
involved in apoptosis induced by HNK in U87 cells.
In conclusion, the present study confirmed that HNK
inhibited proliferation of glioblastoma cells by causing a slight
G0/G1 phase cell cycle arrest and inducing
apoptosis. Here, we demonstrated for the first time that HNK
triggered apoptosis of glioblastoma cells through both
caspase-independent and -dependent pathways; the latter was
initiated by the extrinsic and intrinsic pathway. Moreover, the
inhibition of STAT3 signaling, ERK1/2 as well as activation of p38
MAPK signaling pathway may be involved in apoptosis induced by HNK
in U87 cells. Whether STAT3 and MAPK signaling are necessary for
apoptosis should be further examined.
Acknowledgements
The authors thank Professor Xiao Wang (Shandong
Analysis and Test Center, Shandong Academy of Sciences) for
providing purified Honokiol and Dr Lingzhi Huang for revising the
manuscript. This study was supported by the Natural Science
Foundation of China (81172792 and 81101605), the Natural Science
Foundation of Shandong Province (ZR2011HL045, ZR2010HQ034 and
ZR2011HL050), the Science and Technology Development Grant of the
State Administration of Traditional Chinese Medicine of Shandong
Province (2011-234) and the Doctor’s Foundation of Shandong
Province (BS2009YY008).
References
|
1
|
Dolecek TA, Propp JM, Stroup NE and
Kruchko C: CBTRUS statistical report: primary brain and central
nervous system tumors diagnosed in the United States in 2005–2009.
Neuro Oncol. 14(Suppl 5): v1–v49. 2012.PubMed/NCBI
|
|
2
|
Gauden AJ, Hunn A, Erasmus A, Waites P,
Dubey A and Gauden SJ: Combined modality treatment of newly
diagnosed glioblastoma multiforme in a regional neurosurgical
centre. J Clin Neurosci. 16:1174–1179. 2009. View Article : Google Scholar
|
|
3
|
Jansen M, Yip S and Louis DN: Molecular
pathology in adult gliomas: diagnostic, prognostic, and predictive
markers. Lancet Neurol. 9:717–726. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Arora S, Singh S, Piazza GA, Contreras CM,
Panyam J and Singh AP: Honokiol: a novel natural agent for cancer
prevention and therapy. Curr Mol Med. 12:1244–1252. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ishikawa C, Arbiser JL and Mori N:
Honokiol induces cell cycle arrest and apoptosis via inhibition of
survival signals in adult T-cell leukemia. Biochim Biophys Acta.
1820:879–887. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang T, Chen F, Chen Z, Wu YF, Xu XL,
Zheng S and Hu X: Honokiol induces apoptosis through
p53-independent pathway in human colorectal cell line RKO. World J
Gastroenterol. 10:2205–2208. 2004.PubMed/NCBI
|
|
7
|
Arora S, Bhardwaj A, Srivastava SK, Singh
S, McClellan S, Wang B and Singh AP: Honokiol arrests cell cycle,
induces apoptosis, and potentiates the cytotoxic effect of
gemcitabine in human pancreatic cancer cells. PLoS One.
6:e215732011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wolf I, O’Kelly J, Wakimoto N, et al:
Honokiol, a natural biphenyl, inhibits in vitro and in
vivo growth of breast cancer through induction of apoptosis and
cell cycle arrest. Int J Oncol. 30:1529–1537. 2007.
|
|
9
|
Hahm ER and Singh SV: Honokiol causes
G0–G1 phase cell cycle arrest in human
prostate cancer cells in association with suppression of
retinoblastoma protein level/phosphorylation and inhibition of E2F1
transcriptional activity. Mol Cancer Ther. 6:2686–2695. 2007.
|
|
10
|
Wang Y, Yang Z and Zhao X: Honokiol
induces paraptosis and apoptosis and exhibits schedule-dependent
synergy in combination with imatinib in human leukemia cells.
Toxicol Mech Methods. 20:234–241. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kaushik G, Ramalingam S, Subramaniam D, et
al: Honokiol induces cytotoxic and cytostatic effects in malignant
melanoma cancer cells. Am J Surg. 204:868–873. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang X, Duan X, Yang G, et al: Honokiol
crosses BBB and BCSFB, and inhibits brain tumor growth in rat 9L
intracerebral gliosarcoma model and human U251 xenograft glioma
model. PLoS One. 6:e184902011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jeong JJ, Lee JH, Chang KC and Kim HJ:
Honokiol exerts an anticancer effect in T98G human glioblastoma
cells through the induction of apoptosis and the regulation of
adhesion molecules. Int J Oncol. 41:1358–1364. 2012.
|
|
14
|
Ren X, Zhang Y, Li C, et al: Enhancement
of baicalin by hexamethylene bisacetamide on the induction of
apoptosis contributes to simultaneous activation of the intrinsic
and extrinsic apoptotic pathways in human leukemia cells. Oncol
Rep. 30:2071–2080. 2013.
|
|
15
|
Liu J, Bi G, Wen P, et al: Down-regulation
of CD44 contributes to the differentiation of HL-60 cells induced
by ATRA or HMBA. Cell Mol Immunol. 4:59–63. 2007.PubMed/NCBI
|
|
16
|
Riedl SJ and Shi Y: Molecular mechanisms
of caspase regulation during apoptosis. Nat Rev Mol Cell Biol.
5:897–907. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shimizu S, Narita M and Tsujimoto Y: Bcl-2
family proteins regulate the release of apoptogenic cytochrome
c by the mitochondrial channel VDAC. Nature. 399:483–487.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ishitsuka K, Hideshima T, Hamasaki M, et
al: Honokiol overcomes conventional drug resistance in human
multiple myeloma by induction of caspase-dependent and -independent
apoptosis. Blood. 106:1794–1800. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rahaman SO, Harbor PC, Chernova O, Barnett
GH, Vogelbaum MA and Haque SJ: Inhibition of constitutively active
Stat3 suppresses proliferation and induces apoptosis in
glioblastoma multiforme cells. Oncogene. 21:8404–8413. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yu H and Jove R: The STATs of cancer - new
molecular targets come of age. Nat Rev Cancer. 4:97–105. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhou J, Bi C, Janakakumara JV, et al:
Enhanced activation of STAT pathways and overexpression of survivin
confer resistance to FLT3 inhibitors and could be therapeutic
targets in AML. Blood. 113:4052–4062. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ohtsubo M, Theodoras AM, Schumacher J,
Roberts JM and Pagano M: Human cyclin E, a nuclear protein
essential for the G1-to-S phase transition. Mol Cell Biol.
15:2612–2624. 1995.PubMed/NCBI
|
|
23
|
Ullah Z, Lee CY and Depamphilis ML:
Cip/Kip cyclin-dependent protein kinase inhibitors and the road to
polyploidy. Cell Div. 4:102009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Constantinou C, Papas KA and Constantinou
AI: Caspase-independent pathways of programmed cell death: the
unraveling of new targets of cancer therapy? Curr Cancer Drug
Targets. 9:717–728. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Battle TE, Arbiser J and Frank DA: The
natural product honokiol induces caspase-dependent apoptosis in
B-cell chronic lymphocytic leukemia (B-CLL) cells. Blood.
106:690–697. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Desagher S and Martinou JC: Mitochondria
as the central control point of apoptosis. Trends Cell Biol.
10:369–377. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Reed JC, Jurgensmeier JM and Matsuyama S:
Bcl-2 family proteins and mitochondria. Biochim Biophys Acta.
1366:127–137. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ramos S: Cancer chemoprevention and
chemotherapy: dietary polyphenols and signalling pathways. Mol Nutr
Food Res. 52:507–526. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang W and Liu HT: MAPK signal pathways
in the regulation of cell proliferation in mammalian cells. Cell
Res. 12:9–18. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Glassmann A, Reichmann K, Scheffler B,
Glas M, Veit N and Probstmeier R: Pharmacological targeting of the
constitutively activated MEK/MAPK-dependent signaling pathway in
glioma cells inhibits cell proliferation and migration. Int J
Oncol. 39:1567–1575. 2011.PubMed/NCBI
|
|
31
|
Kyriakis JM and Avruch J: Mammalian
mitogen-activated protein kinase signal transduction pathways
activated by stress and inflammation. Physiol Rev. 81:807–869.
2001.PubMed/NCBI
|
|
32
|
Qiao S, Murakami K, Zhao Q, et al:
Mimosine-induced apoptosis in C6 glioma cells requires the release
of mitochondria-derived reactive oxygen species and p38, JNK
activation. Neurochem Res. 37:417–427. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sharma V, Koul N, Joseph C, Dixit D, Ghosh
S and Sen E: HDAC inhibitor, scriptaid, induces glioma cell
apoptosis through JNK activation and inhibits telomerase activity.
J Cell Mol Med. 14:2151–2161. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Huh JE, Kang KS, Chae C, Kim HM, Ahn KS
and Kim SH: Roles of p38 and JNK mitogen-activated protein kinase
pathways during cantharidin-induced apoptosis in U937 cells.
Biochem Pharmacol. 67:1811–1818. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Konopleva M, Contractor R, Kurinna SM,
Chen W, Andreeff M and Ruvolo PP: The novel triterpenoid CDDO-Me
suppresses MAPK pathways and promotes p38 activation in acute
myeloid leukemia cells. Leukemia. 19:1350–1354. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Han LL, Xie LP, Li LH, Zhang XW, Zhang RQ
and Wang HZ: Reactive oxygen species production and Bax/Bcl-2
regulation in honokiol-induced apoptosis in human hepatocellular
carcinoma SMMC-7721 cells. Environ Toxicol Pharmacol. 28:97–103.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Stearns D, Chaudhry A, Abel TW, Burger PC,
Dang CV and Eberhart CG: c-myc overexpression causes anaplasia in
medulloblastoma. Cancer Res. 66:673–681. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gu J, Li G, Sun T, et al: Blockage of the
STAT3 signaling pathway with a decoy oligonucleotide suppresses
growth of human malignant glioma cells. J Neurooncol. 89:9–17.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Rajendran P, Li F, Shanmugam MK, et al:
Honokiol inhibits signal transducer and activator of
transcription-3 signaling, proliferation, and survival of
hepatocellular carcinoma cells via the protein tyrosine phosphatase
SHP-1. J Cell Physiol. 227:2184–2195. 2012. View Article : Google Scholar
|