Introduction
SH2 domain-containing inositol 5′-phosphatase 1
(SHIP1) is a member of the phosphatidylinositol phosphatase
(PIPase) class that is important in the negative regulation of
proliferation and survival of hematopoietic cells through its
negative regulation of the PI3K/AKT signaling pathway (1,2).
Recent studies have shown that inactivation of SHIP1 could play a
central role in certain hematologic malignancies (1–6).
Inactivation of point mutations of the SHIP1 gene and reduction in
SHIP1 activity have been observed in patients with acute myeloid
leukemia (AML), implicating a possible tumor-suppressor function of
SHIP1 in the pathogenesis of AML (6). However, it appears that SHIP1 gene
mutations are an uncommon cause of reduction in SHIP1 activity in
AML (7). Several other possible
explanations that could account for the loss of SHIP1 function
include epigenetic modification, decreased transcription,
translational repression, and increased protein breakdown. miR-155
has been shown to bind to the 3′UTR of SHIP1 mRNA and inhibit its
translation (8–11). It was demonstrated that SHIP1 is a
primary target of miR-155 in B cells, with high levels of miR-155
and reduced expression of SHIP1 linked to the development of acute
lymphoblastic leukemia in mice (3,12).
Levels of miR-155 were also found to be significantly increased in
human patients with diffuse large B cell lymphoma or NK/T cell
leukemia with decreased SHIP1 expression (13,14).
Further investigation is needed to evaluate the role of miR155 and
SHIP1 in the pathogenesis of AML.
In the present study, we examined the levels of
SHIP1 protein and miR-155 in samples of patients with AML and in
AML cell lines. In addition, we investigated cell proliferation,
apoptosis and expression of SHIP1/PI3K/AKT pathway molecules in the
THP-1 and U937 cell lines after miR-155 inhibitor or mimics were
transfected.
Materials and methods
Bone marrow or blood samples
Bone marrow or blood samples were collected from 30
patients with AML after obtaining informed consent according to our
hospital guidelines. The pathological diagnosis of AML was
established according to the French-American-British (FAB)
classification criteria as M1 (n=4), M2 (n=8), M3 (n=2), M4 (n=6),
M5 (n=9), or M6 (n=1). Bone marrow mononuclear cells (BMMNCs) or
peripheral blood mononuclear cells (PBMNCs) of the patients were
separated by density gradient centrifugation and cryopreserved
until further analysis could be performed. BMMNCs from 10 unrelated
healthy volunteers were also examined for miR-155 and SHIP1 by
quantitative reverse-transcription polymerase chain reaction
(qRT-PCR).
Cell lines
The leukemia cell lines K562, THP-1 and U937 were
purchased from the Blood Institute of Tianjin, China. Cells were
cultured using RPMI-1640 medium containing 10% fetal bovine serum
(FBS), 2 mM glutamine, and streptomycin in a humidified incubator
at 37°C in 5% CO2. Cells were used for experimentation
at a density of 106–107/l.
Cell transfections
U937 and THP-1 cells were transfected using
X-tremeGENE siRNA transfection reagent (Roche, CH) according to the
manufacturer’s protocol. Briefly, cells were cultured in 6-well
plates. Transfection reagent 1 was prepared by mixing 90 μl of
Opti-MEM medium and 10 μl of X-tremeGENE siRNA transfection
reagent. Additionally, reagent 2 containing siRNA (2 μg of
inhibitor) and 100 μl of Opti-MEN medium was prepared. Reagent 2
was mixed with reagent 1 and incubated for 30 min. This mixture was
then carefully added to the plated cells to obtain a final
concentration of miR-155 mimics or miR-155 siRNA of 50 nmol/l.
Cells were cultured for 48 h and then collected for analysis of
gene expression and subsequent experiments. Cells used for the
blank and negative control groups were treated with only serum-free
RPMI-1640 medium or with control reagent (at a final concentration
of 50 nmol/l), respectively.
Measurement of apoptosis
Cellular apoptosis was measured by flow cytometry,
according to the following protocol, 48 h after transfection.
Briefly, ~5–10×104 resuspended cells were collected by
centrifugation at 500 × g for 5 min and resuspended again in 195 μl
of Annexin V-FITC. A total of 5 μl of Annexin V-FITC solution was
added to the cells, mixed gently, and incubated in the dark at room
temperature. Cells were collected again by centrifugation and
resuspended in 190 μl of Annexin V-FITC. A total of 10 μl of
propidium iodide staining solution was added and mixed, and
apoptosis measurement was performed by flow cytometry. Data were
analyzed using FACS Diva software (BD Biosciences, San Jose, CA,
USA).
Measurement of cell proliferation
Cells were collected 48 h after transfection by
centrifugation at 500 × g for 5 min, resuspended, and added to
96-well plates (100 μl/well, approximately 104
cells/well). This time point was defined as zero (0). Then, 10 μl
of CCK-8 was added to the wells separately at 24, 48 and 72 h, and
the absorbance was measured at 450 nm.
RNA isolation and PCR
Total RNA was extracted from cells using TRIzol
reagent (Invitrogen, Carlsbad, CA, USA) following the
manufacturer’s protocol. The quality of total RNA was assessed
using a NanoDrop 2000 Spectrophotometer (Thermo Fisher Scientific
Inc., Waltham, MA, USA).
Measurement of miR-155 gene
expression
Total RNA (2 μg) was reversed-transcribed using an
All-in-One miRNA qRT-PCR detection system (Invitrogen) according to
the manufacturer’s protocol. All-in-One miRNA qRT-PCR detection
system kits were used to measure gene expression using specific
sets of primers (Table I) according
to the protocol (Invitrogen). All reactions were performed with the
ABI Prism 7000 sequence detection system instrument (Applied
Biosystems). Gene expression was normalized to the U6 gene, and the
data were analyzed using the comparative 2−ΔΔCt
method.
 | Table IPrimers and sequences of miR-155
mimics, inhibitors and negative control. |
Table I
Primers and sequences of miR-155
mimics, inhibitors and negative control.
| Primers | Sequence (5′-3′) |
|---|
| miR-155-5p
mimics |
UUAAUGCUAAUCGUGAUAGGGGU
CCCUAUCACGAUUAGCAUUAAUU |
| Negative control for
markup mimics |
UUCUCCGAACGUGUCACGUTT
ACGUGACACGUUCGGAGAATT |
| miR-155-5p
inhibitor |
ACCCCUAUCACGAUUAGCAUUAA |
| Negative control for
miR-155-5p inhibitor |
CAGUACUUUUGUGUAGUACAA |
Measurement of SHIP-1 gene
expression
Total RNA (2 μg) was analyzed for gene expression
using One-Step qRT-PCR kits according to the manufacturer’s
protocol. Briefly, 25 μl of 2X Quant One-Step SYBR qRT-PCR Master
Mix, 2.5 μl of 2.5 U/μl Hot Master Taq polymerase, 0.4 μl of Quant
Rtase, 2 μl of the forward primer (10 pM), 2 μl of the reverse
primer (10 pM) (both by Invitrogen), and 2 μg of total RNA were
mixed and double-distilled water was added to reach a final volume
of 50 μl. The primer sequences of the SHIP1 gene are listed in
Table II. All reactions were
performed in the ABI Prism 7000 sequence detection system. Gene
expression was normalized to the GAPDH gene, and the data were
analyzed using the comparative 2−ΔΔCt method.
| Table IIPrimers for real-time PCR. |
Table II
Primers for real-time PCR.
| Primers | Sequence |
|---|
| SHIP1 | F:
5′-GCGTGCTGTATCGGAATTGC-3′
R: 5′-TGGTGAAGAACCTCATGGAGAC-3′ |
| GAPDH | F:
5′-ACCACAGTCCATGCCATCACT-3′
R: 5′-TCCACCACCCTGTTGCTGTA-3′ |
| miR-155 | F:
5′-CCCCTATCACGATTAGCATTAA-3′ |
| U6 | F:
5′-GCAGGGGCCATGCTAATCTTCTCTGTATCG-3′ |
Western blotting
Cells were digested and proteins were extracted in
RIPA lysis buffer. The concentration of proteins was measured by
the Bradford method according to the standard protocol. A total of
20 μg of total protein extract was electrophoresed on
polyacrylamide/sodium dodecyl sulfate (SDS) gels and transferred by
electroblotting onto polyvinylidene fluoride (PVDF) membranes. The
membranes were blocked for 1 h in 5% (w/v) nonfat milk before
incubation with appropriate dilutions of the primary antibodies
against SHIP-1, AKT, p-AKT and actin (all at 1:1,000; from Cell
Signaling Technology, Boston, MA, USA) at 4°C overnight. The
membranes were then incubated with the corresponding secondary
antibodies (1:10,000; Zymed). The blots were developed using the
enhanced chemiluminescence (ECL) system (Immobilon Western;
Millipore, Billerica, MA, USA) and were analyzed using Quantity One
Software (Bio-Rad Laboratories Inc., Hercules, CA, USA). Protein
contents were normalized to the actin level.
Data analyses
Data are expressed as the means ± SD. Statistical
analyses of experimental groups were performed by Student’s t-test
or one-way analysis of variance (ANOVA). The level of statistical
significance was set at P<0.05. Analyses were performed using
SPSS 13.0 for Windows software (SPSS, Chicago, IL, USA).
Results
Expression of miR-155 and SHIP1 in
patients with AML
The SHIP1 protein content was significantly lower in
the tissue samples from AML patients when compared with that in the
controls (P<0.05). No differences were found in the expression
of SHIP1 at the mRNA level among the 30 patients with different
subtypes of AML according to the FAB classification (P>0.05,
Fig. 1A). However, the levels of
SHIP1 protein varied greatly. A much lower expression than the
average level of SHIP1 protein was detected in 15 patients
classified as FAB-AML-M4 or FAB-AML-M5 compared with the other
subtypes of AML (P<0.05, Fig. 1B and
C). The average level of miR-155 was significantly higher in
the 30 AML patients compared with the average level in the healthy
controls (P<0.05). Additionally, the levels of miR-155 were
significantly higher in patients with AML subtype M4 or M5 compared
with the other AML subtypes (P<0.05, Fig. 1D).
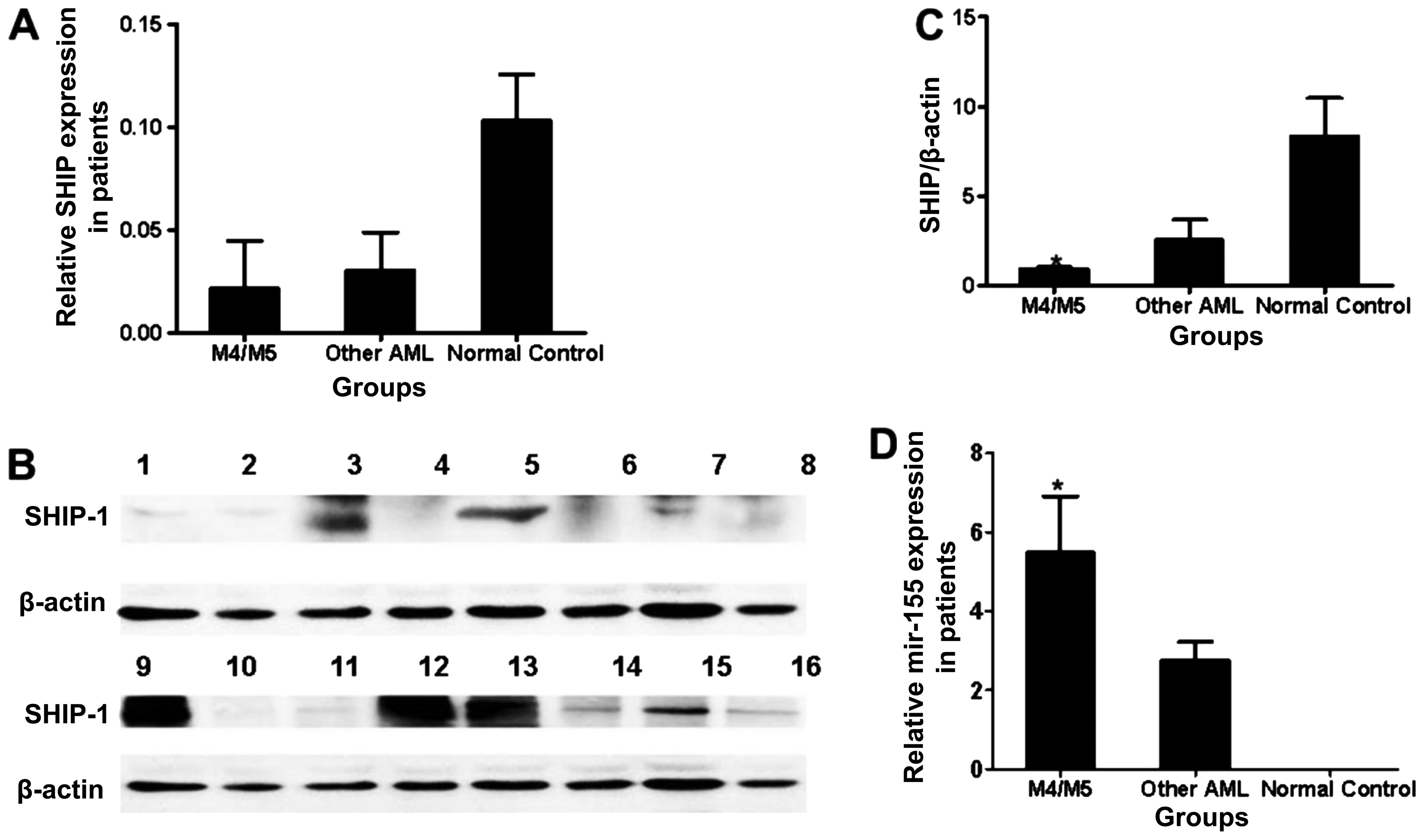 | Figure 1Expression of miR-155 and SHIP1 in
bone marrow cells from the subjects. (A) Expression of SHIP1 at the
mRNA level in tissue samples from patients with M4 or M5 AML
(n=15), with other types of AML according to FAB classification
except M4 or M5 (other AML) (n=15) and in normal controls (n=10).
*P<0.05, M4/M5 vs. other AML. (B) SHIP1 protein
levels in the tissue samples from patients and normal controls.
Lanes 1, 2, 10, 11 and 16 correspond to patients with M4 or M5;
lanes 3, 5, 9, 12 and 13 correspond to normal controls and lanes 4,
6, 7, 8, 14 and 15 correspond to patients with other AML. (C) SHIP1
protein levels in tissue samples from patients with M4 or M5, with
other AML and in normal controls. *P<0.05, M4/M5 vs.
other AML. (D) Relative gene expression of miR-155 at the mRNA
level in tissue samples from patients with M4 or M5, with other AML
and in normal controls. *P<0.05, M4/M5 vs. other
AML. |
Expression of miR-155 and SHIP1 in AML
cell lines
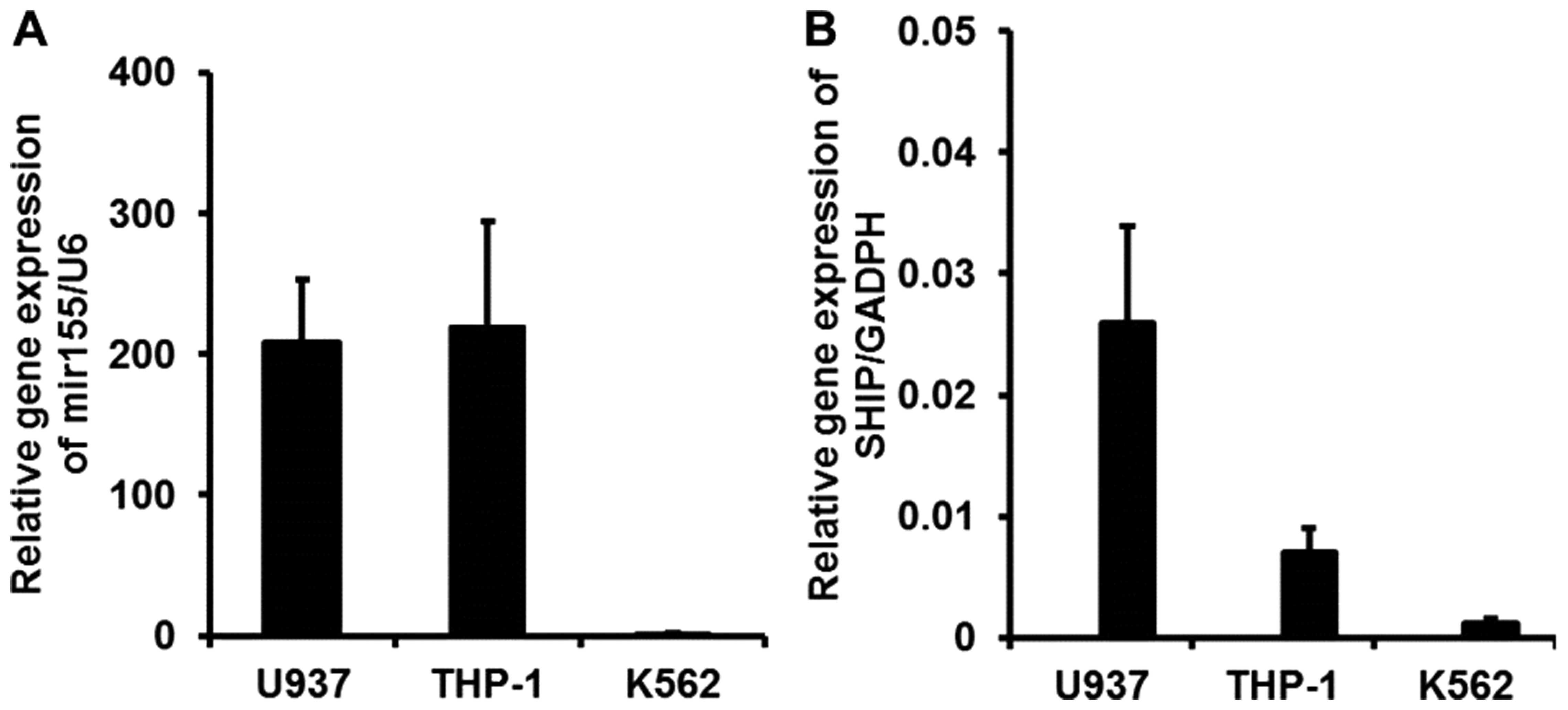
Real-time PCR demonstrated that both miR-155 and
SHIP1 were expressed in the THP-1, K562, and U937 cell lines.
Downregulation of SHIP1 expression in the K562 cells, which are
known to be Philadelphia chromosome-positive, could be the result
of overexpression of BCR/ABL. Thus, the U937 and THP-1 cell lines
were chosen for further study. Specifically, THP-1 cells expressed
a higher miR-155 level but a lower SHIP1 level between the two cell
lines studied (Fig. 2A). In
contrast, U937 cells had a higher SHIP1 level but a lower miR-155
expression (Fig. 2B).
Expression of SHIP1, AKT and pAKT after
miR-155 transfection
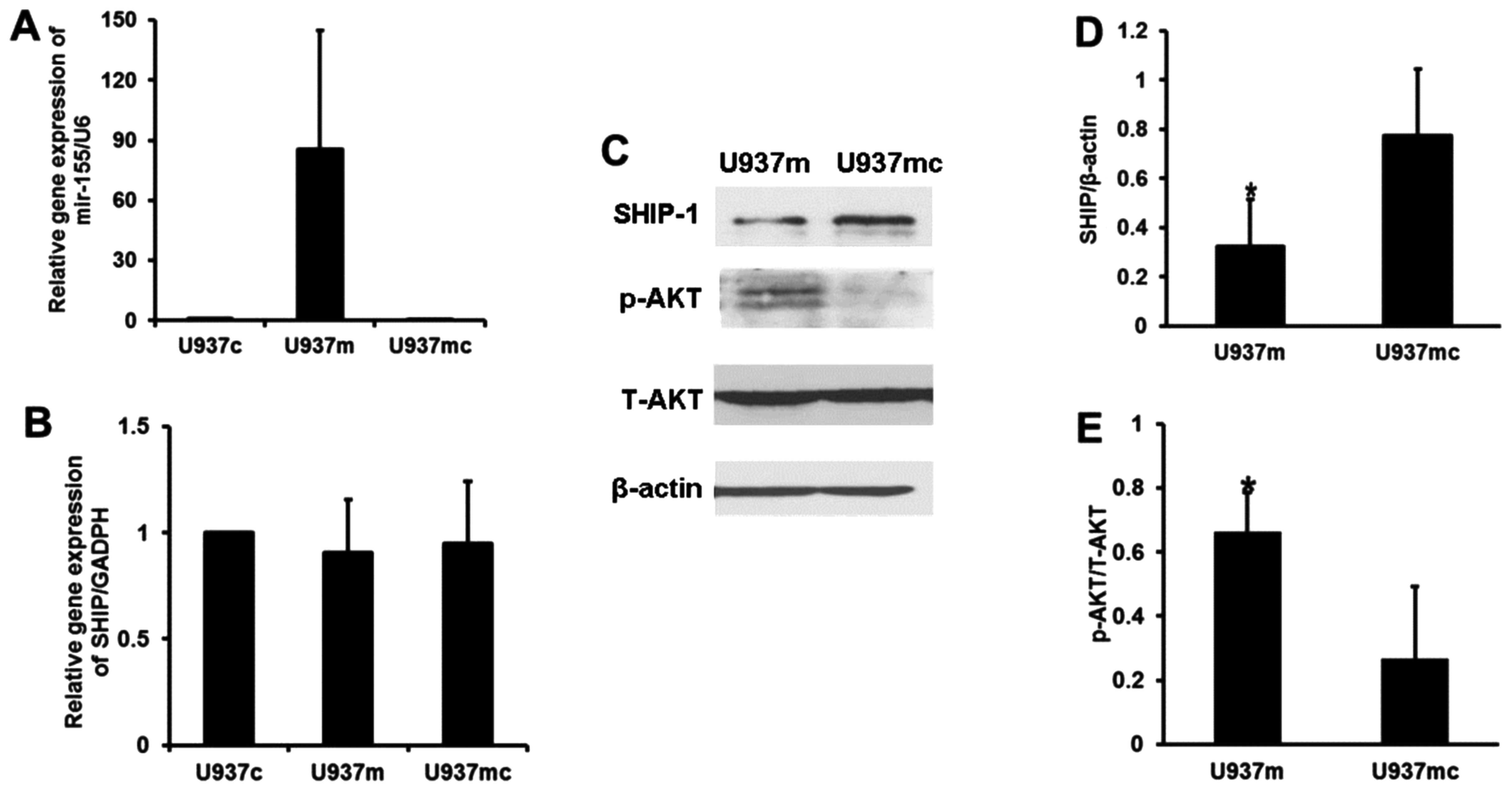
Since SHIP1 is a negative regulator of the PI3K/AKT
signaling pathway, we hypothesized that increased activation of
PI3K/AKT signaling in AML samples with low SHIP1 expression may be
observed after miR-155 transfection. Because the levels of miR-155
expression were lower in the U937 cells than that in the THP-1
cells, we chose to transfect miR-155 mimics into the U937 cells
(Fig. 3A). Transfection of
miRNA-155 mimics in the U937 cells did not alter the expression of
SHIP1 at the mRNA level compared with the expression in the blank
control and negative transfection groups (P>0.05, Fig. 3B). However, U937 cells transfected
with the miRNA-155 mimics had significantly reduced expression of
SHIP1 at the protein level (P<0.05, Fig. 3C and D), associated with unchanged
AKT content but increased p-AKT protein levels (P<0.05, Fig. 3C and E). Consequently, inhibition of
SHIP1 expression by miR-155 would likely increase the activity of
the AKT pathway. Therefore, miR-155 appears to behave as an
onco-miR by reducing SHIP1 expression.
Expression of SHIP1, AKT, and pAKT after
transfection of the miR-155 inhibitor
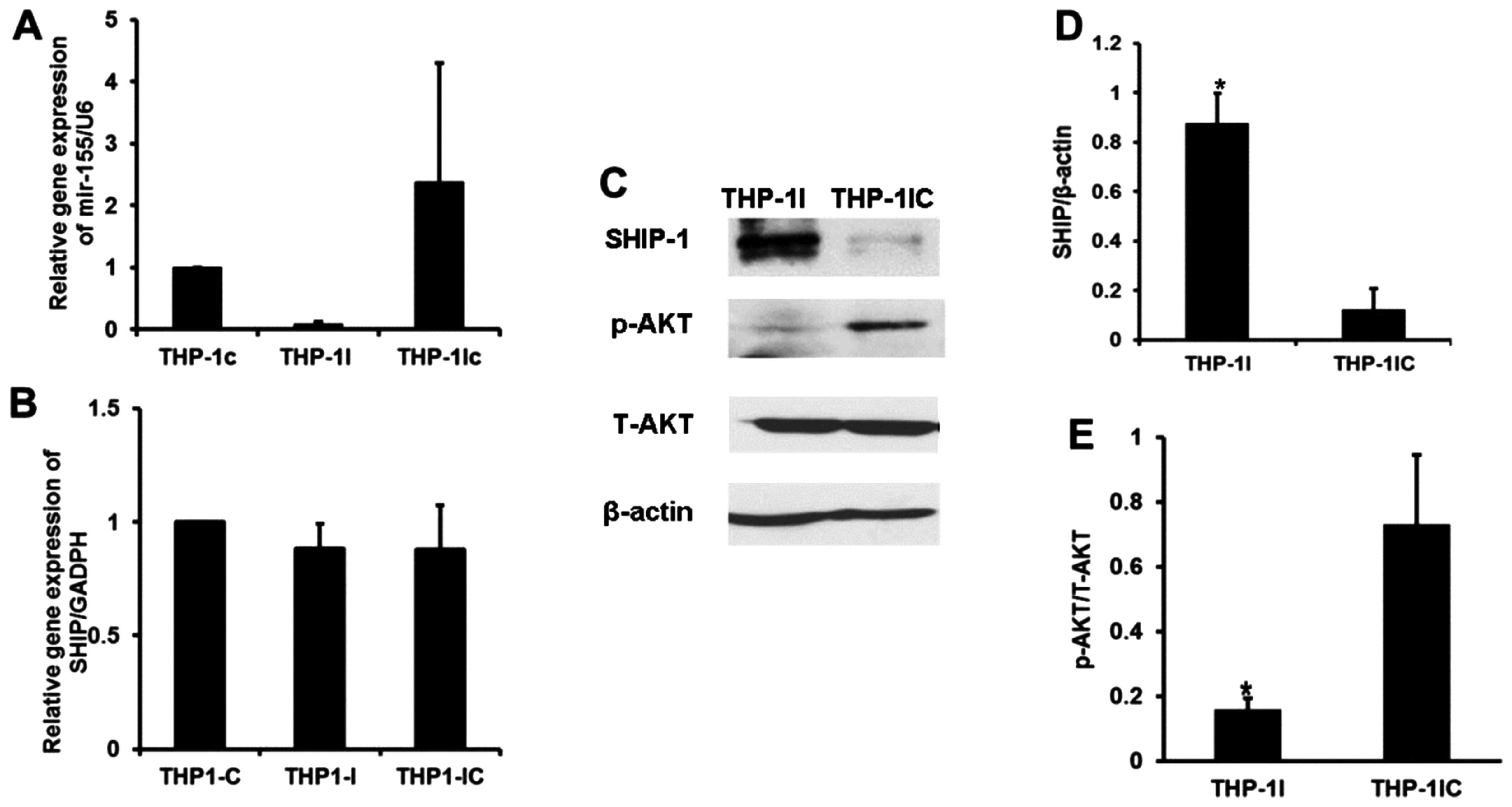
We then used miR-155 siRNA to examine the effects of
restoring SHIP1 protein levels by reducing miR-155 expression in
the THP-1 cells (Fig. 4A), which
are known to overexpress miR-155. When THP-1 cells were transfected
with the miR-155 siRNA, significantly elevated levels of SHIP1
protein were found, although no alterations in SHIP1 gene
expression at the mRNA level could be detected compared with the
expression levels in the blank and negative controls (Fig. 4B). THP-1 cells transfected with
miR-155 siRNA also exhibited no alteration in total AKT content,
but increased SHIP1 protein and decreased p-AKT levels were found
(P<0.05 and P<0.05 respectively; Fig. 4C–E). It appears that suppression of
miR-155 expression leads to inactivation of pAKT without altering
total AKT via restoration of SHIP1 protein. Therefore, SHIP1
negatively regulates signaling in the PI3K/AKT pathway. SHIP1 could
be a tumor suppressor.
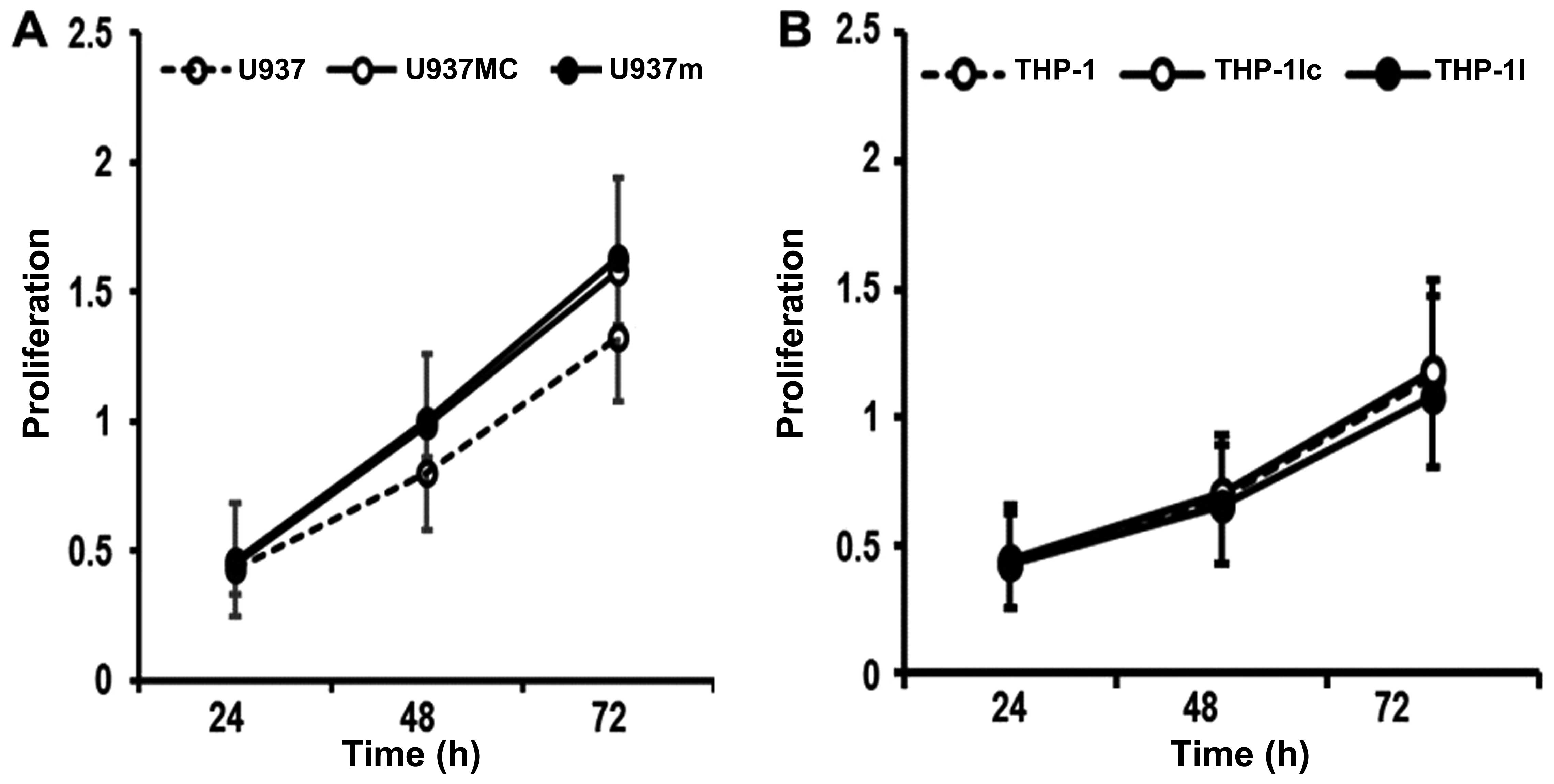
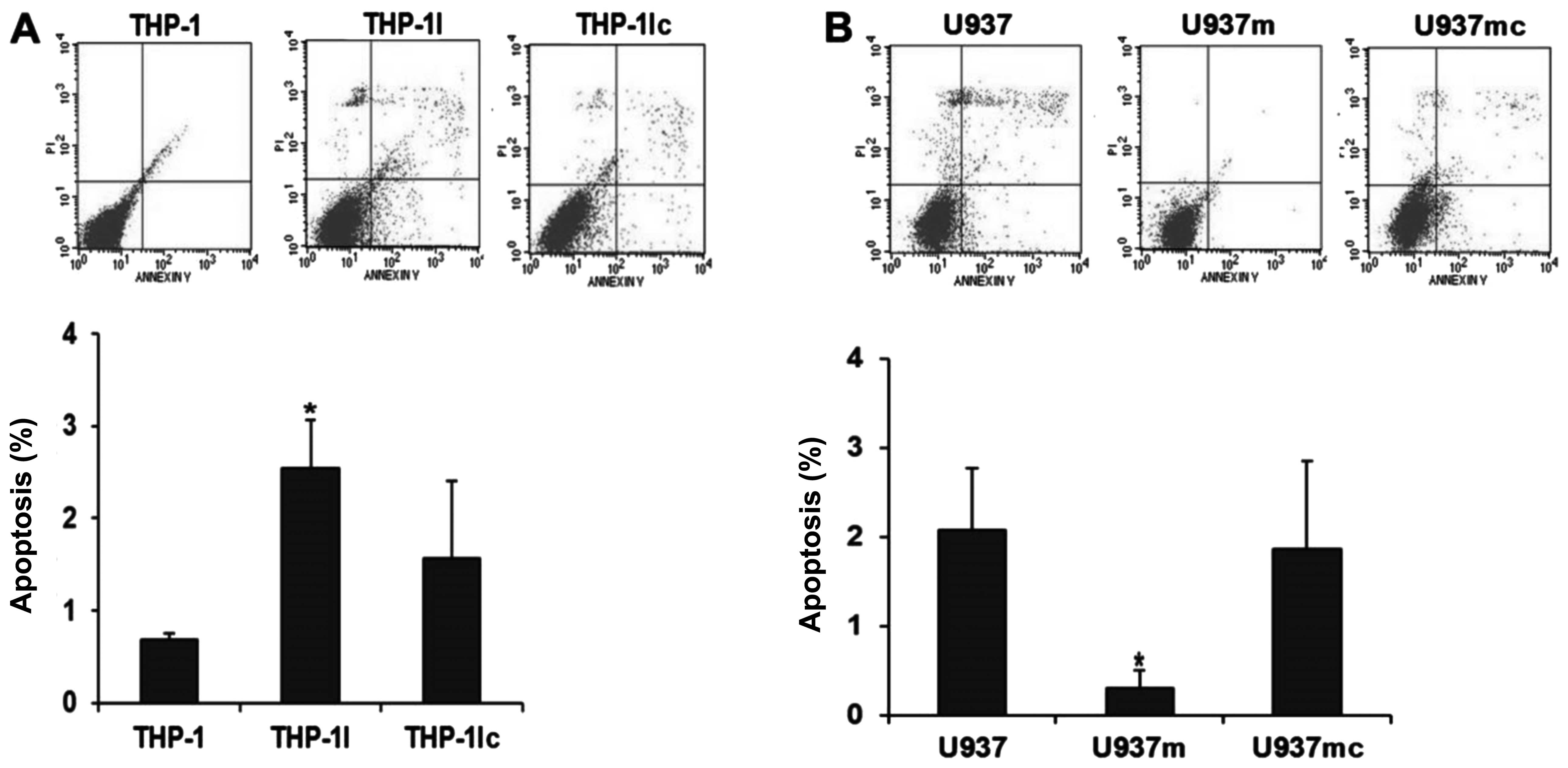
Proliferation and apoptosis in the cell
lines after transfection
To investigate the role of the
miR-155/SHIP1/PI3K/AKT pathway in leukemogenesis, we performed
proliferation and apoptosis assays in cell lines. First, we
analyzed the effect on the autonomous proliferation of cells after
transfection. Compared with the cells transfected with the mimic
control (U937mc), the proliferation rate of the cells transfected
with the miR-155 mimics (U937m) appeared higher but did not reach a
statistically significant difference at any time point (U937m vs.
U937mc; all P>0.05; Fig. 5A).
However, the U937m cells had a significantly lower apoptosis rate
when compared with that of the U937mc cells (U937m vs. U937mc;
P<0.05; Fig. 6A), indicating
that miR-155 might cause AML progression by reducing cell apoptosis
rather than increasing cell proliferation.
Similar results were also noted in the proliferation
rate of THP-1 cells transfected with the miR-155 siRNA (THP-1I) and
transfected with the inhibitor control (THP-1Ic), which indicated
that miR-155 inhibition could not affect the proliferation rate of
THP-1 at any time point (THP-1I vs. THP-1Ic; P>0.05; Fig. 5B). A significantly increased rate of
apoptosis was observed in the THP-1I cells compared with that of
the THP-1Ic cells (THP-1I vs. THP-1Ic; P<0.05; Fig. 6B), indicating miR-155 inhibition may
cause an increased apoptosis rate in AML without interfering with
proliferation.
Discussion
Considering that SHIP1 is a negative regulator of
the PI3K/AKT signaling pathway, inactivation of SHIP1 may result in
Akt activation (1,2,15).
SHIP1-deficient mice develop myeloproliferative disease and
leukemia (11,16). Inactivating point mutations of SHIP1
have been observed in AML. It appears that SHIP1 mutations are an
uncommon cause of the reduction in SHIP1 activity (7). In our study, low SHIP1 protein
expression levels that did not completely correspond to SHIP1 mRNA
levels were detected in a subset of patients with AML. We then
tried to identify the molecular basis for the loss of SHIP1
function at the protein level.
microRNAs are a newly discovered class of short,
noncoding RNA species, which may serve as master switches in gene
networks (8,17–20).
miR-155 was shown to bind to the 3′UTR of SHIP1 mRNA and,
therefore, to inhibit its translation (9–11,16,20,21).
SHIP1 is expressed in the same cell types that express miR-155, and
it plays an opposing role in many cases. Some studies demonstrated
a strong correlation between myeloproliferative disorders (MPDs)
caused by miR-155 expression and specific knockdown of SHIP1
(11,16). Recent studies showed that SHIP1 is a
primary target of miR-155. Therefore, miR-155-mediated suppression
of SHIP1 expression may contribute to the development of human MPDs
and myeloid leukemia, in which miR-155 has been shown to be
overexpressed (8,16,19,20,22,23).
Moreover, it was recently reported that the endogenous expression
of SHIP1 is reduced in hematopoietic cells overexpressing miR-155
in vitro and in vivo, which leads to increased
activation of AKT (3). Our present
findings are consistent with these reports.
We overexpressed miR-155 in U937 cells and found
that the levels of SHIP1 protein were largely reduced in these
cells. Furthermore, in THP-1 cells, we inhibited miR-155 expression
using siRNA targeting miR-155 and found that the level of SHIP1
protein was increased. Following recent studies showing that SHIP1
plays an important role in the pathogenesis of lymphoma and acute
lymphoblastic leukemia that is affected by miR-155 (3,14), we
investigated the existence of an association between SHIP1 and
miR-155 in AML. Our results demonstrated that SHIP1 is also a
direct target of miR-155 in AML cells. We hypothesized that
inhibition of the translation of SHIP1 mRNA by miR-155 could be an
important mechanism for the loss of function of SHIP1.
To evaluate the importance of SHIP1/miR-155 in AML,
we initially examined the levels of expression of SHIP1 and miR-155
in myeloid leukemia cells. We hypothesized that reduced expression
of SHIP1 in leukemia cells could be a result of an elevated miR-155
expression as in other hematological malignancies (2,3,13,14,24).
In fact, the levels of SHIP1 protein were lower in THP-1 and U937
cells than in control cells. In contrast, miR-155 levels were
relatively higher. These results indicate that the expression of
miR-155 is negatively associated with the expression of SHIP1 in
AML cell lines. To test whether this observation could be found in
patients with AML, we examined the expression of SHIP1 and miR-155
in 30 AML patients. We found elevated levels of miR-155 in patients
with acute myelomonocytic leukemia and acute monocytic leukemia,
corresponding to the FAB-AML-M4 and FAB-AML-M5 classifications,
respectively, as well as decreased expression of SHIP1 protein.
These results indicate that miR-155 expression is inversely
correlated with SHIP1 expression in myeloid leukemia cell lines and
in M4 and M5 patients, suggesting that an interaction of SHIP1 and
miR-155 may be biologically significant in these two subtypes of
AML. In addition, SHIP1 expression is downregulated in AML, whereas
miR-155 is commonly overexpressed in subsets of AML patients and
acts as an onco-miR. It is likely that the downregulation of SHIP1
expression by miR-155 is a plausible mechanism of miR-155-mediated
oncogenesis in AML M4 or M5. This study found an association
between the level of miR-155 and a specific leukemic phenotype,
similar to the finding that miR-181 expression is positively
correlated with M1 and M2 subtypes of AML (25), suggesting a possible role in the
developmental lineage and differentiation state of the tumors
(12,20–23,26,27).
Given that miR-155 is overexpressed in AML and that
it may act as an onco-miR, we decided to examine whether miR-155
has oncogenic functions in AML cells in vitro. Our results
indicated that miR-155 may cause AML by reducing cell apoptosis
rather than increasing cell proliferation. The results of our in
vitro assays indicated that miR-155 mimics inhibited the
apoptosis of U937 cells. Conversely, transfection of anti-miR-155
in THP-1 cells increased apoptosis. Western blot analyses confirmed
that the expression of SHIP1 in these cells was modulated by
miR-155. Taken together, our results showed that miR-155 acts as an
onco-miR in AML cells by downregulating SHIP1 expression. In
addition, SHIP1 was also shown to be a tumor suppressor that
regulates cell apoptosis in AML.
SHIP1 has been known to be a negative feedback
regulator of PI3K/Akt signaling, and the antitumoral activity of
SHIP1 is mediated by inhibition of Akt signaling via the PI3K
pathway (1,2,4,5,15,28–30).
Akt activation may affect many cellular processes, including cell
cycle progression, transcription, translation, differentiation, and
apoptosis. Given that SHIP1 expression is inhibited by miR-155, we
hypothesized that miR-155 overexpression in AML may play a role in
Akt signaling. In U937 cells with overexpressed miR-155, Akt was
found to be activated. Conversely, we found that anti-miR-155
reduced p-Akt in THP-1 cells. Meanwhile, the levels of Akt protein
were not affected by miR-155 overexpression or anti-miR-155. These
results demonstrated that overexpression of miR-155 enhanced
oncogenic Akt signaling via the SHIP1/PI3K pathway in AML cells.
Given that the expression of miR-155 was significantly enhanced and
that Akt signaling was active in the AML cell lines and a subset of
patients with AML, we propose that the miR-155/SHIP1/PI3K/Akt
pathway may play an important role in the pathogenesis of AML.
In summary, we hypothesized that inhibition of the
expression of SHIP1 could be a mechanism by which miR-155 acts as
an onco-miR in leukemia. Overexpression of miR-155 alters AKT
signaling by inhibiting SHIP1 expression. The severe adverse
effects and limitations of conventional chemotherapy and
hematopoietic stem cell transplantation underscore the urgent need
for new treatment strategies for patients with AML. Our findings
provide new insight into the pathogenesis of leukemia and suggest
that restoration of SHIP1 expression as a tumor suppressor by
inhibiting miR-155 may represent a useful approach for the
treatment of certain subtypes of AML.
Acknowledgements
This research was supported by the Special Research
Fund of the Ministry of Health of China (no. 201202017) and the
Natural Science Foundation of Hebei Province (no. H2012206139).
References
|
1
|
Hamilton MJ, Ho VW, Kuroda E, et al: Role
of SHIP in cancer. Exp Hematol. 39:2–13. 2011. View Article : Google Scholar
|
|
2
|
Conde C, Gloire G and Piette J: Enzymatic
and non-enzymatic activities of SHIP-1 in signal transduction and
cancer. Biochem Pharmacol. 82:1320–1334. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Costinean S, Sandhu SK, Pedersen IM, et
al: Src homology 2 domain-containing inositol-5-phosphatase and
CCAAT enhancer-binding protein beta are targeted by miR-155 in B
cells of Emicro-MiR-155 transgenic mice. Blood. 114:1374–1382.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kennah M, Yau TY, Nodwell M, et al:
Activation of SHIP via a small molecule agonist kills multiple
myeloma cells. Exp Hematol. 37:1274–1283. 2009. View Article : Google Scholar
|
|
5
|
Lo TC, Barnhill LM, Kim Y, Nakae EA, Yu AL
and Diccianni MB: Inactivation of SHIP1 in T-cell acute
lymphoblastic leukemia due to mutation and extensive alternative
splicing. Leuk Res. 33:1562–1566. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Luo JM, Yoshida H, Komura S, et al:
Possible dominant-negative mutation of the SHIP gene in acute
myeloid leukemia. Leukemia. 17:1–8. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gilby DC, Goodeve AC, Winship PR, Valk PJ,
Delwel R and Reilly JT: Gene structure, expression profiling and
mutation analysis of the tumour suppressor SHIP1 in Caucasian acute
myeloid leukaemia. Leukemia. 21:2390–2393. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
O’Connell RM, Zhao JL and Rao DS: MicroRNA
function in myeloid biology. Blood. 118:2960–2969. 2011.
|
|
9
|
Teng G and Papavasiliou FN: Shhh!
Silencing by microRNA-155. Philos Trans R Soc Lond B Biol Sci.
364:631–637. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tili E, Croce CM and Michaille JJ:
miR-155: on the crosstalk between inflammation and cancer. Int Rev
Immunol. 28:264–284. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
O’Connell RM, Chaudhuri AA, Rao DS and
Baltimore D: Inositol phosphatase SHIP1 is a primary target of
miR-155. Proc Natl Acad Sci USA. 106:7113–7118. 2009.PubMed/NCBI
|
|
12
|
Schwind S, Maharry K, Radmacher MD, et al:
Prognostic significance of expression of a single microRNA,
miR-181a, in cytogenetically normal acute myeloid leukemia: a
Cancer and Leukemia Group B study. J Clin Oncol. 28:5257–5264.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pedersen IM, Otero D, Kao E, et al:
Onco-miR-155 targets SHIP1 to promote TNFalpha-dependent growth of
B cell lymphomas. EMBO Mol Med. 1:288–295. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yamanaka Y, Tagawa H, Takahashi N, et al:
Aberrant overexpression of microRNAs activate AKT signaling via
down-regulation of tumor suppressors in natural killer-cell
lymphoma/leukemia. Blood. 114:3265–3275. 2009. View Article : Google Scholar
|
|
15
|
Metzner A, Precht C, Fehse B, et al:
Reduced proliferation of CD34(+) cells from patients with acute
myeloid leukemia after gene transfer of INPP5D. Gene Ther.
16:570–573. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
O’Connell RM, Rao DS, Chaudhuri AA, et al:
Sustained expression of microRNA-155 in hematopoietic stem cells
causes a myeloproliferative disorder. J Exp Med. 205:585–594.
2008.PubMed/NCBI
|
|
17
|
Sayed D and Abdellatif M: AKT-ing via
microRNA. Cell Cycle. 9:3213–3217. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Volinia S, Galasso M, Costinean S, et al:
Reprogramming of miRNA networks in cancer and leukemia. Genome Res.
20:589–599. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhi F, Cao X, Xie X, et al: Identification
of circulating microRNAs as potential biomarkers for detecting
acute myeloid leukemia. PLoS One. 8:e567182013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Vasilatou D, Papageorgiou S, Pappa V,
Papageorgiou E and Dervenoulas J: The role of microRNAs in normal
and malignant hematopoiesis. Eur J Haematol. 84:1–16. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Faraoni I, Antonetti FR, Cardone J and
Bonmassar E: miR-155 gene: a typical multifunctional microRNA.
Biochim Biophys Acta. 1792:497–505. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang Y, Li Z, He C, et al: MicroRNAs
expression signatures are associated with lineage and survival in
acute leukemias. Blood Cells Mol Dis. 44:191–197. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yuan Y, Kasar S, Underbayev C, Prakash S
and Raveche E: MicroRNAs in acute myeloid leukemia and other blood
disorders. Leuk Res Treatment. 2012:6038302012.PubMed/NCBI
|
|
24
|
Lawrie CH: MicroRNAs and haematology:
small molecules, big function. Br J Haematol. 137:503–512. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Debernardi S, Skoulakis S, Molloy G,
Chaplin T, Dixon-McIver A and Young BD: MicroRNA miR-181a
correlates with morphological sub-class of acute myeloid leukaemia
and the expression of its target genes in global genome-wide
analysis. Leukemia. 21:912–916. 2007.PubMed/NCBI
|
|
26
|
Hyde RK and Liu PP: The role of microRNAs
in acute myeloid leukemia. F1000 Biol Rep. 2:812010.PubMed/NCBI
|
|
27
|
Joyce CE and Novina CD: miR-155 in acute
myeloid leukemia: not merely a prognostic marker? J Clin Oncol.
31:2219–2221. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Baran CP, Tridandapani S, Helgason CD,
Humphries RK, Krystal G and Marsh CB: The inositol 5′-phosphatase
SHIP-1 and the Src kinase Lyn negatively regulate macrophage
colony-stimulating factor-induced Akt activity. J Biol Chem.
278:38628–38636. 2003.
|
|
29
|
Blunt MD and Ward SG: Targeting PI3K
isoforms and SHIP in the immune system: new therapeutics for
inflammation and leukemia. Curr Opin Pharmacol. 12:444–451. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ong CJ, Ming-Lum A, Nodwell M, et al:
Small-molecule agonists of SHIP1 inhibit the phosphoinositide
3-kinase pathway in hematopoietic cells. Blood. 110:1942–1949.
2007. View Article : Google Scholar : PubMed/NCBI
|