1. Introduction
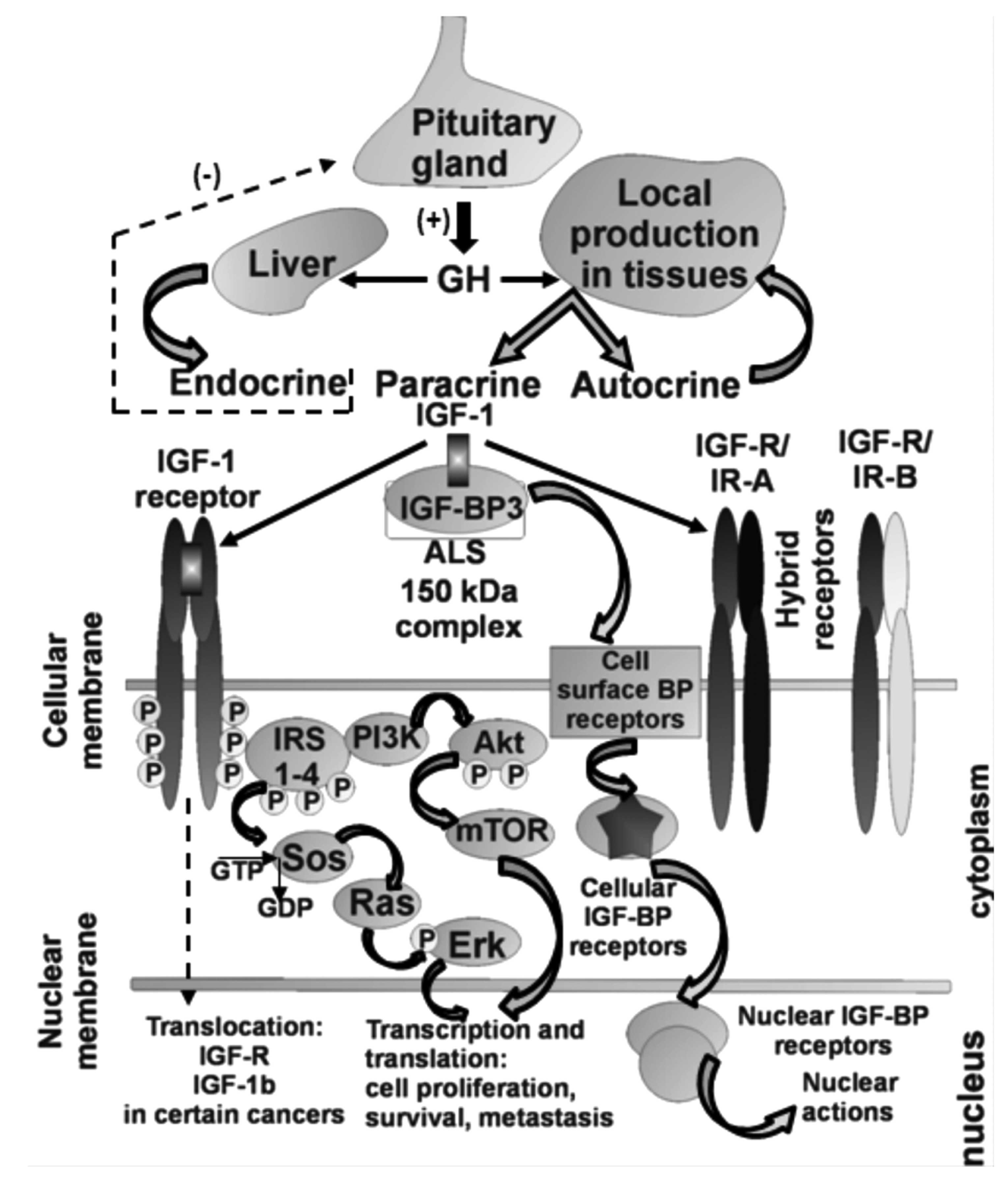
The IGF axis stimulates the growth and proliferation
and involves many key molecules such as insulin-like growth factor
(IGF)-1 and IGF-2, their transmembrane receptors (IGF-1R and
IGF-2R, respectively), the IGF-binding proteins (IGFBPs) and
intracellular signaling proteins such as the insulin receptor
substrate (IRS) family, Akt and many others. Initially, it was
believed that all IGF-1, which is both a growth hormone and a
tissue growth factor of 70-amino acids in length, originated in the
liver and was transported by an endocrine mode to sites of action.
At present, it is recognized that IGF-1 is also produced in other
organs where paracrine and autocrine mechanisms are engaged
(1). Alternative splicing (AS) of
the IGF-1 gene results in multiple isoforms that retain the
identical sequence of mature IGF-1, but also give rise to divergent
C-terminal E-peptides. The peptides may modulate the actions,
stability, or bioavailability of IGF-1, or they may have
independent activity. Six different splice forms can be produced;
from either of the two different promoters P1 and P2 three
isoforms, IGF-1A, IGF-1B and IGF-1C, are transcribed (2). Recent data indicate that the entire
IGF network is even more complicated as in some tissues more than
one form of IGF-1A can be active. In mice, three forms in different
proportions have been detected in muscle: mature IGF-1, pro-IGF-1
(C extension is not cleaved) and glycosylated pro-IGF-1
(C-extension has bound sugars residues and is not cleaved)
(3). Furthermore, it has been shown
in two independent studies that human Eb-peptide cleaved form human
pre-pro-IGF1b, which is 77 amino acids long, localized to the
nuclei of transfected cells and may have IGF-1 independent
mitogenic and bioactive properties (4–6).
Notably, a 10-fold decrease in the IGF-1B transcript level was
observed (7), and a downshift of
the IGF-1B content in favor of the IGF-1A isoform was reported when
non-tumor tissue and colorectal cancer cells were analyzed
(8). On the other hand, an increase
in IGF-1B and decrease in IGF-1A expression were found in cervical
cancer and control cells, respectively (9). It is now clear that it is important to
understand, not only the overall IGF expression level, but also the
entire IGF isoform profile assuring a whole new level of IGF-1
activity regulation in local tissues linked to the presence of
different IGF forms and the presence of different forms of the same
isoform (glycosylated pro-IGF-1A) (Fig.
1).
2. IGF axis and cancer
Recently, accumulating evidence indicates that the
IGF axis is involved in human cancer progression (10). IGF-1 signaling can contribute to
each stage of cancer progression: malignant transformation, tumor
growth, local invasion and distant metastases, and resistance to
treatment. In addition to direct contributions to each of these
stages, IGF-1 may promote cancer indirectly, through interactions
with oncogenes and tumor supressors, with other hormones
(particularly sex steroids in breast and prostate cancers) and with
IGFBPs (11). The findings of
another study suggest that elevated IGF-1 levels may be implicated
in the development of ovarian cancer, diagnosed before age 55 years
(12). Whereas in colorectal
carcinoma, the local expression levels of total IGF-1 mRNA and all
splicing isoforms of IGF-1 mRNA were decreased as compared to
normal colon tissues. The results of this study suggest an
increased regenerative potential in normal colon tissues which, at
least partially, is linked to an elevated expression of total IGF-1
mRNA and its isoform A (8). An
important clue to the essential role of the IGF-1R in cellular
function was uncovered by Sell and co-workers who reported that
IGF-1 signaling is an absolute requirement for viral transformation
of cells (13). Numerous studies
performed over the last 20 years have suggested that transformed
cells express the IGF-1R at higher levels than normal cells.
However, a decade ago the molecular mechanisms by which IGF-1R gene
expression is increased in tumors remained largely unidentified
(14). Further in vitro
studies have demonstrated that a functioning IGF-1R is necessary
for cell transformation by many viral and cellular oncogenes and
appears to be important in expressing the genes that regulate the
cell cycle, cell survival, motility, attachment and metastasis
(15,16). Cell surface IGF-1R translocates to
the nucleus following clathrin-mediated endocytosis, regulated by
IGF levels. The IGF-1R is unusual among transmembrane receptors
that undergo nuclear import, in that both α and β subunits traffic
to the nucleus. Nuclear IGF-1R is phosphorylated in response to
ligands, and undergoes IGF-induced interaction with chromatin,
suggesting direct engagement in transcriptional regulation. Nuclear
IGF-1R is detectable in primary renal cancer cells, formalin-fixed
tumors, preinvasive lesions in the breast, and non-malignant
tissues characterized by a high proliferation rate. In clear cell
renal cancer, nuclear IGF-1R is associated with adverse prognosis.
These findings suggest that IGF-1R nuclear import has biological
significance, and may contribute directly to IGF-1R function
(Fig. 1) (17). It has been pointed out that the
IGF-1R alone does not mediate growth and transforming activities,
but rather the pathway itself, which is administered by IRS-1,
signals to growth promoting and anti-apoptotic pathways. It is
clear that IRS-1 is a key hub overseeing downstream signaling
actions of the IGF-1R, and IRS-1 could be referred to as an
antitumor suppressor acting as an anti-p53 protein (18,19).
The role of the IGF-1R in the progression of epithelial tumors that
are more prevalent in adults is likely to be more complex (20); however, the prevailing notion that
IGF-1R is routinely overexpressed in transformed cells is somewhat
of an overgeneralization (14).
3. IGF-1 deficiency and protection from
cancer
Significantly, IGF-1 deficiency is a major
contributing factor in lifespan prolongation especially in females
and in protection from cancer (21,22).
It has been extensively studied in individuals with Laron syndrome
(LS) who have an inactive GH receptor and IGF-1 deficiency leading
to dwarfism; a recessively inherited syndrome caused by deletions
or mutations in the GH receptor or post-receptor pathways (21,23).
The cohort of LS patients, treated or not treated by recombinant
IGF-1, appears to be protected not only from cancer but also from
diabetes, whereas taller stature is now regarded as a risk for
several types of cancer (24).
Growth disorders are multifactor, complex phenomena with
often-unknown etiology, and in-depth large-scale pooled
next-generation sequencing is used for molecular diagnosis
(25). It has been demonstrated
that GH, GHR, IGF-1 and IGF1-R coding sequences may be altered in
growth disorders (26,27); however, these sequences are not
changed in the genome of children with short stature (28,29)
and the search for other defective genetic backgrounds related to
the IGF-1 axis should be considered. In order to further clarify
the relationship between GH/IGF-1 and cancer more studies are
needed.
4. IGF axis and viruses in cancer
Many studies indicate that the IGF axis and viruses
can combine their actions in cellular transformation leading to
cancer. Several components of the IGF signaling axis, such as
IGF-1, IGF-2 and IGF-1R, are deregulated during HCV-related human
hepatocellular carcinoma (HCC). Only a few investigations have
focused on hepatic expression of IGFs and their receptors at
different stages of chronic hepatitis C infection. The studies
demonstrated an increased IGF-1R synthesis, aberrant IGF-2
expression (decreased/increased), and decreased synthesis of IGF-1
as events in human hepatocarcinogenesis. Recognition of the role
played by HCV in different splicing profiles of the IGF-1
gene in the progres sion of chronic hepatitis C will require
further study. A better understanding of the interactions between
HCV protein and IGF axis component will facilitate the development
of novel approaches to prognose and to treat virus-related HCC
(30).
Large T-antigen from the human John Cunningham
polyomavirus (JCV T-antigen), also present in the SV-40 virus (both
viruses belong to Polyomaviridae), inhibits homologous
recombination directed DNA repair (HRR), which results in an
accumulation of mutations. Following T-antigen-mediated nuclear
translocation, IRS-1 binds Rad51 at the site of damaged DNA. This
T-antigen-mediated inhibition of HRR does not function in cells
lacking IRS-1, and can be reproduced in the absence of T-antigen by
IRS-1 with an artificial nuclear localization signal (31). The interplay described between the
IGF-1R signaling system and JCV T-antigen in the process of DNA
repair could be relevant, since nearly 90% of the human population
is seropositive for JC virus, the JCV T-antigen transforms cells
in vitro, the JCV T-antigen is tumorigenic in experimental
animals, and the presence of the JC virus has been noted in an
increasing number of biopsies of human cancer (32). The family of Papovaviridae
was taxonomically split into the Papillomaviridae (HPV) and
the Polyomaviridae (JCV). John Cunningham virus expresses a
T-antigen that causes malignant transformation through development
of aneuploidy and interaction with some of the same regulatory
proteins as HPV (33).
5. HPV in cervical cancer
Human papillomaviruses (HPVs) constitute a
heterogeneous group of viruses from the Papillomaviridae
family. They are double-stranded circular DNA viruses with an
icosahedral capsid and are able to infect epithelial cells. An HPV
phylo-genetic tree has been designed based on the homologous
nucleotide sequence of the major capsid protein L1 that groups the
different HPV types into genera: α, β, γ, δ, μ and others (34). The HPV genome is approximately 8 kb
in length and is divided into three regions, the non-coding long
control region (LCR), and the coding early (E) and late (L)
regions. The viral genome encodes six early (E1, E2, E4, E5, E6,
E7) and two late proteins (L1 and L2). The transcription of early
and late genes is controlled by the LCR. The viral proteins are
translated from polycistronic mRNAs containing overlapping reading
frames (35). Papillomaviruses have
been extensively studied and more than 100 different types have
been identified (36). The
association between HPV and human cancer was first proposed more
than three decades ago by Herald zur Hausen, and since then
additional studies have fully demonstrated the direct role of HPV
infection in the development of several human cancers (37). Depending on their potential to
induce carcinogenesis HPVs have been divided into low-risk (HPV-6
and -11) and high-risk (HPV-16 and -18) (38). HPVs can also be held responsible for
anogenital, head and neck, skin and other types of cancer (39). The mucosal HPV types preferentially
infect the cervical transformation zone, which is the junction
point of the endocervix columnar cells and the ectocervix
stratified squamous epithelial cells (40). Many HPV types cause only productive
lesions following infection and are not associated with human
cancers. In such lesions, the expression of viral gene products is
carefully regulated, with viral proteins being produced at defined
times and at regulated levels as the infected cell migrates towards
the epithelial surface. The pattern of viral gene expression in
low-grade cervical lesions resembles that observed in productive
warts caused by other HPV types. High-grade neoplasia represents an
abortive infection in which viral gene expression becomes
deregulated, and the normal life cycle of the virus cannot be
completed (41). As is typical of
viruses that co-evolve with their hosts, many PVs produce only
chronic, inapparent infections and produce virions from the surface
of infected epithelium without apparent detriment to the host
(42). The paradox is that the
infection with oncogenic types of HPV is very common and most of
these infections go unnoticed. Malignancy is a rare outcome of a
common HPV infection (43).
However, not all HPV types use the same strategy and it appears
that several of the α PVs, in particular, have acquired
immunoevasion strategies that allow them to cause persistent
visible papillomas (42).
HPV genomes replicate episomally in host cells, but
HPV DNA is frequently found to be integrated into chromosomes in
cervical cancer. The timing of viral integration appears to
correspond to the development of high-grade cervical
intraepithelial neoplasia (CIN) as a consequence of high-level
expression of E6 and E7 (44).
Vinokurova and co-workers suggest that HPV integration is not an
essential event in cervical carcinogenesis. This integration of
oncogenic HPV genomes in cervical lesions is a consequence rather
than the cause of chromosomal instability induced by deregulated
high-risk papillomaviruses (HR-HPVs) E6–E7 oncogene expression. The
integration frequency of various HPV types is strongly correlated
with the age at diagnosis of cancer, suggesting that the malignant
potential of the various HR-HPV types is reflected by their
integration frequency in invasive cervical carcinomas (45). Integration occurs near fragile sites
in the human genome and results in termination of the viral cycle
as large portions of the genome are disrupted and therefore it
becomes functionally inactive (46). The alternative mechanism by-passing
viral integration and E2 gene disruption which enables pure
episomal HPV genomes to maintain an upregulated expression of E6
and E7 oncogenes is methylation of E2 binding sites at the promoter
region of HPV-16 (47).
E6 and E7 have various biological activities in
addition to inactivation of the major tumor suppressors, p53 and
pRB, respectively (48). It has
been suggested that for a lesion to be maintained, the virus must
infect an epithelial stem cell (49). It is generally thought that the
viral E1 and E2 proteins are expressed in order to maintain the
viral DNA as an episome (50) and
to facilitate the correct segregation of genomes during cell
division (51). Expression of E6
and E7 in the lower epithelial layers drives cells into the
S-phase, which creates an environment that is conducive for viral
genome replication and cell proliferation (52). E6 and E7 oncoproteins cooperate in
cellular transformation and evasion of the immune system (40). The PDZ domain-binding motif of E6
[X-(S/T)-X-(V/L/I) where X is any residue, S/T is serine or
threonine and V/L/I is valine, leucine or isoleucine] appears
critical for its transforming activity in cultured cells and
tumorigenicity in xenograft experiments. In contrast, none of the
low-risk HPV E6 proteins have this motif (53). Moreover, a study presented by Sun
et al suggests that trapping of p53 in the cytosol by
HPV-11E6 results in apoptosis and represents a novel mechanism to
explain why low-risk HPV infection does not result in malignant
transformation; this explanation remains tentative, and awaits
further testing (54). The best
characterized high-risk HPV-16 E6 activity is its ability to induce
degradation of the tumor-suppressor protein p53 via the
ubiquitin/proteasome pathway. This cellular protein is a
transcription factor that can trigger cell cycle arrest or
apoptosis in response to a large variety of cellular stresses, such
as hypoxia or DNA damages (55). E6
targeting PDZ domain-containing proteins such as hDlg, hScribble
and p53, further supplemented by induction of the catalytic subunit
of telomerase reverse transcriptase (hTERT) contributes to cellular
immortalization. Targeting pRb and its family members for
degradation by E7 oncoprotein constitutes a major step in
tumorigenesis. Both E6 and E7 bind and inactivate several
transcription factors involved in the immune response, e.g.
interferon regulatory factors (IRFs). This serves to avoid
immune-based destruction of HPV-containing tumors while acquiring
invasive potential through modulation of other pathways (56). The HPV E7 protein shares functional
similarities with such proteins as adenovirus E1A and SV40 large
tumor antigen. In the HPV viral life cycle, E7 disrupts the
intimate association between cellular differentiation and
proliferation in normal epithelium, allowing for viral replication
in cells that would no longer be in the dividing population
(57). HPV-16 is the most prevalent
genotype in cervical carcinoma and is also the most frequently
detected HPV types in head and neck squamous cell carcinoma
(HNSCCs). It is also interesting to note that HPV-associated
oropharyngal squamous cell cancers have a better prognosis than
HPV-negative tumors (58). As
determined by PCR, HPV-16 DNA is present in ~56% of SCCs of the
cervix. HPV-18 is the second most common HPV type associated with
cervical adenocarcinoma, causing 37–41% of SCCs (59). Little is known concerning the
apparent absence of HPV infections in the gastrointestinal
epithelia (60). None of the
individual reports claiming the presence of anogenital HPVs in
cancers of the esophagus, prostate, bladder, lung, demonstrated a
consistent association of these viruses with cancer of these
respective sites (39).
6. IGF axis and HPV-related cervical
cancer
The association between HPV infections and IGF
levels has not been extensively explored in cervical neoplasia;
nevertheless, a growing number of studies have recently
demonstrated an association between serum levels of IGFs and
IGFBP-3 and increased risk for various cancers. For example, it was
shown that the plasma IGF-1 level and IGF-1/IGFBP-3 molar ratio
were significantly associated with CIN, but had no significant
association with cervical cancer. However, it is not clear whether
increased plasma levels of IGF-1 and IGF-1/IGFBP-3 were the cause
or the result of CIN. Despite all these significant associations
observed, the results of this study showed no relationship between
IGF levels and HPV infection status (61). Another much more prospective study
was one of the first to demonstrate a relationship between serum
levels of IGF-1 and precancerous squamous intraepithelial lesions
(SILs). Individuals with either high-grade (HSILs) or low-grade
SILs (LSILs) exhibited significantly higher serum levels of IGF-1,
IGFBP-3 and IGF-1/IGFBP-3 molar ratio than did control subjects.
Furthermore, there was a dose-dependent relationship between risk
of SILs and levels of IGF-1. After adjustment for IGF-1, no
relationship was evident between the IGFBP-3 level and risk of SILs
(62). On the other hand, the
results from another study of preoperative serum total IGF-1 or
IGFBP-3 levels failed to predict cervical cancer mortality and
recurrence. It is difficult to explain why increased serum IGF-1
level may have a protective effect on the risk of cervical cancer
observed, whereas the unfavorable effect of serum IGF-1 is
addressed in certain sex hormone-related cancers, such as prostate
or breast cancer (63). The authors
of the study concluded that more candidates should be enrolled in
further studies to realize the differences in serum levels of IGF-1
between normal individuals, patients with precancer lesions and
cervical cancer (63). In
concordance with these observations, another more prospective study
demonstrated that increased levels of IGF-1 are associated with
reduced risk of HCIN. High IGF-1 concentration was associated with
a reduced risk of being positive for HPV-16 and -18. Levels of
IGFBP-3 were not associated with the risk of HCIN or being HPV
positive among controls. Yet again, it is unclear why increased
levels of IGF-1 would have a protective effect on the risk of
cervical cancer precursors and that the protection would be
stronger among younger women. One possible explanation is that
IGF-1 decreases a woman’s risk of high-grade cervical
intraepithelial neoplasia (HCIN) by decreasing her risk of being
positive for HPV-16 and/or -18, perhaps via increased turnover of
the cervical epithelium, thus reducing the duration of infections
(64). Additionally, it has been
shown that a lower serum IGF-1 level is correlated to increased
risk of cervical cancer (65), and
that the differential expression of IGF components in controls,
LSILs, HSILs and cervical cancer, could be related with the
carcinogenic process in cervical epithelium and could be a
potential marker for progression (66). The same research group demonstrated
that cervical cancer cell lines, positive and negative for HPV,
differ in the type of insulin and IGF-1 receptors expressed, while
SiHa cells expressed IGF-1R, IR-A and IR-B and IR/IGF-1R hybrid
receptors, C33a cells expressed the IR-A only (67). Results showed that median protein
levels of IGF-2 were significantly lower in cervical cancer cases
vs. controls and significantly higher values of IGFBP-3 were found
in HSIL vs. controls, and were not affected by HR-HPV infection.
Meanwhile no significant differences were observed in IGFBP-3
levels between LSILs or cervical cancer as compared to controls.
These significant data suggest that the progression to cervical
cancer is associated with alterations in the IGF system and is not
affected by HR-HPV infection. More studies are needed to understand
the possible role of IGFBP-3 in cervical carcinogenesis (68).
It has been observed that the growth and
invasiveness of cervical cancer cells are dose-dependently
stimulated by IGF-1, whereas the growth and invasiveness of normal
cervical epithelial cells are not. It was pointed out that IGF-1,
acting through IGF-1R, interacted with αvβ3
integrin in cervical cancer cell invasiveness and proliferation
(69). Transformed cells that
overexpress the IGF-1R may subvert growth regulation by minimizing
their dependency on additional growth factors. Data demonstrating
that the IGF-1R is overexpressed in both primary cervical tumor
cells and cervical cell lines are consistent with this concept
(70). Furthermore, in a study by
Kuramoto et al the expression levels of IGF-1R were
significantly higher in CIN and invasive cancer specimens. IGF-1R
was overexpressed in HPV-positive cervical cancer cell lines in
comparison to ovarian cancer cell lines and HPV-negative cervical
cell line C33A. Phosphorylation of IGF-1R was promoted in all CIN
and invasive cancer and its intensity was related to the promotion
of lesions (71). In a
retrospective study of patients with early-stage cervical cancer it
was shown that high overexpression of IGF-1R is an independent
predictor of cervical cancer death and recurrence (63). On the other hand, low levels of
IGFBP-3 and IGF-1R mRNA in cervical scrapes were found to be
associated with progression to cervical cancer. Low levels of
IGF-1R mRNA were already reported in certain types of cancer, for
example breast cancer (72).
The IGF binding proteins (IGFBPs) represent the
third important component of the IGF system, after IGF-1 and
IGF-1R, consisting of a class of six soluble secretory proteins.
They represent a unique class of naturally occurring IGF
antagonists that bind and sequester IGF-1 and IGF-2, limiting their
access to the IGF-1R (73).
Elevated IGFBP-3 levels may have a protective function in ovarian
cancer occurrence (74). In
addition, IGFBP-3 is a proapoptotic agent and has been shown to act
through mechanism(s) independent of IGFs (75). In contrast to early-passage cells,
late-passage cells were found to secrete IGFBP-3 and showed an
increased response to IGF-1 as determined by the IGF-1R and insulin
receptor substrate (IRS) phopshorylation. Thus, the increased
responsiveness of HPV-immortalized cells to IGF-1 could potentially
contribute to their in vivo growth, where IGF-1 is produced
by surrounding stromal cells (76).
The induction of IGFBP-3 during immortalization is somewhat
surprising given previous knowledge of its regulated expression and
activity. Additionally, strong transcriptional activators of the
IGFBP-3 gene must exist and appear in late-passage HPV-16 E6/E7
cervical cells. In situ hybridization results showing
overexpression of IGFBP-3 mRNA in HSIL patient samples supports the
findings of IGFBP-3 upregulation in immortalized cervical cells
(77). In another study, the
influence of circulating IGF-1 levels on the natural history of
oncogenic HPV was prospectively assessed. Women with high serum
IGFBP-3 levels had significantly lower rates of incident oncogenic
HPV detection, and a lower incidence of oncogenic HPV-positive SIL,
than woman with low serum IGFBP-3 levels. The IGF1/IGFBP-3 molar
ratio, in contrast, was positively associated with persistence of
oncogenic HPV infection. Thus, high IGFBP-3 levels could lead to
less replication and/or greater loss of oncogenic HPV-infected
cells. Although in the pilot investigation none of the additional
associations of oncogenic HPV with IGF-1 or IGFBP-3 were
statistically significant, they were fairly similar to the above:
total IGF-1 had a nonsignificantly positive association with
prevalence of oncogenic HPV and oncogenic HPV-positve SIL, whereas
high IGFBP-3 had a nonsignificant inverse association with these
same endpoints. There was one inverse association with IGF-1 in the
study: high total IGF-1 was associated with a lower (not a higher)
prevalence of nononcogenic HPV (78). While it is generally assumed that
properties of HPV E7 depend on its interaction with regulators of
the cell cycle, E7 can also directly bind IGFBP-3, the product of a
p53-inducible gene that is overexpressed in senescent cells.
IGFBP-3 can suppress cell proliferation and induce apoptosis;
IGFBP-3-mediated apoptosis is inhibited by E7, which binds to
IGFBP-3 and triggers its proteolytic cleavage (79). As it is generally assumed that
elevated IGFBP-3 level has a protective function in cancer, it was
reported that serum levels of IGFBP-3 in patients with cervical
cancer are significantly lower than levels in controls, and they
revert to normal following therapy (80).
While comparing data from different studies, a
distinction is needed between results based on an extensive body of
evidence in well-conducted prospective studies and those in small
studies with weak cross-sectional design is important (Table I). It is certain that most of the
studies showed a strong association of IGFs with the HPV status and
cancer risk (61–64,78,80).
It was also indicated in an important review by Pollak et al
that increasing IGF-1 levels are associated with an increased risk
of cancer since somatic cells of individuals with higher levels of
IGF-1 may show slightly higher proliferation rates and have a
slightly increased chance of survival in the presence of genetic
damage because of the antiapoptotic effects of IGF-1 (model of
stepwise accumulation of genetic damage leading to carcinogenesis)
(1). The only unclear aspect of the
association being discussed is that some researchers claim it is
positive (61,62,81)
and others claim the contrary is the case (64–66)
and even though there is a stronger body of evidence supporting the
former, the latter should not be discarded.
 | Table ISummary of the possible correlations
between IGF axis components, HPV status and cervical
cancerogenesis. |
Table I
Summary of the possible correlations
between IGF axis components, HPV status and cervical
cancerogenesis.
| Risk/incidence of
neoplastic change/cancer (increased↑/decreased↓/no
associationΔ) | Correlation with
serum or local tissue level (higher↑/lower↓/no changeΔ) | Correlation with
HPV status (positive↑/negative↓/noneΔ) | Sample size, source
of information | Authors (ref.) |
|---|
| CIN↑, | IGF-1↑a, IGFBP3Δ, | HPVΔ | 126 cases (82 CIN),
40 controls | Lee et al
(61) |
| LSIL↑a, HSIL | IGF-1↑a, IGF-1/IGFBP3↑a | nd | 267 cases, 238
controls | Wu et al
(62) |
| CC | IGF-1↑ | HPV↑a | 50 cases, 40
controls | Sharma et al
(81) |
| SIL↓ | IGFBP-3↑ | oncogenic HPV↓ | | Harris et al
(78) |
| SIL↑ | IGF-1↑ | oncogenic HPV↑ | 137 cases | |
| IGF-1/IGFBP-3 | HPV
persistence↑ | | |
| CCΔ (no
prediction) | IGF-1↓,
IGFBP-3Δ | nd | 72 cases | Huang et al
(63) |
| CC, worse OS | IGF-R↑
(predictor) | | | |
| HCIN↓ | IGF↑, IGFBP-3Δ | HPV-16 and
-18↓ | 329 cases, 621
controls | Schaffer et
al (64) |
| CC↑ | IGF-1↓a, IGF-2↓, | nd | 135 cases, 270
controls | Serrano et
al (65) |
| HSIL↑ and
progression to CC↓ | IGFBP-3↓,
IGF-R↓ | nd | 63 cases, 42
controls | Serrano et
al (66) |
| HSIL vs. control,
LSIL vs CC | IGFBP-3↑a, IGFBP-3Δ | HR-HPVΔ | 93 cases, 51
controls | Serrano et
al (68) |
| CIN III/CC↑ | IGF-R↑ | nd | cases 90, 30
controls | Kuramoto et
al (71) |
| CC↑ | IGF-R↑ | HPV | 72 cases | Huang et al
(63) |
| Control vs. all
stages of CC↑ | IGF-2↑,
IGFBP-3↓ | nd | 160 cases (11
groups), 23 controls | Mathur et al
(80) |
| Breast cancer↑ | IGF-system
components↓ | - | 72 cases | Voskuil et
al (72) |
| Ovarian
cancer↓ | IGFBP-3↑ | nd | 59 cases, 108
controls | Dal Maso et
al (74) |
| HPV E6/E7 induced
cells | IGFBP-3↑ and
IGF-1↑ | HPV↑ | Basic research | Berger et al
(77) |
| Cervical cells with
E6/E7 | IGF-R↑ | | Basic research | Baege et al
(76) |
7. Other factors in cervical cancer
Development of cervical cancer affects a small
percentage of HR-HPV-infected women and often takes decades after
infection, suggesting that HR-HPV is a necessary but not sufficient
cause of cervical cancer (81).
Thus, other cofactors are necessary for progression from cervical
HR-HPV infection to cancer. These factors include long-term use of
hormonal contraceptives, multiparity, smoking, as well as
micronutrient depletion and in particular retinoid deficiency,
which alters epithelial differentiation, cellular growth and
apoptosis of malignant cells. Therefore, early detection of HR-HPV
and management of precancerous lesions together with a profound
understanding of additional risk factors could be a strategy to
avoid this disease (82). Chronic
estrogen exposure is a key factor for the development of this
disease. E6 oncogene was found to synergize with estrogen to induce
cervical cancer after 9 months, indicating that E6 has a weaker but
detectable oncogenic potential in the reproductive tract compared
with the E7 oncogene (83).
Estrogens upregulate HPV E6/E7 oncogene expression, stimulate cell
proliferation, inhibit apoptosis and their metabolites cause DNA
damage. On the other hand, retinoid deficiency is implicated in
cervical squamous metaplasia and the decrease in retinoic acid
receptor β (RARβ2) expression promotes AP1-dependent cellular
proliferation. Synergistic activation of cell proliferation by
viral oncoproteins, estrogen receptor signaling, inhibition of
RARβ2 expression and nutritional status factors may conspire to
support and promote neoplastic progression and cervical cancer
(82). While the uterine cervix is
highly responsive to estrogen, the role of estrogen in cervical
cancer, which is strongly associated with HPV infections, is poorly
understood. Estrogen and estrogen receptor α (ERα) are required for
cervical carcinogenesis, and cervical cancer is often positive for
ERα, although its functionality in this cancer has yet to be
demonstrated (84). Tumors arising
in HPV-16 transgenic mice treated with estrogen for 9 months were
greatly increased in size compared with tumors developing after 6
months of estrogen treatment. It can be concluded that estrogen
plays a critical role not only in the genesis of cervical cancer
but also in its persistence and continued development in this mouse
model (85). A transgenic mouse
model expressing HPV oncogenes E6 and/or E7 has proven useful to
study a mechanism of hormone actions in the context of this common
malignancy. ERα is known to upregulate expression of the
progesterone receptor, which, on activation by its ligands, either
promotes or inhibits carcinogenesis, depending on the tissue
context. These results provide the first experimental evidence that
supports the hypothesis that progesterone signaling has an
inhibitory effect on cervical carcinogenesis in vivo
(86). With regard to apoptosis, it
has been suggested that the stimulatory effects of 17β-estradiol on
E2 and E7-induced cell death are mediated by 16α-hydroxy-estrone in
HeLa cells (87). It was also shown
that cervical malignant cells tend to lose the ERα but maintain the
ERβ actively expressed. Loss of expression of ERα in neoplastic
tissue suggests that the estrogenic effects could be regulated
through the ERβ in human neoplastic cervical tissue (88).
It has been reported that HPV-18 E6 and E7 proteins
directly interact with nuclear receptors (NRs) such as thyroid
receptor (TR), androgen receptor (AR) and ER through a
hormone-independent mechanism (89). HPV-18 E6 protein was found to
generally enhance the reporter activities of these three NRs. In
contrast, HPV-18 E7 protein repressed the reporter activities of
these three NRs either in the absence or presence of cognate
ligands. However, in HeLa cells (compared with HEK 293 cells), in
the absence of an appropriate ligand, no coregulatory effect on AR,
ER and TR was detected, whereas these NR activities increased up to
7-fold in the presence of hormone (89). Folic acid supplementation might be
useful in maintaining cervical health as it upregulates IGFBP-3
(90). Other findings are in
agreement with this observation and indicate that it may be
important to improve the folate status in HR-HPV-infected women and
that folate supplementation should be assessed as a viable option
for reducing the risk of developing CIN ≥2 in women exposed to
HR-HPV, especially HPV-16 (91). It
has been reported that higher circulating concentrations of folate
are independently associated with a lower likelihood of becoming
positive for HR-HPVs and of having a persistent HR-HPV infection
and a greater likelihood of becoming HR-HPV-negative (92).
Alterations in mtDNA both qualitatively (by
mutations) and quantitatively (by mtDNA copy number) are associated
with cervical cancer development. High levels of mtDNA copy with a
4.997 bp deletion in LSIL cells can be associated with the
susceptibility of cells to an HPV-persistent infection and cervical
cancer development (93). The copy
number of mtDNA in cases which carried a D-loop mutation was
significantly higher than that of the negative cases (P<0.05).
These results suggest that the mtDNA D-loop in low-grade squamous
cell carcinoma (LSCC) is an unstable region with a high frequency
of somatic mutations and polymorphisms. Together with the increase
in mtDNA copy number, these factors may play a role in
carcinogenesis of the larynx (94).
Mutations of mtDNA in breast cancer occur both within and outside
of the D-loop, although the mutation rate in the D-loop is more
than 7-fold higher than in coding areas. There have been 26 new
mutation loci identified (25 in regions sequenced by others, one in
an area not sequenced). The high frequency of mtDNA mutations at
polymorphic loci requires further investigation (Fig. 2) (95).
8. Cancer therapy: IGF and HPV
targeting
The IGF axis has emerged as a meaningful therapeutic
target for oncology drug development and is strongly supported by
preclinical studies and promising results from early phase clinical
trials. The 3 major classes of IGF-targeted therapeutic compounds
[i.e., IGF-1R-specific monoclonal antibodies (mAbs), small-molecule
tyrosine kinaze inhibitors (TKIs) targeting IGF-1R and IR kinase
domains, and finally, an IGF-1 and IGF-2 Ligand-neutralizing mAbs)]
differ in the range of target inhibition based on their ability to
block activation of IGF-1R, IGF-1R/IR-A hybrid and IR-A. They also
exhibit different safety profiles, most notably with respect to
modulation of glucose metabolism, as well as through changes in
circulating levels of IGF, insulin, and growth hormone (96). By 2010 there was a list of over 30
drugs under evaluation as single agents or in combination therapies
(73). Recently, targeting therapy
with the IGF-1R antibody has rapidly developed (97,98).
The treatment of blocking IGF-1R with antibodies markedly decreased
IGF-1R phosphorylation and downstream activation of Akt and Erk1/2,
hereby inhibiting tumor growth (96). IGF-1R has been detected in many
studied tumors, regardless of differentiation or proof of EBV or
HPV integration into the genome. These results suggest that IGF-1R
expression in these tumors is capable of transmitting mitogenic
signals to the neoplastic cells (99). The IGF-1R antibodies appear to have
a favorable safety profile and have been demonstrated to reduce
IGF-1R signaling in patients. Concerning the IGF-1R tyrosine kinase
inhibitors, the first published data from clinical trials are still
awaited. Some phase II and III trials have been suspended or
terminated, because of the lack of efficacy of the antibodies. The
identification of predictive biomarkers is of crucial importance
for the further development of anticancer therapies based on
anti-IGF-1R agents (100).
However, the results of targeting the IGF-1R with specific
antibodies for ‘financially attractive tumors’ (breast, colon,
prostate, lung cancers and others) have been unsatisfactory. In
addition, it should be remembered that blocking IGF-1R with
therapeutic antibodies can lead to metabolic toxicity (101). Effective targeting of the IGF
system may require a customized approach in which tumor profiling
guides the selection of the appropriate drugs (102). In the next few years, exciting
clinical trials and translational research will provide information
and explanations in order to identify biomarkers for anti-IGF-1R
treatments, thus allowing the targeting of populations that will
most benefit from anti-IGF-1R mAbs and combined treatments
(103). Indeed, there is very
little to encourage the further use of targeting the IGF-1R as a
single agent in treatment of human cancer, except in a few,
relatively rare tumors. There is more hope in multi-drug therapies
(104) and multiple challenges are
still ahead, including the multiplicity of potential cancer
indications and drug combinations, as well as the need of
biomarkers for resistance and sensitivity (100) and for the time being demonstration
of meaningful clinical benefit remains elusive (96).
Tumorigenesis in nude mice was found to be highly
inhibited in HeLa S3 and SiHa clones transfected with the IGF-1R
antisense RNA. These results indicate that downregulation of IGF-1R
can reverse the transformed phenotype of human cervical cancer
cells, even when harboring malignant type HPVs (105). HPV-associated cancers are prime
candidates for the development of RNA interference-based
therapeutic approaches (106).
Increasing evidence has shown that microRNAs are commonly
deregulated in human malignant cancers, including cervical cancer
(107) but the role of microRNA
(miR)-497 in human cervical cancer still remains unclear. Recently,
miR-497 was demonstrated to bind to the 3′ untranslated regions of
IGF-1R mRNA, and upregulation of miR-497 downregulated IGF-1R
protein expression. Further investigation showed that small
interfering RNA-mediated IGF-1R knockdown could mimic the effect of
enforced miR-497 expression on the malignant phenotypes of cervical
cancer cells (108). Furthermore,
12 highly differentially regulated miRNAs, which distinguished
high-grade CIN specimens from normal cervical epithelium have been
identified. Target prediction analysis revealed that these
deregulated miRNAs mainly control apoptosis signaling pathways and
cell cycle regulation. These findings contribute to understanding
the role of miRNAs in the pathogenesis and progression of cervical
neoplasm at the molecular level (109). Previous studies have demonstrated
that small interference RNAs (siRNAs) can suppress HPV6 and/or E7
expression in various cancer cell lines (110,111). The advantage of using a lentiviral
delivery system compared to synthetic and vector-borne siRNA, used
in previously mentioned studies, is the ability to stably transduce
dividing and non-dividing cells with relatively high efficiency
(112). Data by Gu and co-workers
collectively suggest that lentiviral delivery is an effective way
to achieve stable suppression of E6/E7 oncogene expression and
induce inhibition of tumor growth both in vitro and in
vivo. They demonstrated a high specificity for LV-18E6-1, which
kills only HeLa cells but not other cervical carcinoma cells
without HPV-18 E6 such as C33A and SiHa. These results encourage
further investigation of this form of RNA interference as a
promising treatment for cervical cancer (113). There has been mild success in
treating different diseases with the use of lentiviral vectors
(114). Similar results were
obtained in another study. LV-shRNA specific to HPV-16 oncogenes,
targeting the promoter and the E6-transcript, effectively knocked
down E6 and E7 expression, along with accumulation of p53 and pRB
protein, resulting in markedly reduced abilities of proliferation
and invasiveness of cervical cancer cells in vitro. These
findings may provide important evidence for the application of
LV-shRNA targeting HR-HPV key oncogenes, as a new treatment
strategy, in cervical and other HPV-associated cancer therapy
(115). HPV pseudovirions encoding
shRNA may provide another means of cervical cancer therapy, and
using shRNA/HPV pseudovirions appears to be a more promising
strategy than using siRNA alone. The presence of L1 capsids can
also induce local immunization against L1 neutralizing epitopes,
thus conferring protection in cases of HPV reinfection after the
death of E7-expressing cells. These pseudovirions represent a new
step in designing rational molecular cancer therapy using RNA
interference, at the same time, offering protection against
reinfection by the causative agent of cervical cancer (116). Another new active targeted
immunotherapeutic has been evaluated, Modified Vaccinia Virus
Ankara (MVA) vector, containing the E1 sequence of HPV-16, aimed at
inducing cellular immune responses with potential to help and clear
persistent HPV-16-related infection. It has been shown that
multiple injections of MVA-E1 allowed sustained HPV-16E1-specific
cellular immune responses in vaccinated mice and had no impact on
the exhaustion phenotype of the generated HPV-16E1-specific
CD8+ T cells, but led to the differentiation of
multi-functional effector T cells with high cytotoxic capacity.
This study provides proof of concept that a MVA expressing HPV-16E1
can induce robust and long lasting E1-specific responses, and
further development of this candidate is warranted (117).
The availability of prophylactic HPV vaccines has
provided powerful tools for primary prevention of cervical cancer
and other HPV-associated diseases. By the beginning of 2012, the
HPV vaccine had been introduced into national immunization programs
in at least 40 countries (118).
Prophylactic vaccines induce neutralizing antibodies against HPV L1
structural proteins, which are associated with protection from HPV
infection. However, therapeutic vaccines induce cytotoxic T
lymphocyte (CTL) responses to HPV early regulatory proteins,
possibly leading to eradication of CIN, cervical cancer and other
HPV-associated diseases. The antibodies neutralize infectious HPV
particles, while CTLs recognize and kill HPV-infected epithelial
cells and HPV-associated cancer cells (119). Indeed, E6 and E7 are often the
only viral genes that continue to be expressed in cancerous cells
(120), thus they represent ideal
targets for immunotherapy of cervical cancer. Accordingly, numerous
methodologies to elicit strong anti-E6/E7 cellular immunity have
been explored including peptide immunization (121), DNA immunization (122), immunization with recombinant,
E7-expressing Vaccinia virus (117), adenovirus (123), pathogenic bacteria (124), E7-pulsed dendritic cells (125) or E7-containing virus-like
particles (VLPs) (126). Although
a number of approaches in therapy of cervical cancer have been
developed, none has yet advanced for commercial use. These
approaches are summarized in Table
II.
| Table IIExamples of therapeutic approaches in
IGF and HPV-related cancers. |
Table II
Examples of therapeutic approaches in
IGF and HPV-related cancers.
| Target | Method/Tool | Authors (ref.) |
|---|
| IGF-R | IGF-R specific
mAbs | Shen et al
(69), Miller and Yee (97), Hartog et al (98), Friedrich et al (99) |
| Small molecules
targeting IGF-1R (tyrosine kinase inhibitors) | Gao et al
(96), Arcaro (100) |
| IGF-1 or IGF-2 | Ligand-neutralizing
antibodies | Gao et al
(96) |
| IGF-1R mRNA | Antisense RNA | Nakamura et
al (105) |
| MicroRNA
(miR-497) | Luo et al
(108) |
| Multiple targets
for IGF axis | Multiple drug
therapies | Baserga (104), Arcaro (100), Gao et al (96) |
| HPV E6/E7 | Synthetic
interference RNA | Butz et al
(110), Hall and Alexander
(111) |
| Vector-borne
siRNA | Gu et al
(113), Zhou et al
(115) |
| shRNA via VLPs | Bousarghin et
al (116) |
| Immune system | Prophylactic
vaccines containing VLPs (neutralizing Abs against HPV-L1) | Markowitz et
al (118) |
| Therapeutic
vaccines (inducing cytotoxic T lymphocytes ) | Berraondo et
al (121), Peng et al
(122), Da Silva et al
(126) |
9. Conclusions
This review discusses cervical carcinogenesis as a
multifactor and multistep process. Although substantial progress
has been made in cancer therapies focused on blocking HPV
oncoproteins and IGF axis components, mainly IGF-R, there are many
molecular and clinical studies in progress aimed at the development
of a more effective treatment of cervical neoplasia. Multidrug
combinatorial therapies will greatly help overcome the difficulties
described in the present review.
Acknowledgements
This review article was supported by a Research
Grant from the Polish National Science Center (NN401 555440) to Dr
Julia Durzyńska, 2011-2016.
References
|
1
|
Pollak MN, Schernhammer ES and Hankinson
SE: Insulin-like growth factors and neoplasia. Nat Rev Cancer.
4:505–518. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Barton ER: The ABCs of IGF-I isoforms:
impact on muscle hypertrophy and implications for repair. Appl
Physiol Nutr Metab. 31:791–797. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Durzyńska J, Philippou A, Brisson BK,
Nguyen-McCarty M and Barton ER: The pro-forms of insulin-like
growth factor I (IGF-I) are predominant in skeletal muscle and
alter IGF-I receptor activation. Endocrinology. 154:1215–1224.
2013.PubMed/NCBI
|
|
4
|
Tan DS, Cook A and Chew SL: Nucleolar
localization of an isoform of the IGF-I precursor. BMC Cell Biol.
3:172002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Durzyńska J, Wardziński A, Koczorowska M,
Goździcka-Józefiak A and Barton ER: Human Eb peptide: not just a
by-product of pre-pro-IGF1b processing? Horm Metab Res. 45:415–422.
2013.PubMed/NCBI
|
|
6
|
Siegfried JM, Kasprzyk PG, Treston AM,
Mulshine JL, Quinn KA and Cuttitta F: A mitogenic peptide amide
encoded within the E peptide domain of the insulin-like growth
factor IB prohormone. Proc Natl Acad Sci USA. 89:8107–8111. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kasprzak A, Szaflarski W, Szmeja J, et al:
Expression of various insulin-like growth factor-1 mRNA isoforms in
colorectal cancer. Contemp Oncol. 16:147–153. 2012.
|
|
8
|
Kasprzak A, Szaflarski W, Szmeja J, et al:
Differential expression of IGF-1 mRNA isoforms in colorectal
carcinoma and normal colon tissue. Int J Oncol. 42:305–316.
2013.PubMed/NCBI
|
|
9
|
Koczorowska MM, Kwasniewska A and
Gozdzicka-Jozefiak A: IGF1 mRNA isoform expression in the cervix of
HPV-positive women with pre-cancerous and cancer lesions. Exp Ther
Med. 2:149–156. 2011.PubMed/NCBI
|
|
10
|
Pollak M: Insulin and insulin-like growth
factor signaling in neoplasia. Nat Rev Cancer. 8:915–928. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Grimberg A: Mechanisms by which IGF-I may
promote cancer. Cancer Biol Ther. 2:630–635. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lukanova A, Lundin E, Toniolo P, et al:
Circulating levels of insulin-like growth factor-I and risk of
ovarian cancer. Int J Cancer. 101:549–554. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sell C, Rubini M, Rubin R, Liu JP,
Efstratiadis A and Baserga R: Simian virus 40 large tumor antigen
is unable to transform mouse embryonic fibroblasts lacking type 1
insulin-like growth factor receptor. Proc Natl Acad Sci USA.
90:11217–11221. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
LeRoith D and Roberts CT Jr: The
insulin-like growth factor system and cancer. Cancer Lett.
195:127–137. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Loughran G, Huigsloot M, Kiely PA, Smith
LM, Floyd S, Ayllon V and O’Connor R: Gene expression profiles in
cells transformed by overexpression of the IGF-I receptor.
Oncogene. 24:6185–6193. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gallagher EJ and LeRoith D: Minireview:
IGF, insulin, and cancer. Endocrinology. 152:2546–2551. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Aleksic T, Chitnis MM, Perestenko OV, et
al: Type 1 insulin-like growth factor receptor translocates to the
nucleus of human tumor cells. Cancer Res. 70:6412–6419. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Baserga R: The insulin receptor
substrate-1: a biomarker for cancer? Exp Cell Res. 315:727–732.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Baserga R: Customizing the targeting of
IGF-1 receptor. Future Oncol. 5:43–50. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
DePinho RA: The age of cancer. Nature.
408:248–254. 2000. View
Article : Google Scholar
|
|
21
|
Laron Z: The GH-IGF1 axis and longevity.
The paradigm of IGF1 deficiency. Hormones. 7:24–27. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Steuerman R, Shevah O and Laron Z:
Congenital IGF1 deficiency tends to confer protection against
post-natal development of malignancies. Eur J Endocrinol.
164:485–489. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Laron Z: Laron syndrome (primary growth
hormone resistance or insensitivity): the personal experience
1958–2003. J Clin Endocrinol Metab. 89:1031–1044. 2004.PubMed/NCBI
|
|
24
|
Guevara-Aguirre J, Balasubramanian P,
Guevara-Aguirre M, et al: Growth hormone receptor deficiency is
associated with a major reduction in pro-aging signaling, cancer,
and diabetes in humans. Sci Transl Med. 3:70ra132011.(Epub ahead of
print). View Article : Google Scholar
|
|
25
|
Wang SR, Carmichael H, Andrew SF, et al:
Large-scale pooled next-generation sequencing of 1077 genes to
identify genetic causes of short stature. J Clin Endocrinol Metab.
98:1428–1437. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mullis PE: Genetics of isolated growth
hormone deficiency. J Clin Res Pediatr Endocrinol. 2:52–62. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Forbes BE: Molecular mechanisms underlying
insulin-like growth factor action: How mutations in the GH: IGF
axis lead to short stature. Pediatr Endocrinol Rev. 8:374–381.
2011.PubMed/NCBI
|
|
28
|
Petriczko E, Wikiera B, Horodnicka-Józwa
A, et al: A two year observation of the process of applying
recombinant IGF-1 to treat short stature in children with primary
IGF-1 deficiency - case reports of 3 patients. Pediatr Endocrinol
Diabetes Metab. 17:233–238. 2011.PubMed/NCBI
|
|
29
|
Kędzia A, Durzyńska J, Gabryelczyk B,
Petriczko E and Goździcka-Józefiak A: An analysis of the IGF-I
receptor coding sequence in the genome of children with growth
disorders. Pediatrc Endocrinol, Diabet and Metab. 19:96–99.
2013.PubMed/NCBI
|
|
30
|
Kasprzak A and Adamek A: The insulin-like
growth factor (IGF) signaling axis and hepatitis C virus-associated
carcinogenesis (Review). Int J Oncol. 41:1919–1931. 2012.PubMed/NCBI
|
|
31
|
Trojanek J, Croul S, Ho T, et al:
T-antigen of the human polyomavirus JC attenuates faithful DNA
repair by forcing nuclear interaction between IRS-1 and Rad51. J
Cell Physiol. 206:35–46. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Reiss K, Khalili K, Giordano A and
Trojanek J: JC virus large T-antigen and IGF-I signaling system
merge to affect DNA repair and genomic integrity. J Cell Physiol.
206:295–300. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ramamoorthy S, Devaraj B, Miyai K, et al:
John Cunningham virus T-antigen expression in anal carcinoma.
Cancer. 117:2379–2385. 2011. View Article : Google Scholar
|
|
34
|
de Villiers EM, Fauquet C, Broker TR,
Bernard HU and zur Hausen H: Classification of papillomaviruses.
Virology. 324:17–27. 2004.
|
|
35
|
Jo H and Kim JW: Implications of HPV
infection in uterine cervical cancer. Cancer Ther. 3:419–434.
2005.
|
|
36
|
Bernard HU: The clinical importance of the
nomenclature, evolution and taxonomy of human papillomaviruses. J
Clin Virol. 32:S1–S6. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
zur Hausen H: Papillomaviruses and cancer:
from basic studies to clinical application. Nat Rev Cancer.
2:342–350. 2002.PubMed/NCBI
|
|
38
|
Pietsch EC and Murphy ME: Low risk HPV-E6
traps p53 in the cytoplasm and induces p53-dependent apoptosis.
Cancer Biol Ther. 7:1916–1918. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
zur Hausen H: Papillomaviruses in the
causation of human cancers - a brief historical account. Virology.
384:260–265. 2009.PubMed/NCBI
|
|
40
|
Ghittoni R, Accardi R, Hasan U, Gheit T,
Sylla B and Tommasino M: The biological properties of E6 and E7
oncoproteins from human papillomaviruses. Virus Genes. 40:1–13.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Doorbar J: Molecular biology of human
papillomavirus infection and cervical cancer. Clin Sci.
110:525–541. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Doorbar J, Quint W, Banks L, Bravo IG,
Stoler M, Broker TR and Stanley MA: The biology and life-cycle of
human papillomaviruses. Vaccine. 30:F55–F70. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Whiteside MA, Siegel EM and Unger ER:
Human papillomavirus and molecular considerations for cancer risk.
Cancer. 113:S2981–S2994. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
von Knebel Doeberitz M: New markers for
cervical dysplasia to visualise the genomic chaos created by
aberrant oncogenic papillomavirus infections. Eur J Cancer.
38:2229–2242. 2002.PubMed/NCBI
|
|
45
|
Vinokurova S, Wentzensen N, Kraus I, et
al: Type-dependent integration frequency of human papillomavirus
genomes in cervical lesions. Cancer Res. 68:307–313. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Thorland EC, Myers SL, Gostout BS and
Smith DI: Common fragile sites are preferential targets for HPV16
integrations in cervical tumors. Oncogene. 22:1225–1237. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Cheung JL, Cheung TH, Yu MY and Chan PK:
Virological characteristics of cervical cancers carrying pure
episomal form of HPV16 genome. Gynecol Oncol. 131:374–379. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yugawa T and Kiyono T: Molecular
mechanisms of cervical carcinogenesis by high-risk human
papillomaviruses: novel functions of E6 and E7 oncoproteins. Rev
Med Virol. 19:97–113. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Egawa K: Do human papillomaviruses target
epidermal stem cells? Dermatology. 207:251–254. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Wilson VG, West M, Woytek K and Rangasamy
D: Papillomavirus E1 proteins: form, function, and features. Virus
Genes. 24:275–290. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
You J, Croyle JL, Nishimura A, Ozato K and
Howley PM: Interaction of the bovine papillomavirus E2 protein with
Brd4 tethers the viral DNA to host mitotic chromosomes. Cell.
117:349–360. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Doorbar J: The papillomavirus life cycle.
J Clin Virol. 32:S7–S15. 2005. View Article : Google Scholar
|
|
53
|
Watson RA, Thomas M, Banks L and Roberts
SJ: Activity of the human papillomavirus E6 PDZ-binding motif
correlates with an enhanced morphological transformation of
immortalized human keratinocytes. J Cell Sci. 116:4925–4934. 2003.
View Article : Google Scholar
|
|
54
|
Sun L, Zhang G, Lei T, Huang C, Song T and
Si L: Two different HPV-11E6 fusion proteins trap p53 in the
cytoplasm and induce apoptosis. Cancer Biol Ther. 7:1909–1915.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Tommasino M, Accardi R, Caldeira S, Dong
W, Malanchi I, Smet A and Zehbe I: The role of TP53 in cervical
carcinogenesis. Hum Mutat. 21:307–312. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Ganguly N and Parihar SP: Human
papillomavirus E6 and E7 oncoproteins as risk factors for
tumorigenesis. J Biosci. 34:113–123. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
McLaughlin-Drubin ME and Münger K: The
human papillomavirus E7 oncoprotein. Virology. 384:335–344. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Psyrri A and DiMaio D: Human
papillomavirus in cervical and head-and-neck cancer. Nat Clin Pract
Oncol. 5:24–31. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Clifford GM, Smith JS, Plummer M, Muñoz N
and Franceschi S: Human papillomavirus types in invasive cervical
cancer worldwide: a meta-analysis. Br J Cancer. 88:63–73. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
zur Hausen H: Papillomaviruses causing
cancer: evasion from host-cell control in early events in
carcinogenesis. J Natl Cancer Inst. 92:690–698. 2000.PubMed/NCBI
|
|
61
|
Lee SW, Lee SY, Lee SR, Ju W and Kim SC:
Plasma levels of insulin-like growth factor-1 and insulin-like
growth factor binding protein-3 in women with cervical neoplasia.
Gynecol Oncol. 21:174–180. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Wu X, Tortolero-Luna G, Zhao H, Phatak D,
Spitz MR and Follen M: Serum levels of insulin-like growth factor I
and risk of squamous intraepithelial lesions of the cervix. Clin
Cancer Res. 9:3356–3361. 2003.PubMed/NCBI
|
|
63
|
Huang YF, Shen MR, Hsu KF, Cheng YM and
Chou CY: Clinical implications of insulin-like growth factor 1
system in early-stage cervical cancer. Br J Cancer. 99:1096–1102.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Schaffer A, Koushik A, Trottier H, et al:
Insulin-like growth factor-I and risk of high-grade cervical
intraepithelial neoplasia. Cancer Epidemiol Biomarkers Prev.
16:716–722. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Serrano ML, Romero A, Cendales R,
Sánchez-Gómez M and Bravo MM: Serum levels of insulin-like growth
factor-I and -II and insulin-like growth factor binding protein 3
in women with squamous intraepithelial lesions and cervical cancer.
Biomedica. 26:258–268. 2006.
|
|
66
|
Serrano ML, Sánchez-Gómez M and Bravo MM:
Insulin-like growth factor system gene expression in cervical
scrapes from women with squamous intraepithelial lesions and
cervical cancer. Growth Horm IGF Res. 17:492–499. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Serrano ML, Sánchez-Gómez M, Bravo MM,
Yakar S and LeRoith: Differential expression of IGF-I and insulin
receptor isoforms in HPV positive and negative human cervical
cancer cell lines. Horm Metab Res. 40:661–667. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Serrano ML, Sánchez-Gómez M and Bravo MM:
Cervical scrapes levels of insulin-like growth factor-II and
insulin-like growth factor binding protein 3 in women with squamous
intraepithelial lesions and cervical cancer. Horm Metab Res.
42:977–981. 2010. View Article : Google Scholar
|
|
69
|
Shen MR, Hsu YM, Hsu KF, Chen YF, Tang MJ
and Chou CY: Insulin-like growth factor 1 is a potent stimulator of
cervical cancer cell invasiveness and proliferation that is
modulated by alphavbeta3 integrin signaling. Carcinogenesis.
27:962–971. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Steller MA, Delgado CH, Bartels CJ,
Woodworth CD and Zou Z: Overexpression of the insulin-like growth
factor-1 receptor and autocrine stimulation in human cervical
cancer cells. Cancer Res. 56:1761–1765. 1996.PubMed/NCBI
|
|
71
|
Kuramoto H, Hongo A, Liu YX, et al:
Immunohistochemical evaluation of insulin-like growth factor I
receptor status in cervical cancer specimens. Acta Med Okayama.
62:251–259. 2008.PubMed/NCBI
|
|
72
|
Voskuil DW, Bosma A, Vrieling A, Rookus MA
and van ‘t Veer LJ: Insulin-like growth factor (IGF)-system mRNA
quantities in normal and tumor breast tissue of women with sporadic
and familial breast cancer risk. Breast Cancer Res Treat.
84:225–233. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Rosenzweig SA and Atreya HS: Defining the
pathway to insulin-like growth factor system targeting in cancer.
Biochem Pharmacol. 80:1115–1124. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Dal Maso L, Augustin LS, Franceschi S, et
al: Association between components of the insulin-like growth
factor system and epithelial ovarian cancer risk. Oncology.
67:225–230. 2004.
|
|
75
|
Hong J, Zhang G, Dong F and Rechler MM:
Insulin-like growth factor (IGF)-binding protein-3 mutants that do
not bind IGF-I or IGF-II stimulate apoptosis in human prostate
cancer cells. J Biol Chem. 277:10489–10497. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Baege AC, Disbrow GL and Schlegel R:
IGFBP-3, a marker of cellular senescence, is overexpressed in human
papillomavirus-immortalized cervical cells and enhances
IGF-1-induced mitogenesis. J Virol. 78:5720–5727. 2004. View Article : Google Scholar
|
|
77
|
Berger AJ, Baege A, Guillemette T, et al:
Insulin-like growth factor-binding protein 3 expression increases
during immortalization of cervical keratinocytes by human
papillomavirus type 16 E6 and E7 proteins. Am J Pathol.
161:603–610. 2002. View Article : Google Scholar
|
|
78
|
Harris TG, Burk RD, Yu H, et al:
Insulin-like growth factor axis and oncogenic human papillomavirus
natural history. Cancer Epidemiol Biomarkers Prev. 17:245–248.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Mannhardt B, Weinzimer SA, Wagner M,
Fiedler M, Cohen P, Jansen-Dürr P and Zwerschke W: Human
papillomavirus type 16 E7 oncoprotein binds and inactivates
growth-inhibitory insulin-like growth factor binding protein 3. Mol
Cell Biol. 20:6483–6495. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Mathur SP, Mathur RS, Underwood PB, Kohler
MF and Creasman WT: Circulating levels of insulin-like growth
factor-II and IGF-binding protein 3 in cervical cancer. Gynecol
Oncol. 91:486–493. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Sharma M, Satyam A, Abhishek A, Khan R,
Rajappa M and Sharma A: Molecular and circulatory expression of
insulin growth factors in Indian females with advanced cervical
cancer. Asian Pac J Cancer Prev. 13:6475–6479. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Gariglio P, Gutiérrez J, Cortés E and
Vázquez J: The role of retinoid deficiency and estrogens as
cofactors in cervical cancer. Arch Med Res. 40:449–465. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Shai A, Brake T, Somoza C and Lambert PF:
The human papillomavirus E6 oncogene dysregulates the cell cycle
and contributes to cervical carcinogenesis through two independent
activities. Cancer Res. 67:1626–1635. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Chung SH, Franceschi S and Lambert PF:
Estrogen and ERalpha: culprits in cervical cancer? Trends
Endocrinol Metab. 21:504–511. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Brake T and Lambert PF: Estrogen
contributes to the onset, persistence, and malignant progression of
cervical cancer in a human papillomavirus-transgenic mouse model.
Proc Natl Acad Sci USA. 102:2490–2495. 2005. View Article : Google Scholar
|
|
86
|
Yoo YA, Son J, Mehta FF, Demayo FJ, Lydon
JP and Chung SH: Progesterone signaling inhibits cervical
carcinogenesis in mice. Am J Pathol. 183:1679–1687. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Webster K, Taylor A and Gaston K:
Oestrogen and progesterone increase the levels of apoptosis induced
by the human papillomavirus type 16 E2 and E7 proteins. J Gen
Virol. 82:201–213. 2001.PubMed/NCBI
|
|
88
|
López-Romero R, Garrido-Guerrero E,
Rangel-López A, et al: The cervical malignant cells display a down
regulation of ER-α but retain the ER-β expression. Int J Clin Exp
Pathol. 6:1594–1602. 2013.PubMed/NCBI
|
|
89
|
Wang WM, Chung MH and Huang SM: Regulation
of nuclear receptor activities by two human papillomavirus type 18
oncoproteins, E6 and E7. Biochem Biophys Res Commun. 303:932–939.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Mathur RS and Mathur SP: In vitro
downregulation of growth factors by insulin-like growth factor
binding protein-3 in cervical cancer. Gynecol Oncol. 91:410–415.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Piyathilake CJ, Henao OL, Macaluso M,
Cornwell PE, Meleth S, Heimburger DC and Partridge EE: Folate is
associated with the natural history of high-risk human
papillomaviruses. Cancer Res. 64:8788–8793. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Piyathilake CJ, Badiga S, Paul P, et al:
Indian women with higher serum concentrations of folate and vitamin
B12 are significantly less likely to be infected with carcinogenic
or high-risk (HR) types of human papillomaviruses (HPVs). Int J
Womens Health. 2:7–12. 2010. View Article : Google Scholar
|
|
93
|
Warowicka A, Kwasniewska A and
Gozdzicka-Jozefiak A: Alterations in mtDNA: a qualitative and
quantitative study associated with cervical cancer development.
Gynecol Oncol. 129:193–198. 2013. View Article : Google Scholar
|
|
94
|
Guo W, Yang D and Xu H: Mutations in the
D-loop region and increased copy number of mitochondrial DNA in
human laryngeal squamous cell carcinoma. Mol Biol Rep. 40:13–20.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Zhu W, Qin W, Bradley P, Wessel A, Puckett
CL and Sauter ER: Mitochondrial DNA mutations in breast cancer
tissue and in matched nipple aspirate fluid. Carcinogenesis.
26:145–152. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Gao J, Chang YS, Jallal B and Viner J:
Targeting the insulin-like growth factor axis for the development
of novel therapeutics in oncology. Cancer Res. 72:3–12. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Miller BS and Yee D: Type I insulin-like
growth factor receptor as a therapeutic target in cancer. Cancer
Res. 65:10123–10127. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Hartog H, Wesseling J, Boezen HM and van
der Graaf WT: The insulin-like growth factor 1 receptor in cancer:
old focus, new future. Eur J Cancer. 43:1895–1904. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Friedrich RE, Hagel C and Bartel-Friedrich
S: Insulin-like growth factor-1 receptor (IGF-1R) in primary and
metastatic undifferentiated carcinoma of the head and neck: a
possible target of immunotherapy. Anticancer Res. 30:1641–1643.
2010.PubMed/NCBI
|
|
100
|
Arcaro A: Targeting the insulin-like
growth factor-1 receptor in human cancer. Front Pharmacol.
4:302013. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Pollak M: The insulin
receptor/insulin-like growth factor receptor family as a
therapeutic target in oncology. Clin Cancer Res. 18:40–50. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Samani AA, Yakar S, LeRoith D and Brodt P:
The role of the IGF system in cancer growth and metastasis:
overview and recent insights. Endocr Rev. 28:20–47. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Corvaia N, Beck A, Caussanel V and Goetsch
L: Insulin-like growth factor receptor type I as a target for
cancer therapy. Front Biosci. 5:439–450. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Baserga R: The decline and fall of the
IGF-I receptor. J Cell Physiol. 228:675–679. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Nakamura K, Hongo A, Kodama J, Miyagi Y,
Yoshinouchi M and Kudo T: Downregulation of the insulin-like growth
factor I receptor by antisense RNA can reverse the transformed
phenotype of human cervical cancer cell lines. Cancer Res.
60:760–765. 2000.PubMed/NCBI
|
|
106
|
McLaughlin-Drubin ME, Meyers J and Munger
K: Cancer associated human papillomaviruses. Curr Opin Virol.
2:459–466. 2012. View Article : Google Scholar
|
|
107
|
Martignani E, Miretti S, Accornero P and
Baratta M: miRNAs highlights in stem and cancer cells. Mini Rev Med
Chem. 11:1165–1182. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Luo M, Shen D, Zhou X, Chen X and Wang W:
MicroRNA-497 is a potential prognostic marker in human cervical
cancer and functions as a tumor suppressor by targeting the
insulin-like growth factor 1 receptor. Surgery. 153:836–847. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Cheung TH, Man KN and Yu MY: Dysregulated
microRNAs in the pathogenesis and progression of cervical neoplasm.
Cell Cycle. 11:2876–2884. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Butz K, Ristriani T, Hengstermann A, Denk
C, Scheffner M and Hoppe-Seyler F: siRNA targeting of the viral E6
oncogene efficiently kills human papillomavirus-positive cancer
cells. Oncogene. 22:5938–5945. 2003. View Article : Google Scholar
|
|
111
|
Hall AH and Alexander KA: RNA interference
of human papillomavirus type 18 E6 and E7 induces senescence in
HeLa cells. J Virol. 77:6066–6069. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Kafri T, van Praag H, Gage FH and Verma
IM: Lentiviral vectors: regulated gene expression. Mol Ther.
1:516–521. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Gu W, Putral L, Hengst K, Minto K,
Saunders NA, Leggatt G and McMillan NA: Inhibition of cervical
cancer cell growth in vitro and in vivo with lentiviral-vector
delivered short hairpin RNA targeting human papillomavirus E6 and
E7 oncogenes. Cancer Gene Ther. 13:1023–1032. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Copreni E, Penzo M, Carrabino S and Conese
M: Lentivirus-mediated gene transfer to the respiratory epithelium:
a promising approach to gene therapy of cystic fibrosis. Gene Ther.
11:S67–S75. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Zhou J, Li B, Peng C, et al: Inhibition of
cervical cancer cell growth in vitro and in vivo by
lentiviral-vector mediated shRNA targeting the common promoter of
HPV16 E6 and E7 oncogenes. Antiviral Res. 98:305–313. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Bousarghin L, Touze A, Gaud G, et al:
Inhibition of cervical cancer cell growth by human papillomavirus
virus-like particles packaged with human papillomavirus oncoprotein
short hairpin RNAs. Mol Cancer Ther. 8:357–365. 2009. View Article : Google Scholar
|
|
117
|
Remy-Ziller C, Germain C, Spindler A, et
al: Immunological characterization of a modified vaccinia virus
Ankara vector expressing the human papillomavirus 16 E1 protein.
Clin Vaccine Immunol. 2:147–155. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Markowitz LE, Tsu V, Deeks SL, Cubie H,
Wang SA, Vicari AS and Brotherton JM: Human papillomavirus vaccine
introduction - the first five years. Vaccine. 30:F139–F148.
2012.PubMed/NCBI
|
|
119
|
Han KT and Sin JI: DNA vaccines targeting
human papillomavirus-associated diseases: progresses in animal and
clinical studies. Clin Exp Vaccine Res. 2:106–114. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Frazer IH: Prevention of cervical cancer
through papillomavirus vaccination. Nat Rev Immunol. 4:46–54. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Berraondo P, Nouzé C, Préville X, Ladant D
and Leclerc C: Eradication of large tumors in mice by a tritherapy
targeting the innate, adaptive, and regulatory components of the
immune system. Cancer Res. 67:8847–8855. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Peng S, Ji H, Trimble C, et al:
Development of a DNA vaccine targeting human papillomavirus type 16
oncoprotein E6. J Virol. 78:8468–8476. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Jin HS, Park EK, Lee JM, et al:
Immunization with adenoviral vectors carrying recombinant IL-12 and
E7 enhanced the antitumor immunity to human papillomavirus
16-associated tumor. Gynecol Oncol. 97:559–567. 2005. View Article : Google Scholar
|
|
124
|
Gunn GR, Zubair A, Peters C, Pan ZK, Wu TC
and Paterson Y: Two Listeria monocytogenes vaccine vectors that
express different molecular forms of human papilloma virus-16
(HPV-16) E7 induce qualitatively different T cell immunity that
correlates with their ability to induce regression of established
tumors immortalized by HPV-16. J Immunol. 167:6471–6479. 2001.
|
|
125
|
Chandy AG, Nurkkala M, Josefsson A and
Eriksson K: Therapeutic dendritic cell vaccination with Ag coupled
to cholera toxin in combination with intratumoural CpG injection
leads to complete tumour eradication in mice bearing HPV 16
expressing tumours. Vaccine. 25:6037–6046. 2007. View Article : Google Scholar
|
|
126
|
Da Silva DM, Schiller JT and Kast WM:
Heterologous boosting increases immunogenicity of chimeric
papillomavirus virus-like particle vaccines. Vaccine. 21:3219–3227.
2003.PubMed/NCBI
|