Introduction
Oral and oropharyngeal cancer together constitute
the sixth most common cancer worldwide, with ~400,000 new cases
diagnosed each year (1). In spite
of advances in screening and detection, the 5-year survival rate of
these cancers, which stood at 53% in 1975, has increased only
marginally over the last forty years. The critical need to identify
the mechanisms associated with early tumor development are
underscored by the generally favorable patient outcomes associated
with localized tumors and the precipitous decline in survival rates
associated with regional and distant metastases (2).
In the course of tumor development, normal
epithelial cells undergo a dedifferentiation program that results
in alterations in signaling, gene expression and phenotype, a
phenomenon that is collectively termed epithelial-to-mesenchymal
transition (EMT). During the course of this transition, epithelial
cells lose polarity, exhibit a fibroblastic morphology and begin
expressing mesenchymal genes. These cells may also exhibit
increased motility and invasive capabilities through increased
expression of proteolytic enzymes. EMT also involves alterations in
expression of a group of transmembrane calcium-dependent adhesive
glycoproteins called cadherins. E-cadherin is the best-studied
member of the classical cadherin family and the prototypical
cadherin of normal epithelia. E-cadherin maintains cellular
architecture within the epithelia by linking actin cytoskeletons of
adjacent cells. E-cadherin is also classified as a tumor
suppressor, as expression of E-cadherin is lost relatively early in
tumorigenesis (3,4). The overexpression of E-cadherin in
cell lines has been shown to restrain cell migration and metastatic
signaling (4).
E-cadherin co-resides in the basal and suprabasal
layers of oral epithelia with P-cadherin (5), a classical cadherin that has received
far less study and whose role in epithelial tumor progression is
far more nebulous. Unlike E-cadherin, which is unilaterally lost
during epithelial tumor progression, P-cadherin expression is
elevated in certain advanced malignancies, such as those of breast
and colon, and overexpression of P-cadherin in such cell lines
promotes aggressiveness. In other cancers, such as bladder, it is
the loss of P-cadherin that promotes tumor development (6–8).
The majority of studies regarding the role of
P-cadherin in oral tumor progression are histological in nature and
reveal a rare trend in the expression pattern of P-cadherin during
the course of oral tumor development. Immunohistochemical analyses
of human tissues and carcinogen-induced rodent tumors have
demonstrated an abnormal increase in membrane-resident P-cadherin
protein during oral dysplasia (9,10) and
persistent expression of membranous P-cadherin in
well-differentiated oral tumors (11,12) In
more advanced oral malignancies, however, P-cadherin expression was
decreased or absent (11,12). In the present study, we explored the
hypothesis that the transient increase in P-cadherin during early
oral tumor development is not merely coincidental, but plays an
active role in oral tumor progression.
How then may elevated levels of an endogenous
epithelial cadherin facilitate aggressive cellular behavior? We
addressed this question by investigating the ability of P-cadherin
to modulate ligand-dependent signaling of two growth factor
receptor tyrosine kinases: insulin-like growth factor 1 receptor
(IGF-1R) and epidermal growth factor receptor (EGFR). Precedence
for such a mechanism has been demonstrated for both E-cadherin,
which modulates ligand-dependent signaling of IGF-1R, EGFR and
fibroblast growth factor receptor (FGFR) (13–15)
and N-cadherin, which potentiates ligand-dependent FGFR signaling
(16). Most recently, it has been
demonstrated in ovarian cancer cells that P-cadherin and IGF-1R are
able to form a functional complex upon stimulation of the
gonadotropin-releasing hormone receptor (17).
In the present study, P-cadherin was overexpressed
in both dysplastic and malignant oral cell lines, which were
analyzed for growth factor signaling responses and phenotypic
alterations. The results of this study provide the first evidence
that P-cadherin can potentiate ligand-dependent signaling of both
IGF-1R and EGFR. P-cadherin also modulated mesenchymal signaling,
motility and in the case of dysplastic cells, morphology. These
findings, together with existing histological data, suggest that
transient increases in P-cadherin levels may be a means by which
dysplastic and early neoplastic oral epithelial cells acquire
additional aggressive phenotypes.
Materials and methods
Cell culture
The oral squamous carcinoma cell line UM-SCC22A
(SCC22A) (obtained from Dr Thomas Carey, University of Michigan)
was maintained in Minimum Essential Medium (Hyclone) supplemented
with 10% fetal bovine serum (PAA Scientific) and 1% non-essential
amino acids (Fisher). Dysplastic oral keratinocytes (DOK) (obtained
from the European Collection of Cell Cultures, via Sigma-Aldrich)
were maintained in Dulbecco’s modified Eagle’s medium (DMEM; PAA
Scientific), supplemented with 10% fetal bovine serum and 5 μg/ml
hydrocortisone (Sigma-Aldrich). All cells were maintained at 37°C
and 5% CO2. For retroviral transduction, a cDNA encoding
full-length human P-cadherin (18)
was subcloned into the Sgfi and SfiI sites of the
retroviral expression vector LZRS-MS-Neo (19). Production of amphotropic retrovirus
and subsequent infection of SCC22A and DOK cells with the
LZRS-Ms-Neo (empty control vector) or LZRS-Ms-Neo/P-cadherin
constructs were performed as previously described (20). All transduced cells were selected
and maintained in 400 μg/ml G418 (Santa Cruz Biotechnology). Bright
field photography of subconfluent SCC22A and DOK cell cultures, and
of wounded cell monolayers, was performed utilizing an Axiovert 40
inverted microscope (Zeiss). Immunofluorescence photography was
performed utilizing an Axio Imager Z1 fluorescence microscope with
ApoTome attachment (Zeiss).
Western blotting
For both steady-state and time-course experiments,
cells were grown to equal confluency and harvested in RIPA lysis
buffer (150 mM sodium chloride, 1.0% NP-40, 0.5% sodium
deoxycholate, 0.1% sodium dodecyl sulfate and 50 mM Tris, pH 8.0
TNE) supplemented with HALT phosphatase and protease inhibitor
cocktail (Thermo Fisher Scientific). For signaling analyses of
insulin-like growth factor (IGF) and epidermal growth factor (EGF),
cells were serum starved 24 h prior to administration of growth
factor. Cells were incubated with 10 ng/ml IGF-1 (Peprotech) or 50
ng/ml EGF (Peprotech) in warmed serum-free media at the time-points
indicated. All incubations were performed at 37°C in a humidified
5% CO2 incubator prior to cell lysis.
Lysates were quantitated using a BCA (Pierce) or DC
(BioRad) protein assay. Equal quantities of protein were analyzed
by SDS-PAGE and subjected to western immunoblotting. The bands of
interest were identified utilizing the indicated primary antibody
and HRP-conjugated goat anti-mouse or goat anti-rabbit secondary
antibody (Jackson Laboratories) and the Pierce SuperSignal
Chemiluminescent Reagent. Primary antibodies used in this study
included goat anti-mouse antibodies directed against P-cadherin (BD
Transduction Laboratories), E-cadherin and β-catenin (Zymed),
β-tubulin (Developmental Studies Hybridoma Bank, University of
Iowa) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH;
Sigma-Aldrich). All goat anti-rabbit antibodies were purchased from
Cell Signaling Technology and included antibodies against IGF-1R,
EGFR, Snail, phospho-p42/p44 mitogen-activated protein kinase
(MAPK), phospho-AKT and phospho-glycogen synthase kinase-3β
(phospho-GSK-3β). Blots were also visualized and quantitated on the
Odyssey Imaging System (LI-COR) using DyeLight 680 and DyeLight 800
anti-mouse secondary antibodies. For film analysis, relative band
intensities were quantitated using Image J software (http://rsb.info.nih.gov/ij/index.html)
in accordance with software protocols.
Immunofluorescence staining
For immunofluorescence staining, SCC22A cells were
grown for 48 h on glass cover-slips and were switched to serum-free
media 18 h prior to growth factor treatment. Cells were fixed
immediately or treated with serum-free media containing 10 ng/ml
IGF-1 for 2 h prior to fixation. Cells were fixed using 10%
neutral-buffered formalin for 30 min and permeabilized with 5%
Triton X-100 for 15 min. Coverslips were blocked in 10% goat serum.
Immunofluorescence was performed on cells using a mouse monoclonal
antibody against P-cadherin (BD Biosciences) and an Alexa 488
anti-mouse secondary (Invitrogen) or an anti-IGF-1R rabbit
polyclonal antibody and an Alexa 594 anti-rabbit secondary antibody
(Invitrogen). All slips were mounted in DAPI-containing mounting
media (Vector Labs) and imaged using an ApoTome fluorescence
microscope (Zeiss). Axiovision 4.8 software (Zeiss) was used to
collect fluorescent images and to create merged images for each
image set.
Motility
For the wound healing assays, SCC22A cells were
grown to confluence on grid-etched 60-mm dishes (Fisher), and a
single scratch was created using a pipette tip. Cells were
photographed at the same location of the scratch over the course of
24 h. The area of pixel closure was measured for each cell type by
subtracting the initial scratch area from the final closing area
(24 h). Values reported were normalized to the control and
represent three independent experiments consisting of five plates
for each cell type/time-point studied.
EMT array analysis
The Human EMT RT2 Profiler PCR Array
(Qiagen, Redwood City, CA, USA) was utilized to examine the
expression profile of 84 genes that are known to either regulate or
affect processes related to epithelial-to-mesenchymal transition.
SCC22A cells transduced with control vector or P-cadherin cDNA were
plated at equal densities and serum-starved for 24 h prior to RNA
collection using the High-pure RNA Isolation kit (Roche,
Indianapolis, IN, USA). RNA was isolated according to the
manufacturer’s instructions and quantitated using a Nanodrop
spectrophotometer (Thermo Scientific). cDNA was prepared with the
RT2 First Strand cDNA kit (Qiagen), utilizing 1 μg RNA
per reaction. Samples were run on an Applied Biosystems StepOne
Plus qPCR (Invitrogen) using the RT2 SYBR-Green/ROX qPCR
master mix (Qiagen) utilizing a final cDNA concentration of 0.5
ng/μl (5 ng per reaction). Data were normalized to the average Ct
value of five housekeeping genes. Data analysis was performed
utilizing the 2−ΔΔCt method by means of the
RT2 Profiler PCR Data Analysis Template v4.0, available
from the manufacturer’s website (Qiagen).
Results
Transduction of oral squamous carcinoma
cells with P-cadherin
IGF-1R has been shown to be moderately
over-expressed in dysplastic oral lesions and highly expressed in
malignant tumors (21). A recent
studies by Cheung et al (17) demonstrated in an ovarian cancer cell
model that gonadotropin-releasing hormone stimulates a functional
association between P-cadherin and IGF-1R. To explore the
possibility that P-cadherin may potentiate IGF-1R-related signaling
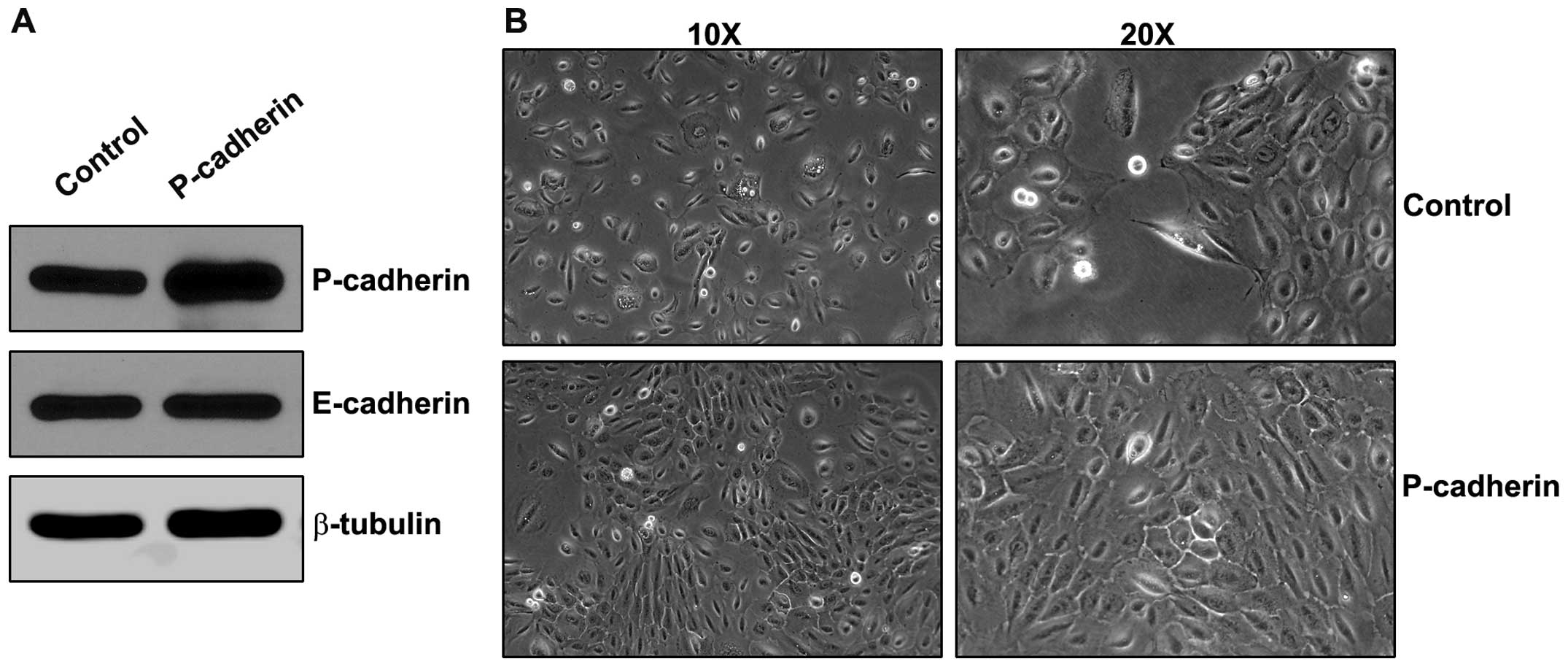
in oral epithelia, we overexpressed P-cadherin in SCC22A oral
squamous carcinoma cells. Cells were analyzed for E-cadherin and
P-cadherin expression as well as the expression of IGF-1R and the
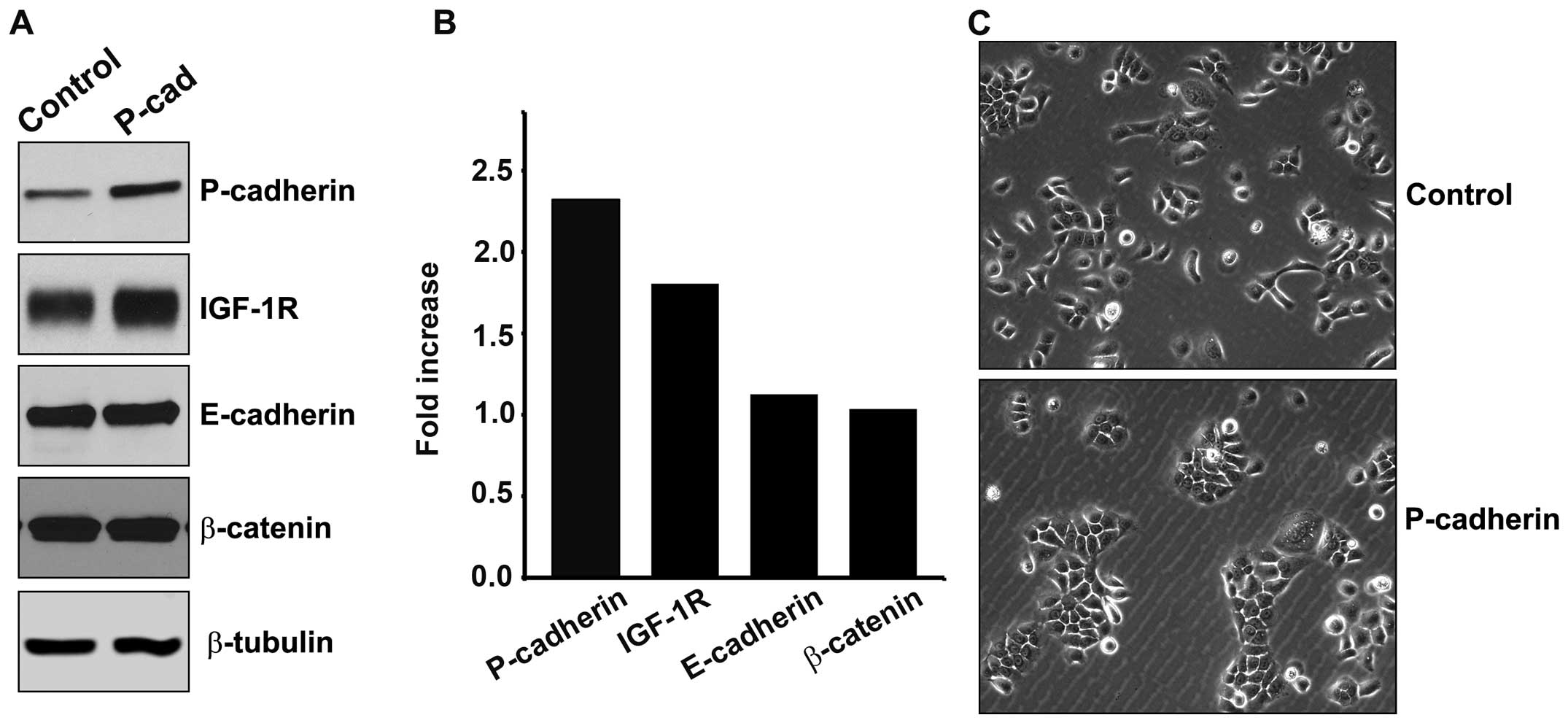
P-cadherin-associated protein β-catenin (Fig. 1A and B). Cells transduced to
overexpress P-cadherin displayed a 2-fold increase in P-cadherin
expression, and also displayed a near 2-fold increase in
steady-state levels of IGF-1R protein. Other resident proteins of
the adherens junction, E-cadherin and β-catenin, were unaltered by
P-cadherin overexpression. SCC22A cells exhibited an
epithelial-like cellular morphology that was unaltered by
expression of P-cadherin. P-cadherin expression did noticeably
reduce cell scattering and favored greater colony formation
(Fig. 1C).
P-cadherin potentiates IGF-1R-stimulated
MAPK activation
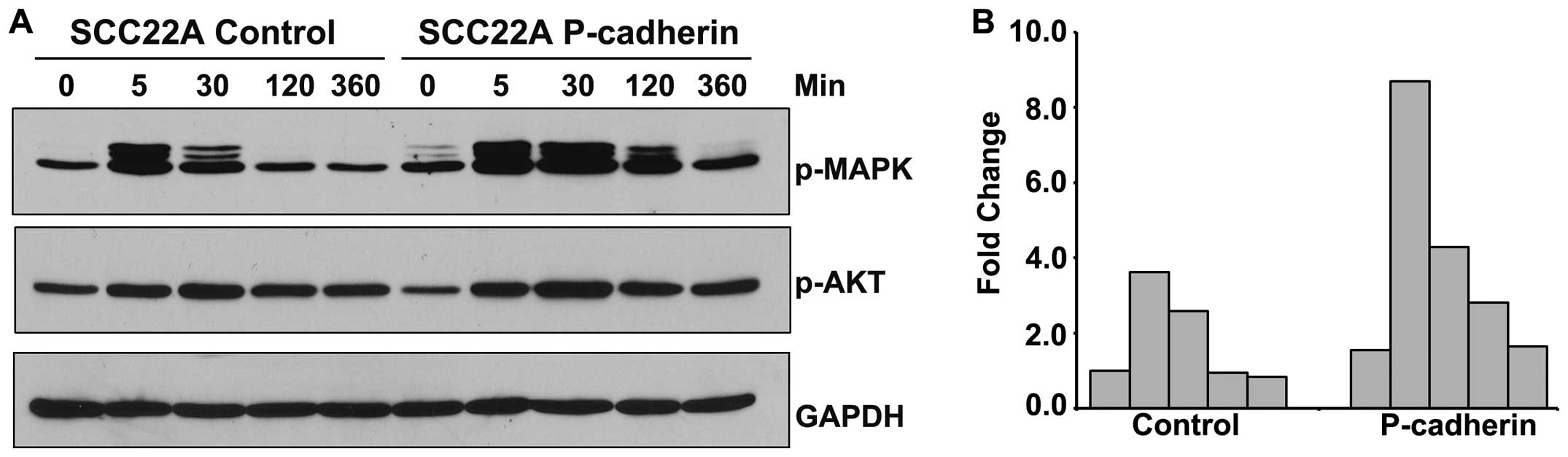
To determine the effect of P-cadherin overexpression
on IGF-1R-mediated signaling, we treated serum-deprived control and
P-cadherin-overexpressing SCC22A cell line with IGF-1 for
increasing time periods and analyzed the activating phosphorylation
of MAPK and AKT (Fig. 2).
P-cadherin increased both basal levels of MAPK phosphorylation (1.5
fold, Fig. 2B) and the magnitude of
ligand-induced MAPK phosphorylation (9-fold compared to control),
and also delayed return to steady-state levels through all
time-points examined (Fig. 2B).
Although ligand-dependent phosphorylation of AKT occurred in all
cell lines examined, no P-cadherin-dependent alterations in the
magnitude of phosphorylation were observed (Fig. 2A).
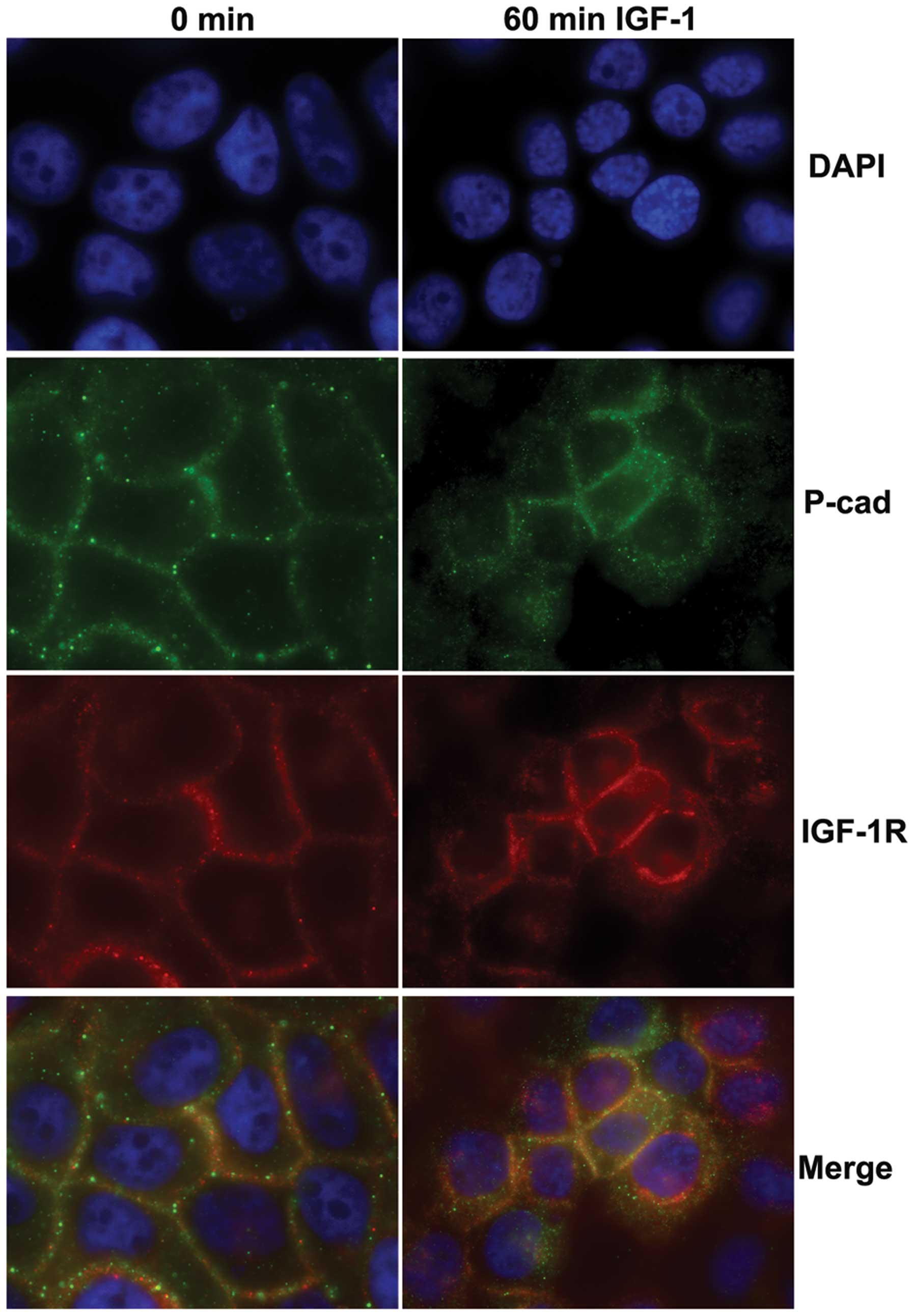
Independent internalization of IGF-1R and
P-cadherin in response to IGF-1 signaling
E-cadherin has been show to alternatively
co-internalize with ligand-stimulated growth-factor receptors
(22) or remain at the cell
membrane during growth factor receptor endocytosis (23). To identify the dynamics of
P-cadherin and IGF-1R interactions and trafficking during IGF-1
stimulation, we examined the localization of each protein by
immunofluorescence in the control and IGF-1-stimulated SCC22A cells
(Fig. 3). In the untreated cells,
both P-cadherin (green) and IGF-1R (red) were detected at points of
cell-cell contact; however co-localization as determined by
synergistic changes in fluorescence intensity was not observed. In
the IGF-1-treated cells, both P-cadherin and IGF-1R were
internalized. Trafficking of these molecules appeared to be
somewhat independent, as P-cadherin staining was found throughout
the cytoplasm, whereas IGF-1R staining was strong at both cell
borders and around the nucleus.
P-cadherin expression increases
transcription of EMT-related transcriptional regulators
The transient increase in P-cadherin expression in
early tumor development suggests that P-cadherin may play an active
role in facilitating epithelial-to-mesenchymal transition. To
identify EMT-related genes that were modulated by increased
P-cadherin expression, an RNA profile array was used to examine the
differential transcript levels of 84 different EMT-related genes.
The panel screened for both effectors and regulators of EMT, and
included genes involved in adhesion, migration, motility, as well
as EMT-related transcriptional regulators. Since the majority of
genes in the panel yielded no variance between the control and
P-cadherin-expressing cells, we report only a relevant subset in
Table I. In agreement with both the
western blot analysis and SCC22A cell morphology, cadherin and
catenin genes (E-cadherin, N-cadherin, β-catenin) showed little
change upon P-cadherin expression (Table I). An increase in gene expression
was detected for all three members of the Snail family of
EMT-associated transcriptional regulatory proteins and the
EMT-associated transcriptional repressor Zeb1 (Table I). Transcript levels of vimentin and
MMP-9, generally associated with mesenchymal phenotypes, were
unaltered.
 | Table IDifferential expression of EMT-related
genes in the control and P-cadherin-overexpressing SCC22A
cells. |
Table I
Differential expression of EMT-related
genes in the control and P-cadherin-overexpressing SCC22A
cells.
| Gene | Protein | Fold change |
|---|
| CDH1 | E-cadherin | 0.94 |
| CDH2 | N-cadherin | 1.13 |
| CTTNNB1 | β-catenin | 1.04 |
| MMP-9 | Matrix
metalloproteinase-9 | 0.99 |
| SNAI1 | Snail homolog 1
(Drosophila) | 1.59 |
| SNAI2 | Snail homolog 2
(Drosophila) | 1.54 |
| SNAI3 | Snail homolog 3
(Drosophila) | 1.41 |
| TWIST1 | Twist homolog 1
(Drosophila) | 1.03 |
| VIM | Vimentin | 1.05 |
| ZEB1 | Zinc finger E-box
binding homeobox 1 | 1.59 |
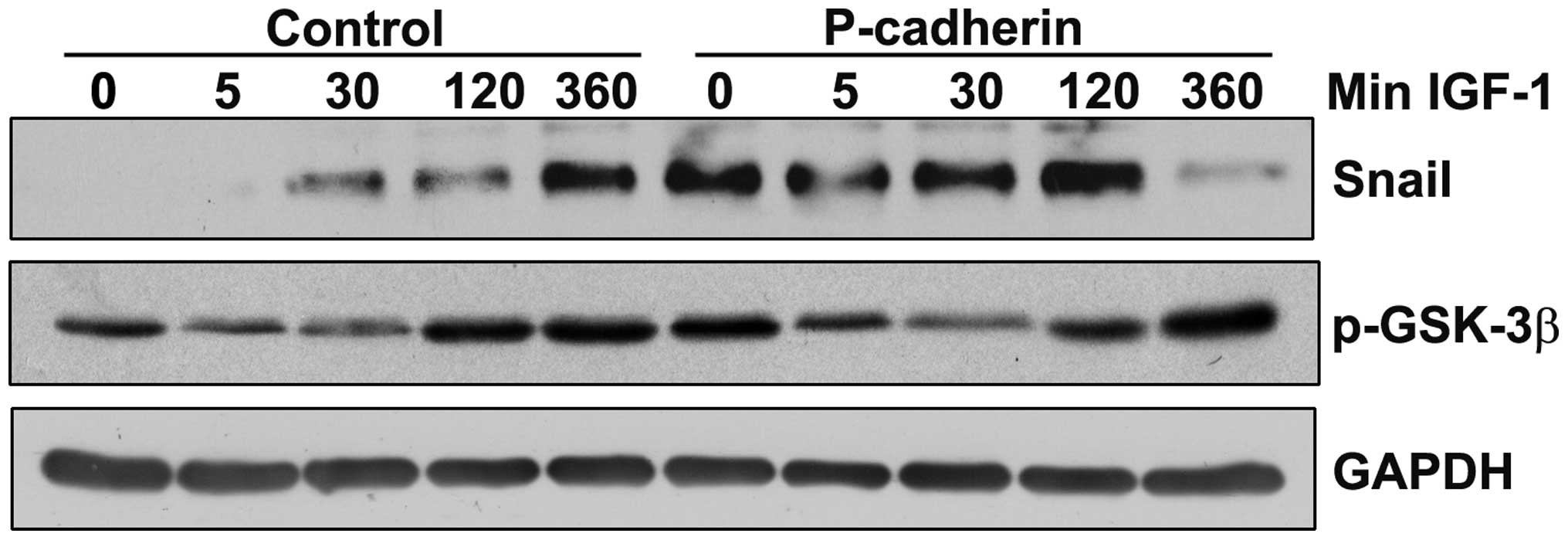
P-cadherin increases Snail protein levels
independent of IGF-1R signaling
The qRT-PCR array analysis suggested that P-cadherin
increased expression of the mesenchymal transcription factor Snail,
which is also a known downstream target of IGF-1R- signaling
(24). Levels of Snail protein were
examined in serum-deprived SCC22A cells treated with IGF-1 for up
to 6 h. In the untreated control cells, Snail levels were
undetectable, but increased significantly by 6 h after IGF-1
administration (Fig. 4). P-cadherin
overexpression alone was sufficient to increase basal levels of
Snail (untreated P-cadherin cells) to levels comparable to the 6 h
IGF-1 time-point in the control cells. IGF-1 was unable to
stimulate further increases in Snail protein in the
P-cadherin-expressing cells.
The 25-minute half-life of Snail protein is
attributed to the phosphorylation of Snail at critical residues by
glycogen synthase kinase-3β (GSK-3β), which results in
ubiquitination of Snail and its subsequent proteosome-mediated
degradation (25). Accumulation of
Snail occurs in response to the Serine-9 phosphorylation of GSK-3β
by multiple kinases, which inactivates GSK-3β and prevents
phosphorylation-dependent ubiquitination of Snail (26). In the SCC22A cells, profiles of
GSK-3β phosphorylation across time-points were consistent between
the control and P-cadherin-expressing cells, suggesting that the
phosphorylation of GSK-3β at Serine-9 is unlikely to be the
mechanism by which Snail levels are increased in
P-cadherin-expressing cells (Fig.
4).
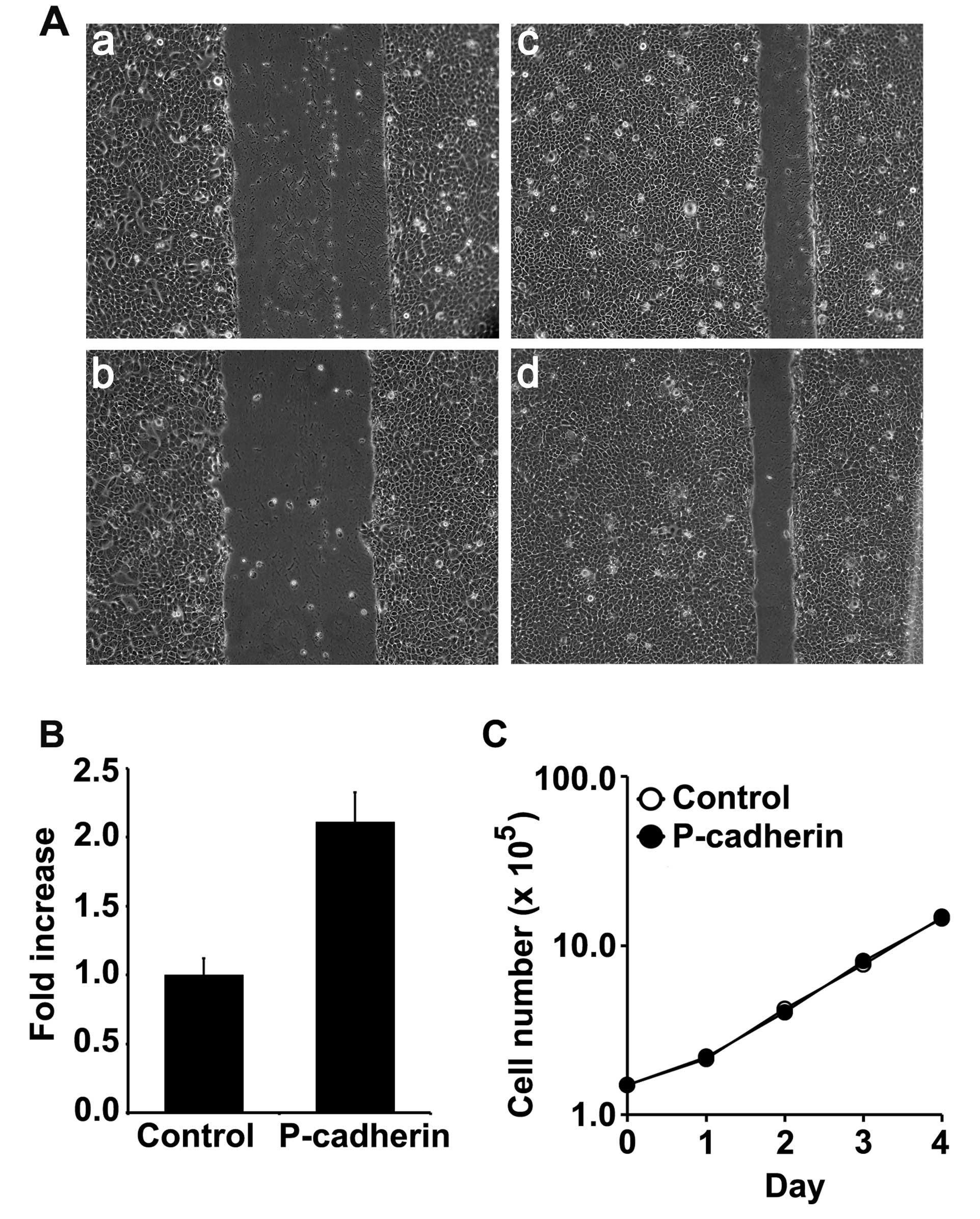
P-cadherin expression increases cell
motility
Snail has been previously shown to be critical for
cell motility in the highly dedifferentiated PCI152 oral squamous
cell line (27). We utilized a
wound healing assay to measure the differences in motility between
the control and the P-cadherin-expressing cells. Overexpression of
P-cadherin conferred a 2-fold increase in motility compared to
control cells (Fig. 5A). We did not
observe P-cadherin-dependent alterations in motility as a result of
IGF-1 administration (data not shown). Although P-cadherin
increased cell motility, we found no evidence of
P-cadherin-dependent alterations in cell proliferation in the
SCC22A cells as measured by growth curve analysis (Fig. 5C).
P-cadherin overexpression induces
mesenchymal morphology in dysplastic oral keratinocytes
The increase observed in P-cadherin expression in
dysplastic oral tissue (10)
suggests the possibility that P-cadherin may play a stage-specific
role in oral tumor progression. To better define the mechanisms by
which P-cadherin may modulate signaling in dysplastic cells, we
overexpressed P-cadherin in the DOK cell line. This cell line
expresses E-cadherin and P-cadherin (Fig. 6A) but does not express the
mesenchymal N-cadherin in quantities detectable by western analysis
(data not shown). Overexpression of P-cadherin resulted in
increased colony formation as observed in the SCC22A cells. In
contrast to the SCC22A cells, however, P-cadherin overexpression in
DOK cells had a profound effect on individual cell morphologies.
DOK cells overexpressing P-cadherin exhibited a more elongated,
fibroblastic phenotype compared to the control cell (Fig. 6B).
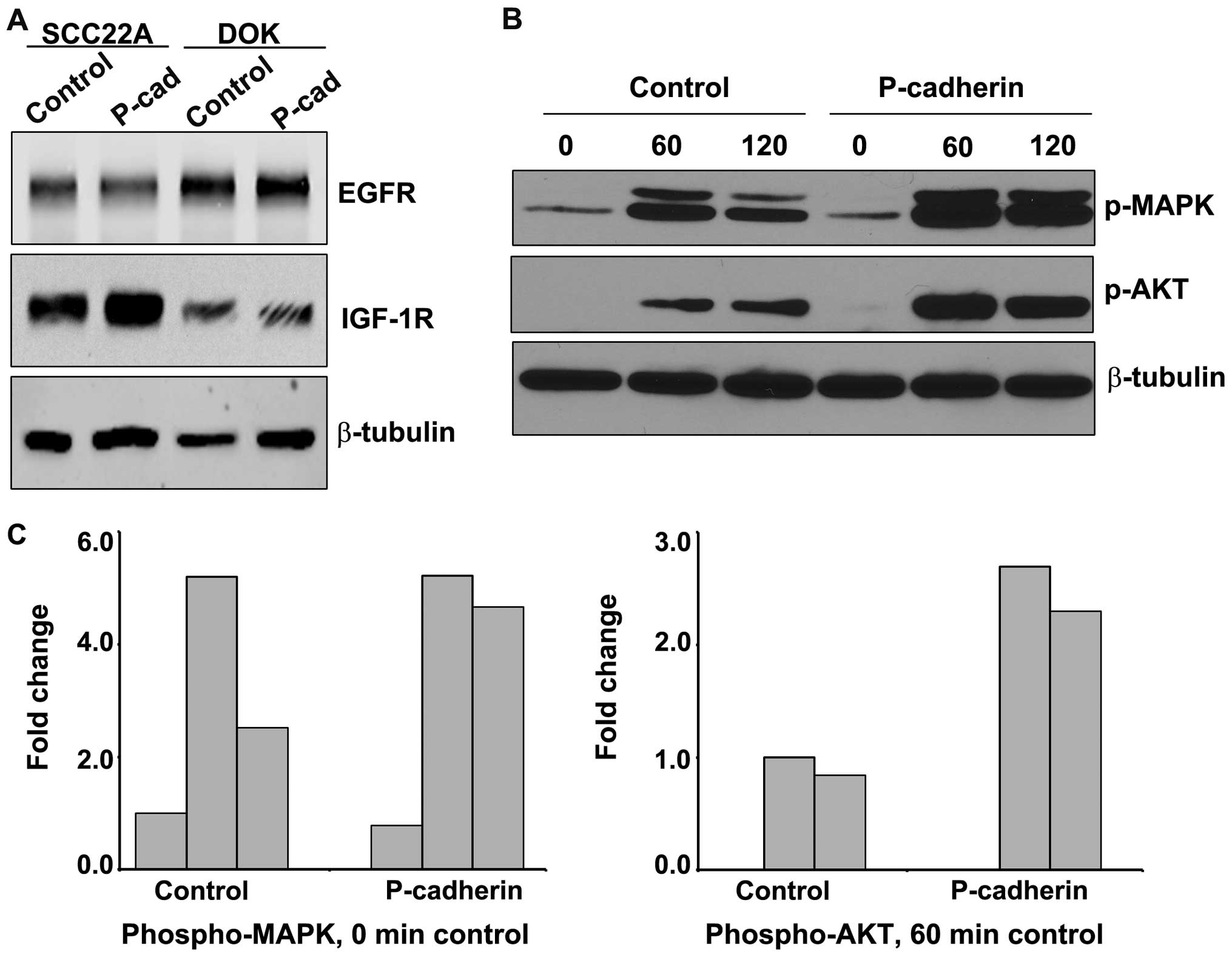
P-cadherin potentiates EGF-dependent MAPK
and AKT signaling in dysplastic oral keratinocytes
In human oral epithelial tumors, increased
expression and activity of EGFR is common, and appears to play a
critical role in the acquisition of mesenchymal characteristics
(28). A comparison of both EGFR
and IGF-1R expression between DOK cells and the malignant carcinoma
SCC22A cell line is shown in Fig.
7A. DOK cells exhibited much greater expression of EGFR and
decreased expression of IGF-1R compared to the SCC22A cells.
We examined the ability of P-cadherin to potentiate
EGF-dependent signaling in DOK cells. Control or
P-cadherin-expressing DOK cells were treated with EGF for 60 and
120 min and analyzed for both MAPK and AKT phosphorylation
(Fig. 7B and C). Representative
quantitation for each time-point analyzed is shown in Fig. 7C. P-cadherin did not appreciably
amplify the magnitude of MAPK phosphorylation compared to control,
but did prolong MAPK signaling at the 120-min time-point.
P-cadherin expression did increase the magnitude of PI3 kinase
signaling (as measured by AKT phosphorylation) by 3-fold, compared
to the 60-min control value. Both control and P-cadherin DOK cells
exhibited an ~20% loss of signal between 60 and 120 min, indicating
that P-cadherin affected the initiation of EGFR signaling but did
not play a protective role in the attenuation of AKT-mediated
signaling.
Discussion
Oral cancer is one of the few tissues in which
levels of membrane-resident P-cadherin is found to aberrantly
increase in dysplasia and well-differentiated carcinomas and is
subsequently lost in more advanced tumors (10,29).
In the present study, we demonstrated that aberrant expression of
P-cadherin may in fact play a causative role during the early
stages of tumor progression, by facilitating increases in motility
and mesenchymal signaling and by strengthening IGF-1R or
EGFR-initiated signal transduction cascades.
The increase in motility noted in
P-cadherin-overexpressing SCC22A cells (Fig. 4) may be due to altered regulation of
the P-cadherin binding partner p120 catenin and its subsequent
modulation of Rho GTPases. These signaling molecules, which include
the GTPases Rac, Rho and cdc42, can increase cell motility via the
reorganization of cytoskeletal architecture and, in certain
circumstances, also promote the dissolution of adherens junctions
(30). In pancreatic cancer cell
lines, P-cadherin overexpression altered p120 localization and, in
a p120-dependent fashion, increased motility via the activation of
both Rac and Cdc42 GTPases (31).
In the present study, P-cadherin overexpression also increased
expression of the Snail transcription factor, which has been shown
by others to increase motility via the RhoA GTPase (32).
We were unable to determine the mechanism by which
P-cadherin increased Snail protein levels. The activity of GSK-3β,
which phosphorylates and targets Snail for degradation, appeared to
be unchanged between the control and P-cadherin cells (Fig. 4). One possible explanation is that
P-cadherin indirectly stimulated the sequestration of active GSK-3β
complexes into multivesicular endosomes, a phenomenon that has been
shown to prolong the half-life of GSK-3β targets that would
otherwise be targeted for degradation (33). Such regulation may have clinical
implications, as Snail has been found at the invasive front of
esophageal tumors and induces motility and invasion in esophageal
squamous carcinoma cell lines (34).
We also demonstrated that overexpression of
P-cadherin is able to increase the downstream signaling of two
different ligand-activated growth factor receptors, IGF-1R and
EGFR. In both dysplastic and malignant cells, P-cadherin expression
prolonged MAPK activation in response to growth factor receptor
signaling (Fig. 2B and C). In DOK
cells, but not SCC22A cells, P-cadherin overexpression greatly
increased the magnitude of growth factor signaling through the PI3
kinase pathway (Fig. 7C). It is
highly likely that the increased susceptibility of DOK cells to
P-cadherin signaling is due to their dysplastic etiology, as they
likely harbor comparatively fewer mutations and exhibit a decreased
number of dysfunctional signaling pathways. The DOK cell line is an
immortalized, nontumorigenic, moderately dysplastic oral
keratinocyte cell line with normal Ha-ras, Ki-ras and N-ras
function (35). Unlike the SCC22A
cells, growth-factor deprived DOK cells do not exhibit detectable
levels of basal AKT phosphorylation (Figs. 2 and 7B). DOK cells, but not SCC22A cells, were
also susceptible to morphological alterations as a result of
P-cadherin overexpression (Fig. 1
and 6B). These data suggest that
P-cadherin overexpression may have a more robust effect in oral
precancers than it does in malignant neoplasms.
The ability of P-cadherin to potentiate IGF-1R and
EGFR signaling may have profound consequences with respect to
dysplastic epithelia. The temporal and spatial occurrence of
aberrantly elevated P-cadherin expression is quite similar to the
pattern of expression of EGFR in early oral tumor development.
EGFR, which undergoes amplification in early oral dysplasia, is
localized to the same basal layers of the epithelia where
elevations in P-cadherin have been detected (5,36).
IGF-1R has also been found to increase during moderate to severe
dysplasia (21). Unlike P-cadherin,
increased expression of both EGFR and IGF-1R persists during tumor
progression and thus, the increased downstream signaling responses
conferred P-cadherin may occur only during dysplasia and early
neoplasia. The MAPK and AKT signaling pathways mediate a number of
tumor-associated activities, including growth, survival, cell
motility and invasion (37–39). The increased activity of these
pathways provided by the augmentation of growth factor signaling by
P-cadherin may enhance tumor development at a critical stage and as
such, may also provide new strategies for therapeutic
intervention.
Acknowledgements
Support for this study was received from the Kenneth
A. Suarez Summer Research Fellowship Program, Midwestern University
College of Health Sciences and intramural funding from Midwestern
University (Glendale, AZ, USA).
References
|
1
|
Warnakulasuriya S: Global epidemiology of
oral and oropharyngeal cancer. Oral Oncol. 45:309–316. 2009.
View Article : Google Scholar
|
|
2
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar
|
|
3
|
Santos-García A, Abad-Hernández MM,
Fonseca-Sánchez E, et al: E-caderin, laminin and collagen IV
expression in the evolution from dysplasia to oral squamous cell
carcinoma. Med Oral Path Oral Cir Bucal. 11:E100–E105. 2006.(In
English and Spanish).
|
|
4
|
Birchmeier W and Behrens J: Cadherin
expression in carcinomas: role in the formation of cell junctions
and the prevention of invasiveness. Biochim Biophys Acta.
1198:11–26. 1994.PubMed/NCBI
|
|
5
|
Shimoyama Y, Hirohashi S, Hirano S, et al:
Cadherin cell-adhesion molecules in human epithelial tissues and
carcinomas. Cancer Res. 49:2128–2133. 1989.PubMed/NCBI
|
|
6
|
Van Marck V, Stove C, Jacobs K, Van den
Eynden G and Bracke M: P-cadherin in adhesion and invasion:
Opposite roles in colon and bladder carcinoma. Int J Cancer.
128:1031–1044. 2011.PubMed/NCBI
|
|
7
|
Van Marck V, Stove C, Van Den Bossche K,
et al: P-cadherin promotes cell-cell adhesion and counteracts
invasion in human melanoma. Cancer Res. 65:8774–8783.
2005.PubMed/NCBI
|
|
8
|
Paredes J, Albergaria A, Oliveira JT,
Jerónimo C, Milanezi F and Schmitt FC: P-cadherin overexpression is
an indicator of clinical outcome in invasive breast carcinomas and
is associated with CDH3 promoter hypomethylation. Clin Cancer Res.
11:5869–5877. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bagutti C, Speight PM and Watt FM:
Comparison of integrin, cadherin and catenin expression in squamous
cell carcinomas of the oral cavity. J Pathol. 186:8–16. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Williams HK, Sanders DS, Jankowski JA,
Landini G and Brown AM: Expression of cadherins and catenins in
oral epithelial dysplasia and squamous cell carcinoma. J Oral
Pathol Med. 27:308–317. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Muñoz-Guerra MF, Marazuela EG,
Fernández-Contreras ME and Gamallo C: P-cadherin expression reduced
in squamous cell carcinoma of the oral cavity: an indicatior of
poor prognosis. Cancer. 103:960–969. 2005.PubMed/NCBI
|
|
12
|
Lo Muzio L, Pannone G, Mignogna M, et al:
P-cadherin expression predicts clinical outcome in oral squamous
cell carcinomas. Histol Histopathol. 19:1089–1099. 2004.PubMed/NCBI
|
|
13
|
Bedzhov I, Liszewska E, Kanzler B and
Stemmler MP: IGF1R signaling is indispensable for preimplantation
development and is activated via a novel function of E-cadherin.
PLoS Genet. 8:e10026092012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bryant DM, Wylie FG and Stow JL:
Regulation of endocytosis, nuclear translocation and signaling of
fibroblast growth factor receptor 1 by E-cadherin. Mol Biol Cell.
16:14–23. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Qian X, Karpova T, Sheppard AM, McNally J
and Lowy DR: E-cadherin-mediated adhesion inhibits ligand-dependent
activation of diverse receptor tyrosine kinases. EMBO J.
23:1739–1784. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Suyama K, Shapiro I, Guttman M and Hazan
RB: A signaling pathway leading to metastasis is controlled by
N-cadherin and the FGF receptor. Cancer Cell. 2:301–314. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cheung LW, Mak AS, Cheung AN, Ngan HY,
Leung PC and Wong AS: P-cadherin cooperates with insulin-like
growth factor-1 receptor to promote metastatic signaling of
gonadotropin-releasing hormone in ovarian cancer via p120 catenin.
Oncogene. 30:2964–2974. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Johnson KR, Lewis JE, Li D, et al: P- and
E-cadherin are in separate complexes in cells expressing both
cadherins. Exp Cell Res. 207:252–260. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ireton RC, Davis MA, Van Hengel J, et al:
A novel role for p120 catenin in E-cadherin function. J Cell Biol.
159:465–476. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Maeda M, Johnson E, Mandal SH, et al:
Expression of inappropriate cadherins by epithelial tumor cells
promotes endocytosis and degradation of E-cadherin via competition
for p120(ctn). Oncogene. 25:4595–4604. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Joseph BK and Sundaram DB: Insulin-like
growth factor-1 receptor expression in oral squamous cell
carcinoma. J Clin Exp Invest. 2:354–361. 2011. View Article : Google Scholar
|
|
22
|
Morali OG, Delmas V, Moore R, Jeanney C,
Thiery JP and Larue L: IGF-II induces rapid beta-catenin relocation
to the nucleus during epithelium to mesenchyme transition.
Oncogene. 20:4942–4950. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chavez MG, Buhr CA, Petrie WK,
Wandinger-Ness A, Kusewitt DF and Hudson LG: Differential
downregulation of E-cadherin and desmoglein by epidermal growth
factor. Dermatol Res Pract. 2012:3095872012.PubMed/NCBI
|
|
24
|
Kim HJ, Litzenburger BC, Cui X, et al:
Constitutively active type I insulin-like growth factor receptor
causes transformation and xenograft growth of immortalized mammary
epithelial cells and is accompanied by an epithelial-to-mesenchymal
transition mediated by NF-κB and Snail. Mol Cell Biol.
27:3165–3175. 2007.PubMed/NCBI
|
|
25
|
Zhou BP, Deng J, Xia W, et al: Dual
regulation of Snail by GSK-3beta-mediated phosphorylation in
control of epithelial-mesenchymal transition. Nat Cell Biol.
6:931–940. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sutherland C, Leighton IA and Cohen P:
Inactivation of glycogen synthase kinase-3 beta by phosphorylation:
new kinase connections in insulin and growth-factor signalling.
Biochem J. 296:15–19. 1993.PubMed/NCBI
|
|
27
|
Bauer K, Dowejko A, Bosserhoff AK,
Reichert T and Bauer RJ: P-cadherin induces an epithelial-like
phenotype in oral squamous cell carcinoma by GSK-3beta-mediated
Snail phosphorylation. Carcinogenesis. 30:1781–1788. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hiraishi Y, Wada T, Nakatani K, Negoro K
and Fujita S: Immunohistochemical expression of EGFR and p-EGFR in
oral squamous cell carcinomas. Pathol Oncol Res. 12:87–91. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lo Muzio L, Campisi G, Farina A, et al:
P-cadherin expression and survival rate in oral squamous cell
carcinoma: an immunohistochemical study. BMC Cancer.
5:632005.PubMed/NCBI
|
|
30
|
Lozano E, Betson M and Braga VM: Tumor
progression: small GTPases and loss of cell-cell adhesion.
Bioessays. 25:452–463. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Taniuchi K, Nakagawa H, Hosokawa M, et al:
Overexpressed P-cadherin/CDH3 promotes motility of pancreatic
cancer cells by interacting with p120ctn and activating rho-family
GTPases. Cancer Res. 65:3092–3099. 2005.PubMed/NCBI
|
|
32
|
Zhang A, Wang Q, Han Z, et al: Reduced
expression of Snail decreases breast cancer cell motility by
downregulating the expression and inhibiting the activity of RhoA
GTPase. Oncol Lett. 6:339–346. 2013.PubMed/NCBI
|
|
33
|
Taelman VF, Dobrowolski R, Plouhinec JL,
et al: Wnt signaling requires sequestration of glycogen synthase
kinase 3 inside multivesicular endosomes. Cell. 143:1136–1148.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Usami Y, Satake S, Nakayama F, et al:
Snail-associated epithelial-mesenchymal transition promotes
oesophageal squamous cell carcinoma motility and progression. J
Pathol. 215:330–339. 2008. View Article : Google Scholar
|
|
35
|
Chang SE, Foster S, Betts D and Marnock
WE: DOK, a cell line established from human dysplastic oral mucosa,
shows a partially transformed non-malignant phenotype. Int J
Cancer. 52:896–902. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tsui IF, Poh CF, Garnis C, Rosin MP, Zhang
L and Lam WL: Multiple pathways in the FGF signaling network are
frequently deregulated by gene amplification in oral dysplasias.
Int J Cancer. 125:2219–2228. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Grille SJ, Bellacosa A, Upson J, et al:
The protein kinase Akt induces epithelial mesenchymal transition
and promotes enhanced motility and invasiveness of squamous cell
carcinoma lines. Cancer Res. 63:2172–2178. 2003.PubMed/NCBI
|
|
38
|
Reddy KB, Krueger JS, Kondapaka SB and
Diglio CA: Mitogen-activated protein kinase (MAPK) regulates the
expression of progelatinase B (MMP-9) in breast epithelial cells.
Int J Cancer. 82:268–273. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Klemke RL, Cai S, Giannini AL, Gallagher
PJ, De Lanerolle P and Cheresh DA: Regulation of cell motility by
mitogen-activated protein kinase. J Cell Biol. 137:481–492. 1997.
View Article : Google Scholar : PubMed/NCBI
|