Introduction
Although medical technology has experienced great
advances, to date cancer still remains an unsolved major health
issue. In recent years, the most promising chemotherapy for the
treatment of cancer is known to induce apoptosis. Cell mutations
and their unrepaired DNA damage not only adversely affect the
surrounding normal cells but also initiate diseases such as cancer
(1). For escaping these risks,
cells operate self death systems such as apoptosis. Cancer cells
have the ability to evade signal stimulation inducing apoptosis.
Therefore, apoptosis induction may represent a new therapeutic
strategy for the treatment of cancer. Apoptosis is proceeded by
various death-associated proteins such as the caspase family, the
Bcl-2 family, mitogen-activate protein kinase (MAPK) and
tumor-suppressor genes such as p53. The caspase family is divided
into initiator caspases and effector caspases (2). When killer T cells detect damaged
cells, procaspase-8 is activated by CFasL/D95L a death ligand that
binds to the surface of death receptors such as Fas, tumor necrosis
factor receptor-1 (TNFR-1) and TNFR-2 (3). Caspase-8, an initiator caspase,
induces the break-down of proteins, the actin cytoskeleton and
lamin protein, a protein of the nuclear membrane, by activation of
caspase-3 and other effector caspases, and then causes cell death
through DNA fragmentation (2,4,5). In
contrast, the mitochondrial apoptotic pathway is initiated by the
release of cytochrome c from mitochondria into the cytosol
by external death stimulation (6).
The apoptosome formed by binding the cytochrome c to the
complex of apoptotic protease activation factor-1 (Apaf-1) and
procaspase-9 activates capase-9 and caspase-3, then finally
resulting in apoptosis. This mitochondrial pathway is controlled by
the Bcl-family (7) that includes
both proapoptotic factors and antiapoptotic factors. In normal
cells, the antiapoptotic factors inhibit the release of cytochrome
c from mitochondria. However, once external death signals
are transmitted to them, they cause the release of cytochrome
c. When DNA damage is not successfully repaired, it induces
apoptosis through activation of Bax, a proapoptosis factor as
previously described (2). Hypoxia
and a malnutrition condition strongly induce apoptosis as cancer
cells proliferate at a rapid rate. Moreover, cancer cells evade
apoptosis through downregulation of proapoptotic factors and
upregulation of antiapoptotic factors. Thus, apoptosis is a
valuable target for the development of anticancer drug since
apoptotic cells form apoptotic bodies that are removed by
macrophages proceeding inflammation.
Ginkgo biloba is deciduous castle chaplain,
and numerous studies have reported the physiological effects of
Ginkgo biloba leaves. The major classes of constituents in
Ginkgo biloba leaves include flavonoids, diterpenes,
sesquiterpenes, polyphenol, organic acid, and polycaccharide
(8). Among these, the major
components in Ginkgo biloba are flavonoids such as
quercetin, kaempferol, rutin and robinin. In particular, most of
the physiological effects of Ginkgo biloba are the result of
flavonoids and terpenes. The extract of Ginkgo biloba leaves
has been used as a therapeutic agent for ischemic stroke, ischemic
heart disease and atherosclerosis, and has been reported to have
antioxidant, memory and blood circulation effects (9). Particularly, the extract of Ginkgo
biloba leaves was found to inhibit amyloid-β fibril formation
and activate caspase-3 and was demonstrated to have an Alzheimer’s
disease protective effect through repressing the apoptosis of
neuronal cells (10). In contrast,
other studies have found that kaempferol contained in extracts of
Ginkgo biloba leaves induced the apoptosis of pancreatic
cancer cells (11). However, the
physiological effects of Ginkgo biloba fallen leaves has not
been investigated to date. Therefore, the aim of this study was to
investigate the potential of Ginkgo biloba fallen leaves as
a cancer therapeutic agent by studying the inductive effect of
apoptosis on a murine melanoma cell line.
Materials and methods
Materials
Dulbecco’s modified Eagle’s medium (DMEM),
trypsin-EDTA, penicillin/streptomycin/amphotericin (10,000 U/ml,
10,000 g/ml and 2,500 g/ml, respectively) and fetal bovine serum
(FBS) were obtained from Gibco-BRL, Life Technologies (Grand
Island, NY, USA). B16F1 (ATCC no. CRL-6323) cells were purchased
from the American Type Culture Collection (ATCC). MTT reagent,
agarose, and other materials were purchased from Sigma Chemical Co.
(St. Louis, MO, USA).
Extract preparation
For producing the methanol extracts of Ginkgo
biloba fallen leaves, air-dried Ginkgo biloba leaves
(MEGL) and fallen leaves (MEGFL) underwent extraction with 95%
methanol, respectively. The solvent was evaporated in vacuo to
yield 50.0 g of MEGFL as a dark brown solid material. MEGFL (1 g)
was suspended in 10 ml of methanol, and was subjected to membrane
(0.45 μm) filtration. The extracts were dissolved in DMSO for this
study.
Spectrophotometric determination of
flavonoid
The total flavonoid content in the MEGFL was
determined according to a modified version of the Folin-Ciocalteu
method (12) using phloroglucinol
as the standard. Samples were diluted taking into account the
measurable range of the spectrophotometer (a 0.005-ml aliquot of
extract of soluble phenolics was mixed with 0.495 ml water). A
0.1-ml aliquot of the diluted sample was mixed in a test tube with
1.0 ml of 1 N Folin-Ciocalteu reagent. The mixture was allowed to
stand for 3 min following addition of 2.0 ml of 20%
Na2CO3. The samples were incubated in the
dark at room temperature for 45 min and centrifuged at 1,600 × g
for 8 min. The optical density (OD) of the supernatant was measured
at 730 nm using a GENios® microplate reader (Tecan
Austria GmbH, Austria). The total flavonoid content was calculated
using a standard plotted graph and was expressed as a
percentage.
Cell line and culture
The cell lines were separately grown as monolayers
in 5% CO2 and at 37°C in a humidified atmosphere using
appropriate media supplemented with 5% FBS, 2 mM glutamine and 100
g/ml penicillin-streptomycin. DMEM was used as the culture medium
for the B16F1 cell line. Cells were passaged 3 times a week by
treatment with trypsin-EDTA.
MTT assay
Cytotoxic levels of MEGFL were measured using
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
method. The B16F1 cell line was grown in 96-well plates at a
density of 5×103 cells/well. After 24 h, the cells were
washed with fresh medium and were treated with MEGFL at 1, 2, 4, 6,
8, 10, 50 and 100 μg/ml. After 48 h of incubation, the cells were
rewashed, and 20 μl of MTT (5 mg/ml) was added and incubation was
carried out for 4 h. Finally, DMSO (150 μl) was added to solubilize
the formazan salt formed, and the amount of formazan salt was
determined by measuring the OD at 540 nm using a GENios®
microplate reader (Tecan Austria GmbH). The relative cell viability
was determined by the amount of MTT converted into formazan salt.
The viability of the cells was quantified as a percentage compared
to the control (OD of treated cells/OD of blank ×100) and the dose
response curves were developed. The data are expressed as the means
from at least three independent experiments, and P<0.05 was
considered to indicate a statistically significant result.
Reducing power
The reducing power of MEGFL was determined using a
method described previously (13).
The absorbance of this mixture was measured at 700 nm. The level of
reducing power was calculated by the absorbance and expressed as a
percentage: Reducing power = (OD of MEGFL/OD of blank) × 100.
DNA oxidation
Genomic DNA was extracted from the B16F1 cells using
a standard phenol/proteinase K procedure. The purity of genomic DNA
was spectrophotometrically determined at 260/280 nm. DNA oxidation
mediated by the Fenton reaction was determined by a method
described elsewhere (14). One
hundred microliters of the DNA reaction mixture was prepared by
adding pre-determined concentrations of the test sample (or the
same volume of 10% FBS/DMEM with Fenton reaction as the control
group), 200 μM final concentration of FeSO4, 2 mM final
concentration of H2O2 and 50 μg/ml final
concentration of genomic DNA in the same order. Then the mixture
was incubated at room temperature for 30 min, and the reaction was
terminated by adding a final concentration of 10 mM of EDTA. An
aliquot (20 μl) of the reaction mixture containing ~5 μg of DNA was
electrophoresed on 1% agarose gel for 30 min at 100 V. The gels
were then stained with 1 mg/ml ethidium bromide and visualized by
UV light using AlphaEase® gel image analysis software
(Alpha Innotech, San Leandro, CA, USA). The protective effect of
MEGFL was quantified as a percentage compared to the blank group
(density of the genomic DNA band treated with MEGFL/density of the
intact genomic DNA band × 100).
DNA ladder assay
For the DNA fragmentation analysis, the cells were
treated with different concentrations of MEGFL. DNA was isolated
from the cells, as follows. Cells were washed twice in
phosphate-buffered saline (PBS), resuspended in lysis buffer (10 mM
EDTA, 20 mM Tris, pH 8.0, 0.5% Triton X-100) and incubated at 50°C
for 1 h. RNase A was added to a final concentration of 0.5 mg/ml,
and incubation was continued at 50°C for 1 h. Samples were then
extracted with phenol-chroloform-isoamyl alchohol (25:24:l). High
molecular weight DNA was then pelleted at 13,000 × g for 10 min,
and the low molecular weight DNA in the supernatant was removed and
precipitated overnight in two volumes of ice-cold ethanol at
−70°C.
Analyses of protein expression using
western blot analysis
Western blotting was performed according to standard
procedures. Cells treated with different concentrations of MEGFL
were lysed with RIPA lysis buffer (Sigma Chemical Co.). Cell
lysates were resolved on a 4–20% Novex® gradient gel
(Invitrogen, USA), electrotransferred onto a nitrocellulose
membrane and blocked with 10% skim milk. The primary antibodies
including p53, p-p53, Ac-p53, p-p21, caspase-3, caspase-9, Bcl-2,
cytochrome c, β-actin and their secondary antibodies (Santa
Cruz Biotechnology, Inc., Santa Cruz, CA, USA) were used to detect
the respective proteins using a chemiluminescent ECL assay kit
(Amersham Pharmacia Biotech, Piscataway, NJ, USA) according to the
manufacturer’s instructions. Protein bands were visualized using
AlphaEase® gel image analysis software (Alpha
Innotech).
Statistics
Data were analyzed using the Student’s t-test for
paired data (comparison of the control group and MEGFL). Data are
expressed as means ± SD from three independent experiments.
P<0.05, P<0.01 and P<0.001 are indicative of statistically
significant results, and are indicated in the figures and
legends.
Results
Flavonoid contents in MEGL and MEGFL
Flavonoids are produced entirely by polymerization
of phloroglucinol, which is a product of the acetate-malonate
pathway, also known as the polyketide pathway. Following extraction
with methanol, harvest yields of MEGL and MEFGL were 13.40 and
26.22%, respectively, from starting material at dry weight basis.
MEGFL consisted of 5.50±0.21% of flavonoids at dry weight basis. In
contrast, the methanol extract of Ginkgo biloba leaves
consisted of 1.87±0.17% of flavonoids at dry weight basis.
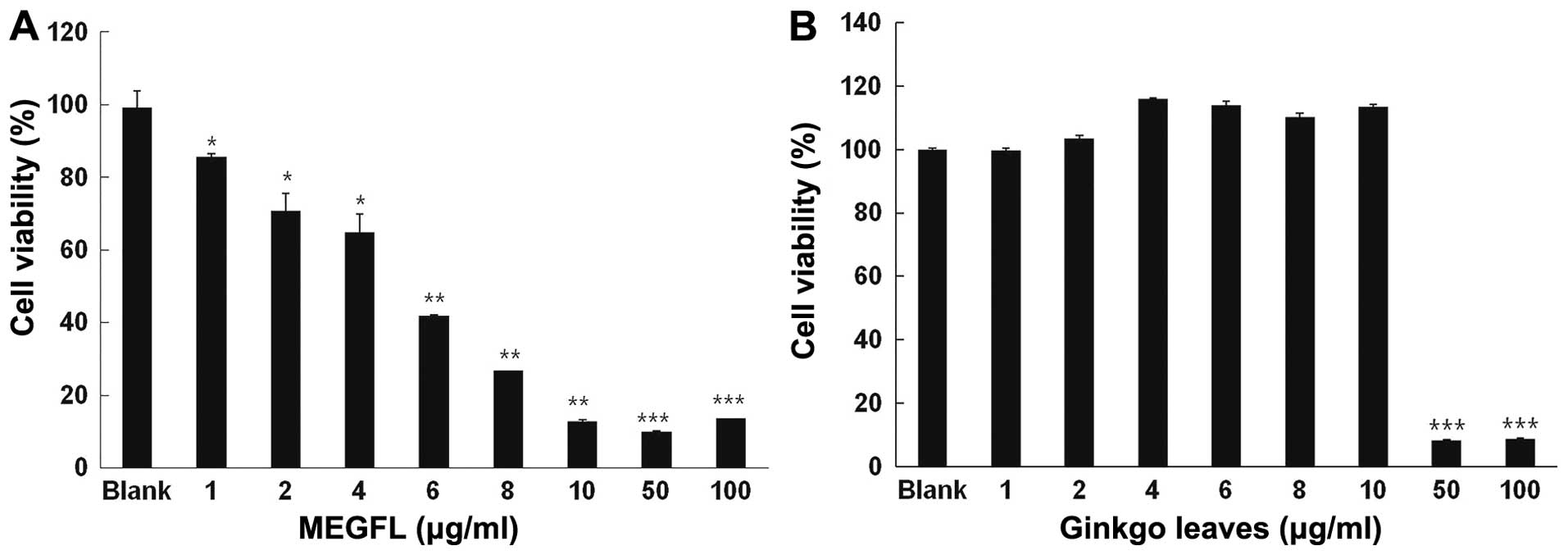
Effect of MEGFL on cell viability
To investigate the cytotoxic effect of MEGFL, MTT
assay was carried out in the B16F1 cell line. The cells were
treated with MEGFL at the indicated doses for 48 h. MEGFL exhibited
a cytotoxic effect in a dose-dependent manner in the B16F1 cell
line (Fig. 1A). MEGFL at 100 μg/ml
inhibited cell viability by 80% (P<0.001). However, MEGL did not
exhibit any cytotoxicity at a concentration <10 μg/ml. It was
found that MEGL at a concentration >50 μg/ml showed cytotoxicity
in B16F1 cells (Fig. 1B).
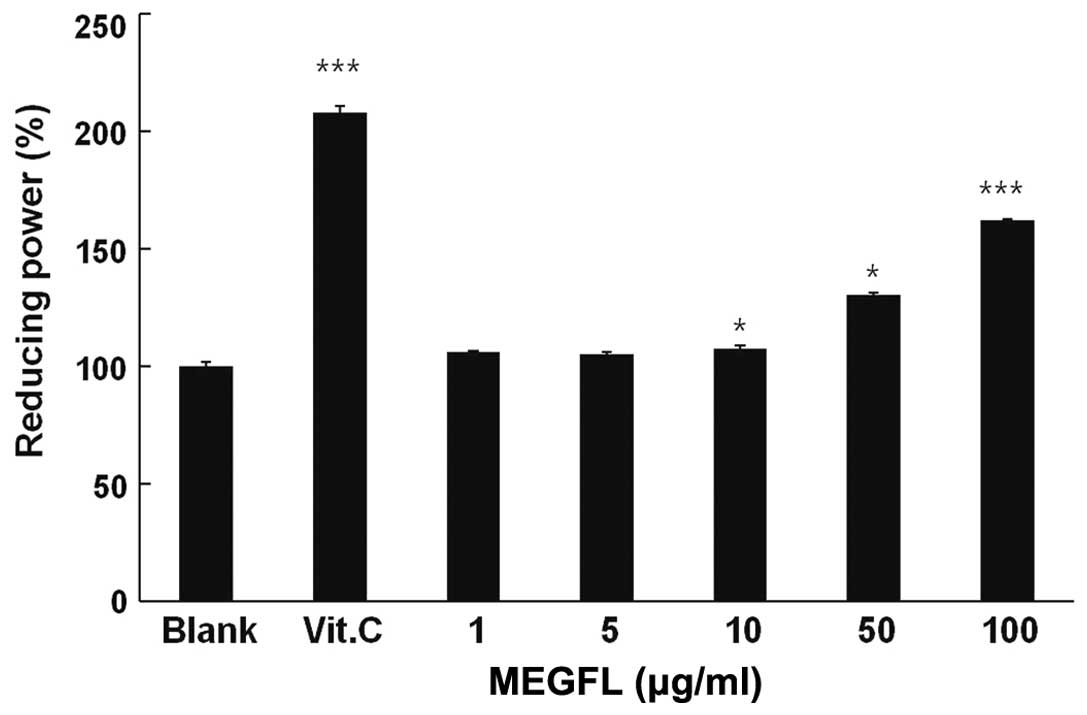
Reducing power of MEGFL
The reducing ability of a compound generally depends
on the presence of a reducing agent which exhibits antioxidative
potential by breaking the free radical chain, donating a hydrogen
atom. Vitamin C at 100 μg/ml significantly displayed reducing power
compared with the blank group (P<0.001) (Fig. 2). MEGFL significantly showed
reducing power in a dose-dependent manner. In particular, it was
observed that MEGFL increased the reducing power by 60% compared
with the blank group without MEGFL treatment.
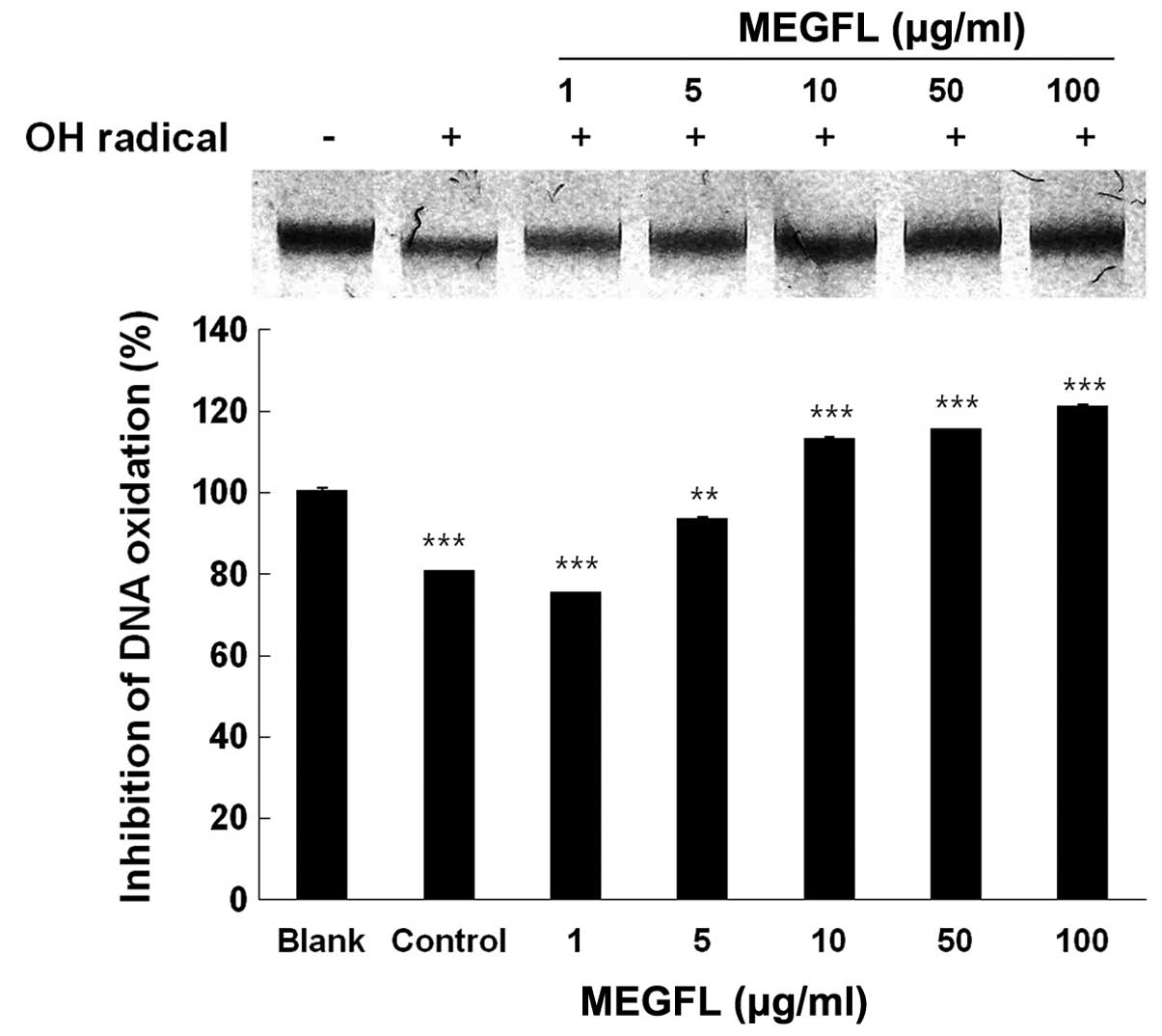
Inhibition of radical-mediated DNA
oxidative damage by MEGFL
In a subsequent experiment, genomic DNA was isolated
from B16F1 cells to study the protective effect of MEGFL against
DNA oxidative damage induced by hydroxyl radical. The genomic DNA
of the control group was completely degraded by the hydroxyl
radical produced by the Fenton reaction, compared with the blank
group without the Fenton reaction (Fig.
3). Treatment with MEGFL at 10 μg/ml or more significantly
inhibited the oxidative damage of DNA (P<0.001). The DNA damage
was inhibited by 80% in the presence of MEGFL at 10 μg/ml, compared
with the control group treated with the same volume of 5%
FBS/DMEM/instead of the test sample with the Fenton reaction.
However, it was found that MEGFL at 1 μg/ml could not clearly
protect the oxidative damage of DNA by the hydroxyl radical.
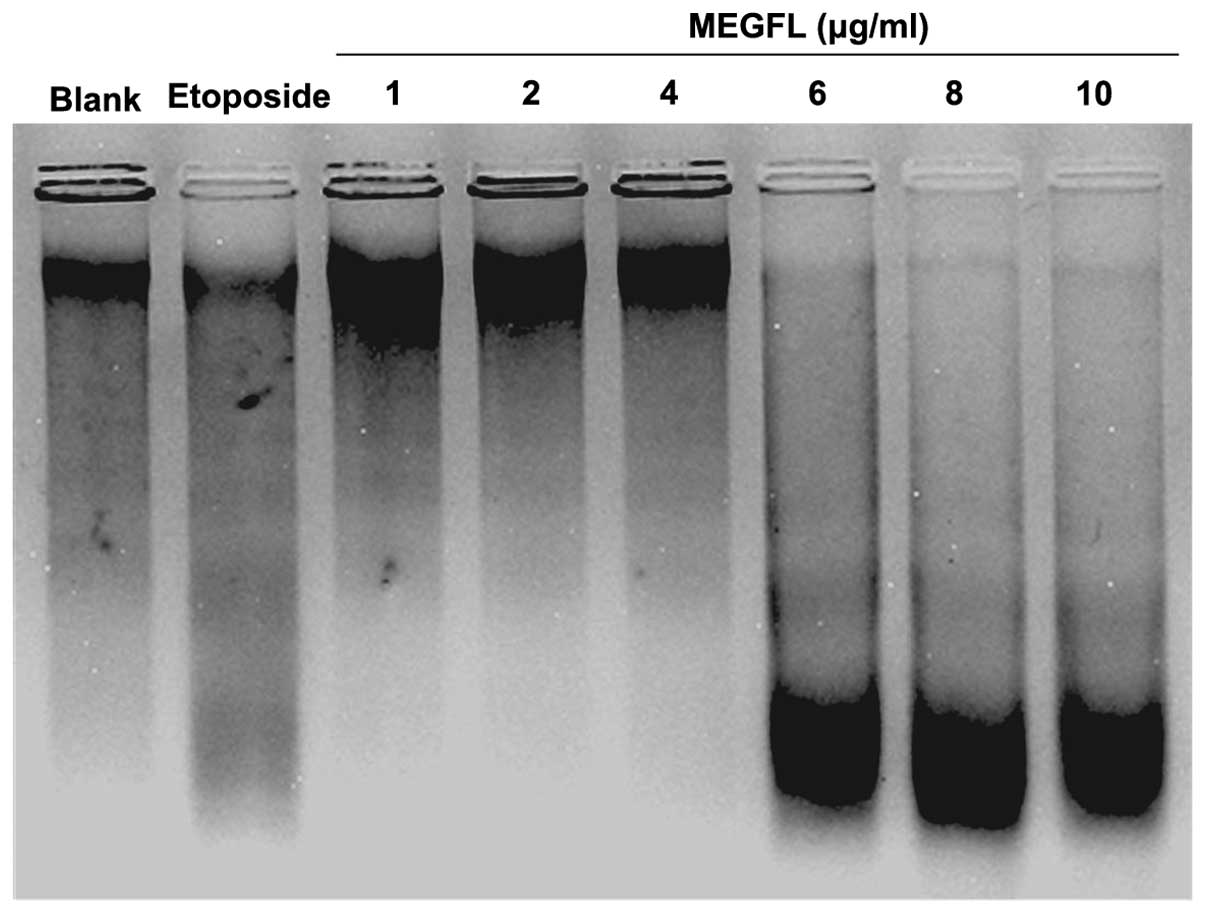
Effect of MEGFL on DNA fragmentation
To investigate the effect of MEGFL on DNA
fragmentation, DNA of B16F1 cells treated with MEGFL was observed
using the technique of DNA electrophoresis. Etoposide at 100 μM was
used as a positive control for DNA fragmentation. DNA of cells
treated with etoposide was cleaved compared to the blank group
without any stimulation (Fig. 4).
Furthermore, MEGFL had an inductive effect on DNA fragmentation in
a dose-dependent manner and showed apoptosis at a concentration of
MEGFL >6 μg/ml.
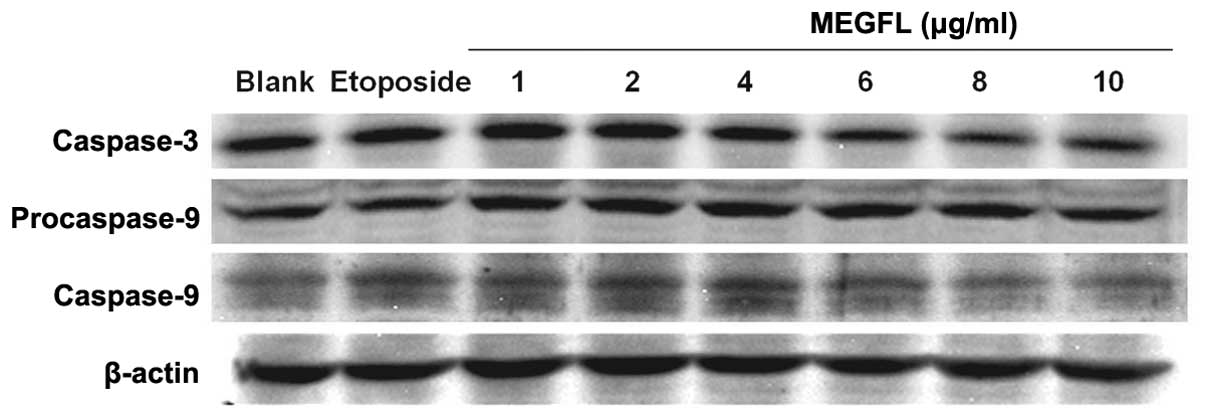
Effect of MEGFL on the protein expression
levels of caspase-3 and caspase-9
During the apoptosis process, caspases play a key
role in protein cleavage. To investigate the effect of MEGFL on the
protein expression levels of caspase-3 and caspase-9, western blot
analysis was carried out in B16F1 cells. Etoposide at 100 μM was
used as a positive control. The protein expression levels of
caspase-3 and caspase-9 were significantly increased in the
presence of etoposide and MEGFL in the range from 1 to 4 μg/ml
compared with the blank group (Fig.
5). However, MEGFL decreased the protein expression levels of
caspase-3 and caspase-9 in the presence of MEGFL at 6 μg/ml.
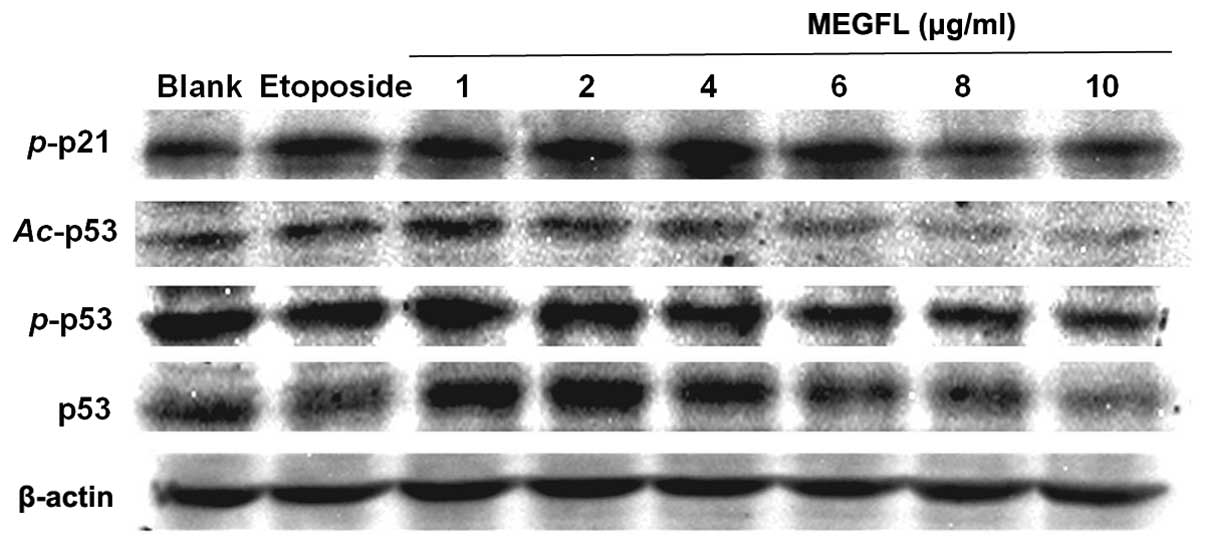
Effect of MEGFL on the protein expression
levels of p53, phosphorylated p53, acetylated p53 and
phosphorylated p21
When DNA damage occurs, p53 is activated to repair
DNA or to induce apoptosis. The activation of p53 is controlled by
p53 phosphorylation and acetylation. In addition, p53, a
transcription factor, binds to the promoter of p21. p21 is closely
associated with cell cycle arrest. As shown in Fig. 6, consistent with the results shown
in Fig. 5 the protein expression
levels of p53, phosphorylated p53 (p-p53 and Ser15), acetylated p53
(Ac-p53 and Lys373–382) and phosphorylated p21 (p-p21 and
Ser146) were increased in the presence of etoposide and MEGFL at a
range from 1 to 4 μg/ml. MEGFL treatment at concentrations >6
μg/ml caused a reduction in the protein expression levels. These
results suggest that the altered expression levels of p53 and p-p21
are related to cell death.
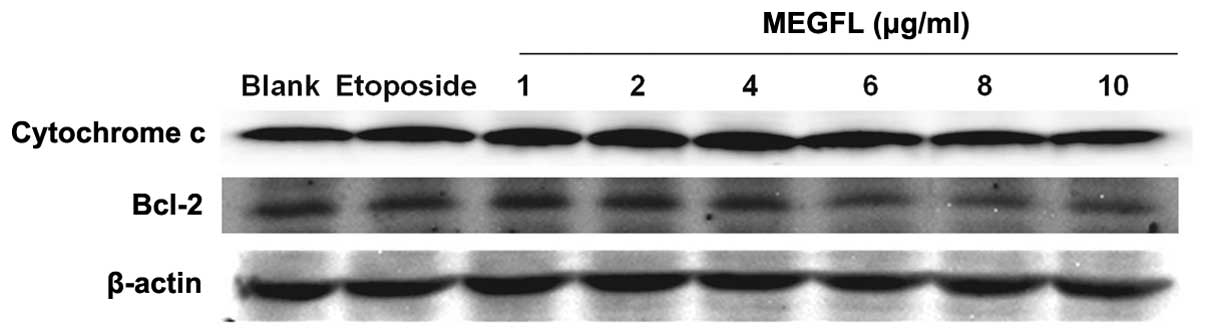
Effect of MEGFL on the protein expression
levels of cytochrome c and Bcl-2
During the apoptosis process, the cytochrome
c released from mitochondria activates caspase leading to
cell death. This mitochondrial pathway is regulated by the Bcl-2
family. It was found that the protein expression levels of
cytochrome c and Bcl-2 were not altered in the presence of
etoposide and MEGFL when compared with the blank group (Fig. 7). These results indicate that MEGFL
does not induce apoptosis through the mitochondrial-dependent
pathway.
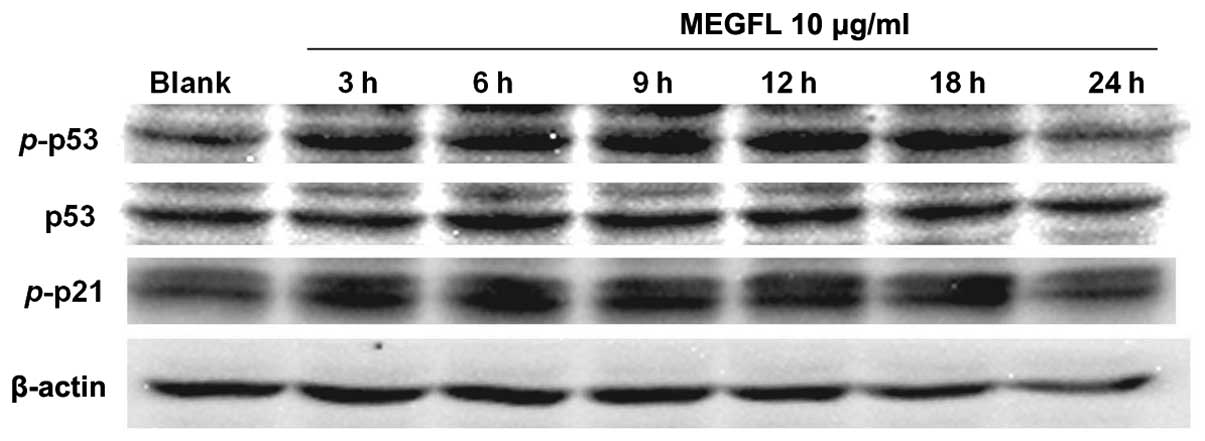
Effect of MEGFL on the protein expression
levels of p53, phosphorylated p53 and phosphorylated p21 with
time
To confirm that these protein expression levels are
associated with cell death, cells were treated with MEGFL at 10
μg/ml for increasing treatment times. The protein expression levels
of p53, p-p53 and p-p21 were increased at 18 h (Fig. 8). But their expression levels were
reduced at 24 h due to cell death.
Discussion
The aim of the present study was to investigate the
potential of MEGFL to induce apoptosis through modulation of p53
activation in the B16F1 cell line. Apoptosis, programmed cell death
by signal interaction, is not only a necessary process to ensure
body shape in ontogeny but also a protective reaction against a
detrimental influence on healthy cells surrounding diseased cells
when found to be seriously damaged (15,16).
In contrary with necrosis, apoptosis dose not release toxic
substance out of cells. Therefore, it was suggested that apoptosis
can be used as a target with which to treat cancer (17). In the present study, an MTT assay
was initially carried out to investigate the effect of MEGFL on
induction of apoptosis. Consequently, MEGFL caused cell death of
the B16F1 cell line. MEGFL exhibited apoptosis at a much lower
concentration when compared with other plant extracts. We carried
out various characteristic experiments concerning apoptosis to
support that cell death was the result of the apoptosis induced by
MEGFL. Apoptosis exhibits DNA laddering through cut linker region
between nucleosome as 180–200 base pair length while necrosis
causes random DNA cleavage. In order to examine the DNA ladder
effect of MEGFL, a DNA ladder assay was performed. Etoposide, a
well-known positive control, and MEGFL clearly indicated DNA
laddering compared with the blank group without treatment.
Especially, MEGFL caused marked DNA laddering at 6 μg/ml or more.
This result was associated with the tendency of protein expression
related to apoptosis. However, most cells were dead in the presence
of MEGFL at 6 μg/ml. Apoptosis is initiated by various proteins
such as the caspase family and Bcl-2 family (18,19).
The caspase family, cystein-dependent aspartate-specific protease,
is divided into initiator caspases and effector caspases. During
the cell death process, caspase-8, -9 and -10, initiator caspases,
transmit apoptotic signals, and caspase-3, -6 and -7, effector
caspases, exert the effect to degrade protein (4,20). Our
results suggest that MEGFL activated caspase-9 through cleavage of
procaspase-9 in the presence of MEGFL. Furthermore, the protein
expression level of caspase-3 was increased in the presence of
MEGFL. In general, the apoptosome is a complex formed with Apaf-1
that is activated by cytochrome c released from mitochondria
into the cytosol. It subsequently activates caspase-3 leading to
apoptosis. Both the expression levels of cytochrome c and
Bcl-2, an antiapoptotic factor, were not altered at a nontoxic
concentration of MEGFL. p-p21 arrests the cell cycle not only
through inhibition of cyclin D/CDK4 and cyclin E/CDK2 complexes in
early G1 phase but also inhibiton of cyclin A/CDK2 complex prior to
the S phase/G2 phase transition (2,21).
MEGFL highly increased the protein expression level of p-p21
(Ser146) at nontoxic concentrations. Therefore, our findings
revealed that MEGFL inhibits cell proliferation by disturbing
progression of the cell cycle. p53, a transcription factor binding
to the promoter of p21, is a normal short-lived protein that is
maintained at low levels, yet p53 is transiently accumulated when
serious DNA damage occurs in cells. p53 modulates the cell cycle
through induction or inhibition of WAF1, AFN and MDM (22,23).
When DNA damages are induced, the cell cycle is arrested and p53 is
activated for DNA repair. If DNA repair is not successful, p53
causes apoptosis by induction of Bax (24). Stability and site-specific
DNA-binding activity of p53 are associated with phosphorylation and
acetylation of p53 (25). The
phosphorylation of p53 is caused by protein kinase C (PKC) at
Ser378 and by casein kinase 2 (CK2) at Ser392. The carboxy-terminal
region of p53, including phosphorylation by CK2 and PKC kinases and
truncation of the last 30 residues, modulate the ability of p53 to
bind its recognition site through the central sequence-specific
DNA-binding domain (23,26). On the other hand, acetylation of p53
is formed by histone acetyl transferases (HATs) such as p300 and
PCAF. The p300 acetylates Lys382 in the carboxy-terminal region of
p53, whereas PCAF acetylates Lys320 in the nuclear localization
signal. Acetylations at either site enhance sequence-specific DNA
binding site of p21 (27). Our
results suggest that MEGFL increased the protein expression level
of p53 at nontoxic concentrations. Notably, the expression levels
of all proteins were decreased following treatment of MEGFL above a
concentration of 6 μg/ml. This was caused by cell death. Like the
preceding results, MEGFL increased both the protein expression
levels of phosphorylate p53 at the C terminal region (Ser15) and
acetylated p53 at the C terminal region (Lys373–382) at nontoxic
concentrations. To support these results, we investigated the
protein expression levels of p53, p-p53 and p-p21 with increasing
treatment times. While the expression level of p53 was constant at
a nontoxic exposure time with MEGFL, up to 18 h, both the
expression levels of p-p53, an activated form, and p-p21 were
increased. However their expression levels were reduced at 24 h due
to cell death. These results suggest that MEGFL induced activation
of p21 through modulation of p53, CDK inhibitor. Furthermore, our
findings revealed that MEGFL induced apoptosis by inhibiting cell
proliferation as a result of disturbing progression of the cell
cycle and increasing the expression levels of caspase-3 and
caspase-9. In addition MEGFL has an antioxidant effect through
enhancing reducing power and DNA protection. Therefore, this study
provides evidence that MEGFL has the potential to cause apoptosis
induction, and represents a new therapeutic strategy for the
treatment of cancer.
Acknowledgements
This study was supported by the Basic Science
Research Program through the National Research Foundation of Korea
(NRF) funded by the Ministry of Science, ICT and Future Planning
(no. 2013R1A1A1A05005160).
References
|
1
|
Maynard S, Schurman SH, Harboe C, de
Souza-Pinto NC and Bohr VA: Base excision repair of oxidative DNA
damage and association with cancer and aging. Carcinogenesis.
30:2–10. 2009. View Article : Google Scholar :
|
|
2
|
Singhal S, Vachani A, Antin-Ozerkis D,
Kaiser LR and Albelda SM: Prognostic implications of cell cycle,
apoptosis, and angiogenesis biomarkers in non-small cell lung
cancer: a review. Clin Cancer Res. 11:3974–3986. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Aamodt HM, Clifford A, Ellingson CC,
Burnett SH, Murray BK and O’Neill KL: Apoptosis through
Fas-mediated suicide gene induction in mouse melanoma. AACR.
2005:4142005.
|
|
4
|
Mashima T, Naito M and Tsuruo T:
Caspase-mediated cleavage of cytoskeletal actin plays a positive
role in the process of morphological apoptosis. Oncogene.
18:2423–2430. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mashima T, Naito M, Noguchi K, Miller DK,
Nicholson DW and Tsuruo T: Actin cleavage by CPP-32/apopain during
the development of apoptosis. Oncogene. 14:1007–1012. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Caroppi P, Sinibaldi F, Fiorucci L and
Santucci R: Apoptosis and human diseases: mitochondrion damage and
lethal role of released cytochrome c as proapoptotic protein. Curr
Med Chem. 16:4058–4065. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Estaquier J, Vallette F, Vayssiere JL and
Mignotte B: The mitochondrial pathways of apoptosis. Adv Exp Med
Biol. 942:157–183. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
van Beek TA and Montoro P: Chemical
analysis and quality control of Ginkgo biloba leaves, extracts, and
phytopharmaceuticals. J Chromatogr A. 1216:2002–2032. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yoshikawa T, Naito Y and Kondo M: Ginkgo
biloba leaf extract: review of biological actions and clinical
applications. Antioxid Redox Signal. 1:469–480. 1999. View Article : Google Scholar
|
|
10
|
Luo Y, Smith JV, Paramasivam V, et al:
Inhibition of amyloid-beta aggregation and caspase-3 activation by
the Ginkgo biloba extract EGb761. Proc Natl Acad Sci USA.
99:12197–12202. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang Y, Chen AY, Li M, Chen C and Yao Q:
Ginkgo biloba extract kaempferol inhibits cell proliferation and
induces apoptosis in pancreatic cancer cells. J Surg Res.
148:17–23. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Waterman PG and Mole S: Analysis of
Phenolic Plant Metabolites. Blackwell Scientific Publications;
Oxford, UK: 1994
|
|
13
|
Yen GC and Duh PD: Scavenging effect of
methanolic extracts of peanut hulls on free-radical and
active-oxygen species. J Agr Food Chem. 42:629–632. 1994.
View Article : Google Scholar
|
|
14
|
Imlay JA, Chin SM and Linn S: Toxic DNA
damage by hydrogen peroxide through the Fenton reaction in vivo and
in vitro. Science. 240:640–642. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Orrenius S: Apoptosis: molecular
mechanisms and implications for human disease. J Intern Med.
237:529–536. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Eguchi R, Toné S, Suzuki A, et al:
Possible involvement of caspase-6 and -7 but not caspase-3 in the
regulation of hypoxia-induced apoptosis in tube-forming endothelial
cells. Exp Cell Res. 315:327–335. 2009. View Article : Google Scholar
|
|
17
|
Ghobrial IM, Witzig TE and Adjei AA:
Targeting apoptosis pathways in cancer therapy. CA Cancer J Clin.
55:178–194. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Martinou JC and Youle RJ: Mitochondria in
apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev
Cell. 21:92–101. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Youle RJ and Strasser A: The BCL-2 protein
family: opposing activities that mediate cell death. Nat Rev Mol
Cell Biol. 9:47–59. 2008. View
Article : Google Scholar
|
|
20
|
Fan TJ, Han LH, Cong RS and Liang J:
Caspase family proteases and apoptosis. Acta Biochim Biophys Sin
(Shanghai). 37:719–727. 2005. View Article : Google Scholar
|
|
21
|
Besson A, Dowdy SF and Roberts JM: CDK
inhibitors: cell cycle regulators and beyond. Dev Cell. 14:159–169.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Levav-Cohen Y, Goldberg Z, Tan KH,
Alsheich-Bartok O, Zuckerman V, Haupt S and Haupt Y: The p53-Mdm2
loop: A critical juncture of stress response. Subcell Biochem.
85:161–186. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shieh SY, Ikeda M, Taya Y and Prives C:
DNA damage-induced phosphorylation of p53 alleviates inhibition by
MDM2. Cell. 91:325–334. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xu J, Lian LJ, Wu C, Wang XF, Fu WY and Xu
LH: Lead induces oxidative stress, DNA damage and alteration of
p53, Bax and Bcl-2 expressions in mice. Food Chem Toxicol.
46:1488–1494. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sakaguchi K, Herrera JE, Saito S, et al:
DNA damage activates p53 through a phosphorylation-acetylation
cascade. Genes Dev. 12:2831–2841. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Teufel DP, Bycroft M and Fersht AR:
Regulation by phosphorylation of the relative affinities of the
N-terminal transactivation domains of p53 for p300 domains and
Mdm2. Oncogene. 28:2112–2118. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fang J-Y and Lu Y-Y: Effects of histone
acetylation and DNA methylation on p21WAFI regulation.
World J Gastroenterol. 8:400–405. 2002.PubMed/NCBI
|