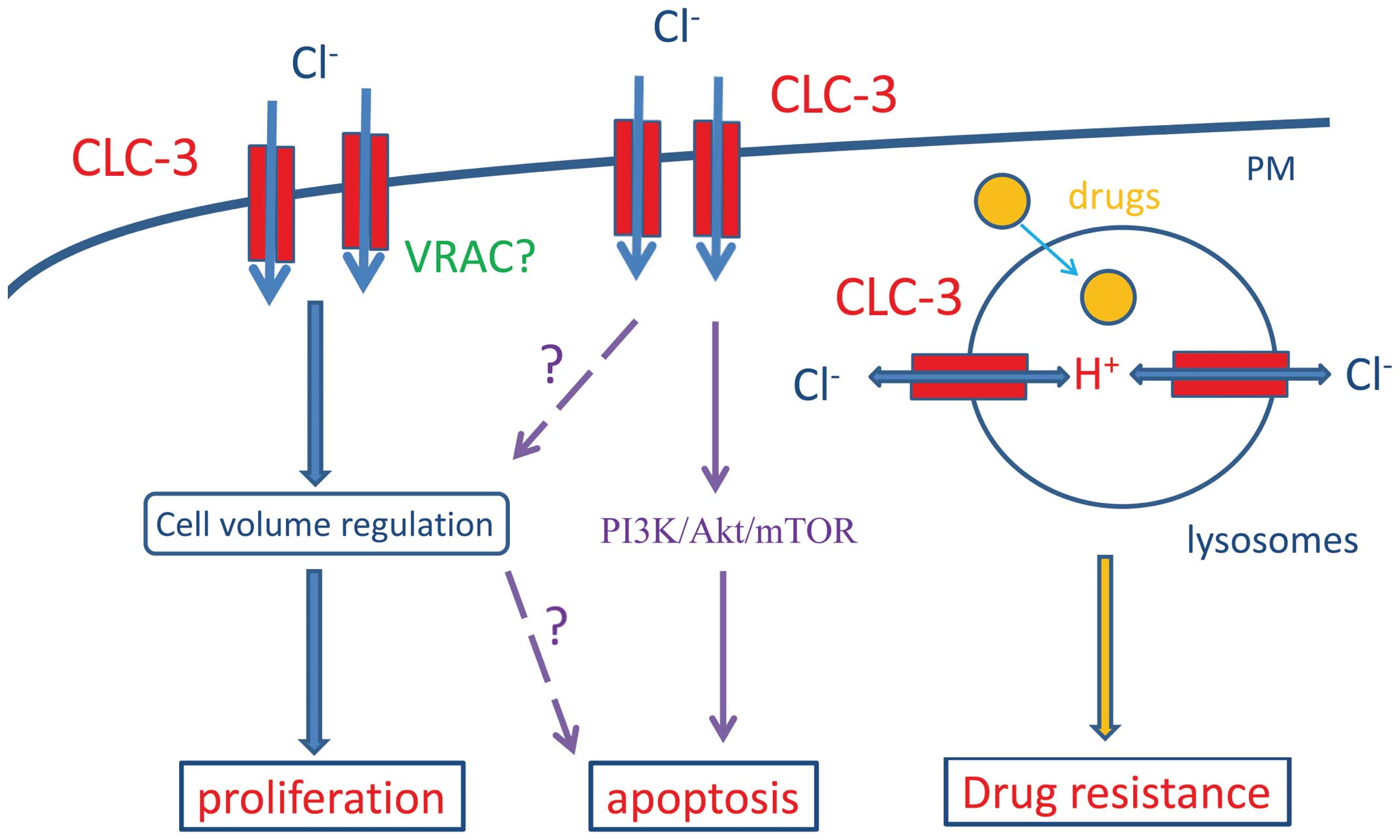
1. Introduction
Ion channels have various and important roles in
cellular functions, ranging from the control of cell excitability
to the regulation of cell volume. Channel dysfunctions have been
associated with arrhythmias (1),
skeletal muscle disorders (2),
neurological disorders including epilepsy and migration (3), cystic fibrosis (4) and endocrine disorders such as diabetes
(5). There is increasing evidence
that ion channel dysfunction is also involved in cancer (6,7); in
particular, potassium, sodium, calcium and chloride channels have
been shown to contribute to cancer development, metastasis and drug
resistance (8,9). Targeting ion channels is a promising
strategy for the treatment of cancer (10–12).
Recent studies have shown that ion channels and
transporters have crucial roles in regulating cell proliferation,
differentiation and apoptosis (13–16).
The functions of ion channels include maintenance of membrane
potential, cell cycle regulation and control of cell volume and
thus contribute to the regulation of cellular processes (16–19).
Cancer cells are characterized by the malignant transformation of
cells resulting in increased proliferation, aberrant
differentiation and reduced apoptosis. Although the mechanisms that
control ion channels in cancer are not well understood, it is known
that they contribute to cancer pathology by way of changes in the
cell cycle and cell volume (6,7,13).
Chloride channels contribute to the regulation of
the cell cycle and volume (7,11,20).
The process of cell proliferation requires an increase in cell
volume for generating daughter cells, while apoptosis is
characterized by cell shrinkage (17). The volume-regulated anion channel
(VRAC) is a major mechanism through which cells maintain a
relatively constant cell volume (6). The VRAC also participates in cell
proliferation, differentiation, migration and apoptosis (21,22).
Although the identity of the molecule responsible for
VRAC-activated cellular swelling has not been established, the
protein CLC-3 is a strong candidate (23). CLC-3 encodes a key component of the
native VRAC in many cancer cells including that of prostate cancer
epithelial, nasopharyngeal carcinoma and malignant glioma (24–26).
Knockdown of CLC-3 inhibits endogenous VRAC currents in many cells
such as cardiomyocytes, vascular smooth muscle cells and
non-pigmented ciliary epithelial cells (27–29).
Inhibition of CLC-3 by specific anti-CLC9-3 antibodies reduces the
swelling activated by chloride currents in human prostate cancer
epithelial cells (25).
Furthermore, CLC-3 mediates upregulation of VRAC by the
anti-apoptotic B-cell lymphoma 2 (BCL2) protein (22,25).
Therefore, CLC-3 may function as a VRAC in cancers, participating
in cell proliferation and apoptosis.
CLC-3 belongs to the CLC voltage-gated chloride
channel superfamily, which includes 2 distinct functional groups:
voltage-gated chloride channels and Cl−/H+
antiporters (30). Similar to CLC-4
and CLC-5, CLC-3 functions as a Cl−/H+
transporter in intracellular membranes (30,31).
CLC-4 and CLC-5 have been shown to have an important role in
acidification of endosomes and lysosomes (32,33).
Knockout of CLC-3 impairs acidification and chloride accumulation
in the endosome (34). Increased
acidification of intracellular compartments can sequester basic
anticancer drugs, thereby decreasing effective concentrations of
anticancer drugs and inducing drug resistance (35). Several studies have shown that CLC-3
contributes to the resistance of cancer cells to chemotherapeutic
drugs such as etoposide and cisplatin (36–38).
Therefore, inhibition of CLC-3 is a potential strategy for
increasing sensitivity to chemotherapy.
In this review, we summarize the function of CLC-3
in cancer and discuss the mechanisms by which CLC-3 contributes to
proliferation, apoptosis and drug resistance in cancer cells
(Fig. 1). We further explore
whether CLC-3 may be a potential strategy for the treatment of
cancer.
2. CLC-3 in cancer
Several studies have recently shown that CLC-3
participates in cell proliferation, apoptosis, the cell cycle and
metastasis in many cancers. These include nasopharyngeal carcinoma,
glioma, endometrial cancer and prostate cancer epithelial cells
(25,39–43).
Here, we review the role of CLC-3 in the proliferation, apoptosis
and drug resistance of these cancer cells (Fig. 1 and Table I).
 | Table IRoles of CLC-3 in cancer. |
Table I
Roles of CLC-3 in cancer.
| Cancer type | Channel
function | Roles in
cancer | References |
|---|
| Glioma | Resting outwardly
rectifying chloride channel; Ca2+-activated chloride
channel mediated by CaMKII | Cell
migration
Cell invasion | (41,45,46,51) |
| Outwardly
rectifying chloride channel associated with premitotic
condensation | Cell volume
regulation
Regulation of premitotic condensation
Cell cycle regulation
Cell mitosis
Cell division | (26,53) |
| Nasopharyngeal
carcinoma | Background chloride
channel; ATP-induced chloride channel; volume-regulated chloride
channel; acid-activated chloride channel | Cell volume
regulation
Cell proliferation
Cell migration
Cell apoptosis
Cell cycle progression | (24,39,40,42,56,57,89) |
| Prostate
cancer | Volume-regulated
chloride channel | Cell volume
regulation
Cell apoptosis? | (25) |
| Neuroendocrine
tumors |
Cl−-H+
transporter | Increased acidity
of intracellular vesicles
Drug resistance | (36) |
Glioma
Glioma is a lethal brain tumor characterized by
strong invasion into surrounding brain tissues (41,44).
Glioma cell invasion requires cell volume changes that are
regulated by many ion channels, including potassium channels and
chloride channels (41). CLC-3 has
been shown to have an important role in the invasiveness of human
glioma cells (41,45). CLC-3 is abundantly expressed on the
cytoplasmic membrane and in intracellular vesicles of glioma cells
(26,41). Knockdown of CLC-3 reduces resting
outwardly-rectifying chloride currents in glioma cells (41,46),
suggesting that CLC-3 mediates resting chloride currents in the
plasma membrane and thus may be involved in cell shrinkage during
invasion.
The migration of glioma cells is associated with
changes in the intracellular Ca2+ concentration
(47), and bradykinin has been
shown to increase the intracellular Ca2+ concentration
via bradykinin B2 receptors and to induce cell migration along
cerebral vasculature (48).
Interestingly, CLC-3 can be activated by
Ca2+/calmodulin-dependent kinase II (CAMKII) (49,50).
Knockout of CLC-3 was found to reduce Ca2+-activated
chloride currents mediated by CaMKII in glioma cells and to
decrease bradykinin-induced migration of human glioma cells
(51).
CLC-3 also is implicated in premitotic condensation
(PMC), a process that involves a decrease in cytoplasmic volume as
glioma cells retract processes, round up and progress with mitosis
(52). PMC is a crucial step in
cell division and is linked to chromatic condensation (52). Knockout of CLC-3 by shRNA reduced
PMC-associated outwardly rectifying chloride currents, inhibited
the rate of PMC and impaired DNA condensation (26). Furthermore, similar to knockout of
CLC-3 by shRNA, the chloride channel blocker
5-nitro-2-3-phenylpropylamino benzoic acid (NPPB) produces a
similar effect on PMC, suggesting that the chloride channel
function of CLC-3 on the plasma membrane determines the rate at
which glioma cells undergo PMC and progress through mitosis
(26). Furthermore, activation of
CLC-3 by CaMKII is involved in PMC and accelerates cytoplasmic
condensation during glioma cell division (53), suggesting that CaMKII-mediated
activation of CLC-3 contributes to PMC.
Nasopharyngeal carcinoma
Nasopharyngeal carcinoma cells abundantly express
CLC-3 (39,54–56).
CLC-3 is located in the plasma membrane, cytoplasm and nuclei in
nasopharyngeal carcinoma (CNE-2Z) cells. Under isotonic conditions,
knockdown of CLC-3 was found to reduce background chloride currents
and to inhibit ATP-induced chloride channels, accompanied by
increased cell volume. These findings suggest that CLC-3
constitutes the major background chloride channel under isotonic
conditions and may be responsible for maintenance of basal cell
volume (56).
The role of CLC-3 in the regulation of cell volume
is further supported by several studies showing that knockout of
CLC-3 reduced volume-regulated chloride currents in CNE-2Z cells
(39,40,57).
In addition, CLC-3 was identified as the acid-activated chloride
channel in CNE-2Z cells, which can be inhibited by hypertonic
solutions (42) and inhibition of
the volume-activated chloride channel by CLC-3 knockout was found
to be positively correlated with inhibition of cell proliferation
and migration in CNE-2Z cells (24,39).
This suggests that CLC-3 is involved in cell proliferation and
migration via modulation of volume-regulated chloride currents.
Prostate cancer
CLC-3 is expressed in lymph node carcinoma of
prostate (LNCaP) cancer epithelial cells (25). Inhibition of CLC-3 by its specific
antibodies effectively prevented activation of swelling-activated
chloride currents in LNCaP cells. This suggests that CLC-3 may be
the swelling-activated chloride channel in these cells.
Furthermore, BCL2 increased the expression of CLC-3, accompanied by
an increase in swelling-activated chloride currents, indicating
that CLC-3 mediates BCL2-dependent modulation of swelling-activated
chloride currents. It has been reported that BCL2 increases the
ability of cells to regulate cell volume via upregulation of
swelling-activated chloride currents in MDCK (Madin-Darby canine
kidney) cells (22). Since cell
proliferation is associated with cell volume changes, CLC-3 is
likely to contribute to cell volume changes during the
proliferation of LNCaP cells.
Neuroendocrine tumors
CLC-3 has been found to be expressed in several
neuroendocrine cell lines including BON (a human pancreatic
neuroendocrine carcinoma), CC-18 (a human
neuroendocrine-differentiated colonic carcinoma) and QGP-1 (a human
pancreatic carcinoma of islet origin) (36). The expression of CLC-3 is
predominantly located in the late endosome and lysosome, and
overexpression of CLC-3 increases the acidity of intracellular
vesicles. In addition, overexpression of CLC-3 decreased the
cytotoxicity of the chemotherapeutic drug etoposide in human
carcinoid BON cells. Since weakly basic chemotherapeutic drugs such
as etoposide can be sequestered to acidic intracellular membrane
compartments, the expression of CLC-3 may confer drug resistance to
basic chemotherapeutic agents such as etoposide by increasing the
acidity of intracellular compartments (58).
3. CLC-3 and VRACs in cell
proliferation
Cancer cells are characterized by unlimited
proliferation, which is associated with substantial changes in cell
volume. During the early phase of cell proliferation, cells
synthesize a large amount of proteins, which can result in
intracellular hyperosmolarity, thus leading to water influx and
cell swelling. Cancer cells maintain their normal size and avoid
excessive cell volume changes that can damage structural integrity
and cellular functions (59,60).
Cell swelling initiates a regulatory volume decrease through
activation of many ion channels (K+ and Cl−)
to return the cell volume to its normal size. The VRAC is important
in the regulation of cell volume (61) and is associated with cell
proliferation in various types of cells. These include hepatocytes
(62), endothelial cells (63), pulmonary artery smooth muscle cells
(64) and cancer cells (7,65).
The molecular characterization of the VRAC is still
unclear. CLC-3 has been proposed as the native VRAC in various
cells including cardiac myocytes, vascular smooth muscle cells and
non-pigmented ciliary epithelial cells (27–29,66).
Knockdown of CLC-3 by siRNA and anti-CLC-3 antibodies inhibits
swelling-activated chloride currents, suggesting that CLC-3 is an
integral component of the VRAC (27–29,66).
However, the role of CLC-3 as a native VRAC remains controversial.
Several other studies have shown that CLC-3 is predominantly
expressed in intracellular compartments and is not the
swelling-activated chloride channel (64,61).
In addition, the swelling-activated chloride current remains
present in CLC-3-knockout mice, arguing against the role of CLC-3
as a native VRAC (67). However,
since the properties of native swelling-activated chloride currents
are altered in CLC-3-knockout mice, accompanied by the altered
expression of many proteins (68),
it is possible that CLC-3 may be a regulatory component of the VRAC
and another protein compensates for the loss of CLC-3 in these
knockout mice. Furthermore, a recent study found that endogenous
swelling-activated chloride currents are eliminated in inducible
cardiac-specific CLC-3 knockout mice (70), suggesting that CLC-3 may be a key
component of VRAC in the heart.
CLC-3 has been found in the plasma membrane and is
involved in the regulation of cell volume in many types of cancers
such as glioma, nasopharyngeal carcinoma and prostate cancer
epithelial cells (25,39–41,57).
Several lines of evidence have shown that CLC-3 has an important
role in cell proliferation in vascular smooth muscle cells via
control of the cell cycle (70,71).
It has been reported that levels of CLC-3 vary according to the
phase of the cell cycle in nasopharyngeal carcinoma cells, i.e. low
in the G1 phase and high in the S phase (54). Furthermore, CLC-3 can promote
passage of the cells through the G1 phase into the S (39). Suppression of CLC-3 was demonstrated
to inhibit swelling-activated chloride currents and decrease
regulatory volume, which was correlated with cell proliferation in
nasopharyngeal carcinoma cells (39). Therefore, CLC-3 is believed to be
involved in cell proliferation and cell cycle progression through
regulation of cell volume via the swelling-activated chloride
channel (39) (Fig. 1).
Changes in cell volume was correlated with the
regulation of cell cycle progression, which during proliferation is
generally stimulated by cell swelling and inhibited by cell
shrinkage (72,73). In addition, cell volume is greatest
in the M phase and least in the G1 phase and increases in cell
volume are associated with the G1-S transition (59). Although it is known that cell volume
is associated with cell cycle progression, the regulatory
mechanisms remain unclear. Recently, Zhang et al (40) found that in nasopharyngeal carcinoma
cells, CLC-3 is a downstream target of cyclin D1, which controls
cell cycle progression through regulation of cyclin-dependent
kinases (CDKs). Cyclin D1 promotes the protein expression of CLC-3,
associated with an increase in volume-activated chloride channels,
suggesting that cyclin D1 regulates volume-activated chloride
currents via upregulation of CLC-3 expression. In addition,
inhibition of CDK4 activated volume-activated chloride currents,
whereas blockade of CDK6 reduced these currents (40). These results suggest that the cyclin
D1-CDK4 complex may inhibit the VRAC and the cyclin D1-CDK6 complex
may activate the VRAC. It appears that cyclin D1 can mediate cell
cycle progression and regulate cell volume via CLC-3. Furthermore,
it has been reported that cell swelling stimulates extracellular
signal regulated kinase (ERK1/2) (59,74,75),
and ERK activity regulates the induction of cyclin D1 (76–78).
Therefore, it appears that cell volume regulates cell cycle
progression via control of cyclin D1.
CLC-3 in cell apoptosis
A characteristic hallmark of apoptosis is cell
shrinkage or apoptotic volume decrease (AVD). AVD occurs early in
apoptosis in response to apoptotic stimuli and may be a
prerequisite (59,79). Most commonly, subsequent to AVD is
regulatory volume increase, which allows cells to return to their
original cell volume (59). The
VRAC has been shown to be involved in AVD (80–82).
CLC-3 has been found to contribute to the VRAC in prostate cancer
epithelial cells and the anti-apoptotic BCL2 increases
swelling-activated chloride currents via upregulation of CLC-3
expression (25). However, since
inhibition of VRAC can effectively prevent apoptotic events
(83), the finding that BCL2
mediates upregulation of VRAC suggests that BCL2 promotes
VRAC-mediated AVD and induces apoptosis. This clearly contradicts
the anti-apoptotic function of BCL2. It is possible that CLC-3 may
not contribute to the apoptosis-inducing VRAC. Consistent with this
idea, Okada et al (84)
concluded that CLC-3 is not the volume-sensitive outwardly
rectifying chloride channel involved in AVD.
In basilar arterial smooth muscle cells, knockout of
CLC-3 increases apoptosis induced by hydrogen peroxide via the
intrinsic mitochondrial pathway (85). In human bronchial epithelial cells,
overexpression of CLC-3 inhibited transforming growth factor
(TGF)-β 1-induced apoptosis, which was suppressed by overexpression
of BCL2 (86). Knockout of CLC-3
facilitated apoptosis induced by thapsigargin, a specific inhibitor
of the endoplasmic reticulum calcium ATPase, in
pheochromocytoma-derived PC12 cells (87). These findings suggest that CLC-3 is
likely to promote apoptosis in these cells via a different
signaling pathway.
Accordingly, in nasopharyngeal carcinoma cells, the
expression of CLC-3 was upregulated during early apoptosis and
after treatment with paclitaxel. Overexpression of CLC-3 associated
with microtubules was involved in paclitaxel-induced apoptosis
(88). Furthermore, in
nasopharyngeal carcinoma cells activation of CLC-3 by a novel class
of CLC-3 activators (bufadienolides) induced apoptosis via
inhibition of the PI3K/Akt/mTOR signaling pathway (89) (Fig.
1).
4. CLC-3 in drug resistance
Multidrug resistance is the main obstacle in the
treatment of cancer. Ion channels and transporters that are
involved in promotion of cell proliferation and evasion of
apoptosis contribute to the development of multidrug resistance
(8,81). As discussed above, overexpression of
CLC-3 is associated with increased cell proliferation. Thus, CLC-3
may be related to multidrug resistance in cancer cells via
increased cell proliferation. Consistent with this idea,
suppression of CLC-3 was found to increase the sensitivity of human
glioma U251 cells to cisplatin via inhibition of Akt and autophagy
(37).
CLC-3 is expressed in late endosomes and lysosomes
and contributes to their acidity. Increasing the acidity of
intracellular compartments confers drug resistance to weakly basic
chemotherapeutic drugs. Therefore, overexpression of CLC-3 may
increase sequestration of basic drugs in acidic compartments, thus
leading to drug resistance (Fig.
1). This hypothesis has been proven by the finding that
overexpression of CLC-3 increases drug resistance to etoposide by
increasing acidification of the late endocytic compartment in BON
cells (36). Similarly, Xu et
al (38) reported that in
erythroleukemia K562 and RK562 cells, upregulation of CLC-3 by NPPB
increased acidification of intracellular compartments and promoted
sequestration of cisplatin, conferring drug resistance to
cisplatin.
5. Targeting CLC-3 in the treatment of
cancer
As described above, increased expression of CLC-3 in
cancer cells stimulates proliferation and migration, but also
increases apoptosis. It appears to be paradoxical that cancer cells
can manage to upregulate CLC-3 to promote proliferation and
migrate, while at the same time avoiding its pro-apoptotic effect.
Because the expression of CLC-3 is cell cycle-dependent (high in
the S and low in the G1 phase) (39), transient upregulation of CLC-3 at
specific stages between the G1 and S phases may promote cell cycle
progression, leading to cancer cell proliferation. In contrast,
persistent upregulation of CLC-3 may lead to apoptosis.
The mechanisms by which cancer cells evade apoptosis
induced by upregulation of CLC-3 remain unclear. Since CLC-3 is
regulated by anti-apoptotic BCL2 in prostate cancer cells (25) and BCL2 upregulation is required for
the progression of prostate cancer cells (90), it may be that cancer cells can
upregulate anti-apoptotic proteins such as BCL2 to inhibit the
pro-apoptotic effect of CLC-3.
CLC-3 is involved in cell proliferation and
apoptosis and contributes to drug resistance in many cancer cells
(Fig. 1). CLC-3 inhibitors can be
used for blocking cancer cell proliferation and increasing
chemosensitivity, whereas CLC-3 activators can promote cancer cell
apoptosis. Therefore, developing novel CLC-3 inhibitors and
activators may be a strategy for the treatment of cancer (Table II).
| Table IICLC-3 inhibitors and activators in
the treatment of cancer. |
Table II
CLC-3 inhibitors and activators in
the treatment of cancer.
| Targets | Results | Mechanisms | References |
|---|
| CLC-3
inhibitors |
| Nonspecific
inhibitors (tamoxifen, NPPB) | Inhibition of cell
proliferation | Inhibits chloride
currents | (42,56,57,65,89) |
| Specific CLC-3
antibodies | | Inhibits chloride
currents | (27,68,92) |
| CLC-3
knockdown | Inhibition of
proliferation and apoptosis | Inhibits chloride
currents | (41,42,46,56,57,89) |
| Chlorotoxin | Inhibition of cell
migration
Phase I clinical trial | Inhibits CLC-3
chloride currents via the binding with MMP2 | (95–97) |
| CLC-3
activators |
|
Bufadienolides | Antitumor
activities | Inhibits CLC-3
chloride currents via inhibition of the PI3K/Akt/mTOR signaling
pathway | (89,101) |
| Promotion of
apoptosis | | |
CLC-3 inhibitors
A highly specific CLC-3 inhibitor is preferred for
use in cancer therapy, to reduce nonspecific side effects.
Unfortunately, specific CLC-3 inhibitors have not been developed.
Several chloride channel inhibitors such as tamoxifen and NPPB have
been shown to inhibit CLC-3 currents and reduce proliferation of
many cancer cells (42,56,57,65,89).
However, these inhibitors are not specific for CLC-3. They can
block other chloride channels such as Ca2+-activated
chloride channel anoctamin 1 (also known as transmembrane member
16A or TMEM16A) (91) and therefore
are not used as specific CLC-3 blockers. As far as cancer therapy
is concerned, these inhibitors are not suitable to use because
chloride channel inhibition is wide-ranging. Developing specific
compounds for a chloride channel such as CLC-3 remains a major
challenge.
Specific CLC-3 targeting can be affected by
developing CLC-3-specific antibodies. Several studies have shown
that CLC-3 antibodies can effectively inhibit CLC-3 currents
(27,68,92).
It has been reported that antibodies against human Eag1 potassium
channels display antitumor activity both in vitro and in
vivo (93). However, it remains
to be determined whether antibodies against CLC-3 can be used for
the treatment of cancer.
Specific inhibition of CLC-3 can be obtained by
knockdown of CLC-3 with siRNAs. Knockdown of CLC-3 by specific
siRNA has been used in many in vitro studies and effectively
inhibits CLC-3 currents and CLC-3-mediated proliferation and
apoptosis (41,42,46,56,57,89).
However, the use of siRNA in cancer therapy has not been tested in
animals or clinical studies.
Chlorotoxin, a 36-amino acid peptide that is
isolated from a scorpion toxin, was originally regarded as a
chloride channel inhibitor (94).
Chlorotoxin targets glioma cells specifically by binding to matrix
metalloproteinase-2 (MMP-2) (95).
A recent study by Qin et al (96) confirmed that MMP-2 mediates the
target delivery from chlorotoxin-modified liposomes to tumors. This
study also indicated that chlorotoxin inhibited CLC-3 chloride
currents by binding with MMP-2, resulting in inhibition of cell
migration in gliomas (96) and
suggested that chlorotoxin may also target other cancers with high
MMP-2 expression. A phase I clinical trial of a synthetic
chlorotoxin derivative labelled with iodine-131 was completed with
adult patients with recurrent high-grade glioma and a single dose
of this compound was well tolerated (97).
CLC-3 activator
Recently, bufadienolides were discovered to be CLC-3
activators with antitumor activities (89). Of the 7 compounds tested, bufalin
exhibited the most potent antitumor activity and induced the
largest chloride currents, which were blocked by CLC-3 siRNA.
Furthermore, bufalin induced apoptosis via inhibition of the
PI3K/Akt/mTOR signaling pathway. Knockout of CLC-3 reduced
bufalin-induced apoptosis. These findings suggest that bufalin
exerts its antitumor activity via activation of CLC-3 chloride
currents, leading to inhibition of the PI3K/Akt/mTOR signaling
pathway.
In addition, it has been reported that
bufadienolides are associated with antitumor activity through
various other mechanisms, including p21-dependent cell cycle
arrest, induction of mitochondria-dependent and death
receptor-mediated apoptosis and inhibition of anti-apoptotic
proteins such as BCL2 (98). It
remains to be determined whether CLC-3 is involved in these
antitumor mechanisms affected by bufadienolides. However,
bufadienolides may not be safe to use, due to high toxicity
(99,100). A recent study showed that
bufadienolides in poloxamer-modified liposomes improved antitumor
efficacy and reduced toxicity (101) and these may provide a safe way to
deliver bufadienolides for the treatment of cancer.
6. Conclusions and perspectives
CLC-3 participates in the process of proliferation,
apoptosis and drug resistance in many types of cancers, including
nasopharyngeal carcinoma, glioma, endometrial cancer and prostate
cancer epithelial cells (25,39–43).
Whether CLC-3 is the native VRAC is debatable, but CLC-3 has been
found to be a component of the VRAC in many cancer cells.
Inhibition of CLC-3 currents by channel blockers, CLC-3 antibodies
and CLC-3 siRNA can effectively reduce cancer cell proliferation.
Furthermore, CLC-3 contributes to the acidity of intracellular
compartments, in which basic chemotherapeutic drugs can be
sequestered, with subsequent chemoresistance. Inhibition of CLC3
can increase the sensitivity of cancer cells to chemotherapeutic
drugs. In addition, CLC-3 contributes to the apoptosis of cancer
cells and CLC-3 activators can promote this. Taken together,
targeting CLC-3 could be a therapeutic strategy for the treatment
of cancer.
There are many challenges to overcome before CLC-3
can be used in the treatment of cancer. Although CLC-3 currents
have been associated with proliferation and apoptosis (39,88,89),
the specific signaling pathway responsible for this is unclear. It
is also essential that the different roles of CLC-3 in apoptosis
and proliferation should be elucidated - it remains to be
determined whether a specific CLC-3 activator that promotes cell
apoptosis increases cell proliferation, thus leading to increased
tumor growth, migration and invasion and whether a specific CLC-3
blocker that inhibits cell proliferation could reduce cell
apoptosis. There is also very little knowledge regarding the
expression of CLC-3 in tumor tissues of either animal models or
cancer patients or the role of CLC-3 at different stages of cancer
development. Finally, highly specific blockers and activators for
CLC-3 are not available.
A major obstacle in implementing anti-CLC-3 therapy
is that targeting CLC-3 may inhibit the normal functions of CLC-3
in non-cancer tissues. It is known that CLC-3 is widely expressed
in many tissues, including the brain, and mice with disrupted CLC-3
exhibit signs of neurodegeneration (67,102).
This problem needs to be solved before CLC-3 inhibitors can be used
in the treatment of cancer. Further research into the mechanisms of
CLC-3 in carcinogenesis may result in the development of novel
drugs targeting CLC-3, which could serve as therapeutic tools in
the treatment of cancer.
References
|
1
|
Marban E: Cardiac channelopathies. Nature.
415:213–218. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Platt D and Griggs R: Skeletal muscle
channelopathies: new insights into the periodic paralyses and
nondystrophic myotonias. Curr Opin Neurol. 22:524–531. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Catterall WA, Dib-Hajj S, Meisler MH and
Pietrobon D: Inherited neuronal ion channelopathies: new windows on
complex neurological diseases. J Neurosci. 28:11768–11777. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Planells-Cases R and Jentsch TJ: Chloride
channelopathies. Biochim Biophys Acta. 1792:173–189. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rolim AL, Lindsey SC, Kunii IS, et al: Ion
channelopathies in endocrinology: recent genetic findings and
pathophysiological insights. Arq Bras Endocrinol Metabol.
54:673–681. 2010. View Article : Google Scholar
|
|
6
|
Lehen’kyi V, Shapovalov G, Skryma R and
Prevarskaya N: Ion channels and transporters in cancer. 5 Ion
channels in control of cancer and cell apoptosis. Am J Physiol Cell
Physiol. 301:C1281–C1289. 2011. View Article : Google Scholar
|
|
7
|
Kunzelmann K: Ion channels and cancer. J
Membr Biol. 205:159–173. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hoffmann EK and Lambert IH: Ion channels
and transporters in the development of drug resistance in cancer
cells. Philos Trans R Soc Lond B Biol Sci. 369:201301092014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lang F and Stournaras C: Ion channels in
cancer: future perspectives and clinical potential. Philos Trans R
Soc Lond B Biol Sci. 369:201301082014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li M and Xiong ZG: Ion channels as targets
for cancer therapy. Int J Physiol Pathophysiol Pharmacol.
3:156–166. 2011.PubMed/NCBI
|
|
11
|
Arcangeli A, Crociani O, Lastraioli E,
Masi A, Pillozzi S and Becchetti A: Targeting ion channels in
cancer: a novel frontier in antineoplastic therapy. Curr Med Chem.
16:66–93. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Conti M: Targeting ion channels for new
strategies in cancer diagnosis and therapy. Curr Clin Pharmacol.
2:135–144. 2007. View Article : Google Scholar
|
|
13
|
Bortner CD and Cidlowski JA: Ion channels
and apoptosis in cancer. Philos Trans R Soc Lond B Biol Sci.
369:201301042014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Becchetti A, Munaron L and Arcangeli A:
The role of ion channels and transporters in cell proliferation and
cancer. Front Physiol. 4:3122013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Prevarskaya N, Skryma R, Bidaux G,
Flourakis M and Shuba Y: Ion channels in death and differentiation
of prostate cancer cells. Cell Death Differ. 14:1295–1304. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lang F, Foller M, Lang KS, et al: Ion
channels in cell proliferation and apoptotic cell death. J Membr
Biol. 205:147–157. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lang F, Foller M, Lang K, et al: Cell
volume regulatory ion channels in cell proliferation and cell
death. Methods Enzymol. 428:209–225. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Urrego D, Tomczak AP, Zahed F, Stuhmer W
and Pardo LA: Potassium channels in cell cycle and cell
proliferation. Philos Trans R Soc Lond B Biol Sci.
369:201300942014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Blackiston DJ, McLaughlin KA and Levin M:
Bioelectric controls of cell proliferation: ion channels, membrane
voltage and the cell cycle. Cell Cycle. 8:3527–3536. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Abdul M and Hoosein N: Expression and
activity of potassium ion channels in human prostate cancer. Cancer
Lett. 186:99–105. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nilius B, Eggermont J, Voets T and
Droogmans G: Volume-activated Cl− channels. Gen
Pharmacol. 27:1131–1140. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shen MR, Yang TP and Tang MJ: A novel
function of BCL-2 overexpression in regulatory volume decrease.
Enhancing swelling-activated Ca(2+) entry and Cl(−) channel
activity. J Biol Chem. 277:15592–15599. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Duan DD: The CLC-3 chloride channels in
cardiovascular disease. Acta Pharmacol Sin. 32:675–684. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mao J, Chen L, Xu B, et al: Suppression of
CLC-3 channel expression reduces migration of nasopharyngeal
carcinoma cells. Biochem Pharmacol. 75:1706–1716. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lemonnier L, Shuba Y, Crepin A, et al:
Bcl-2-dependent modulation of swelling-activated Cl−
current and CLC-3 expression in human prostate cancer epithelial
cells. Cancer Res. 64:4841–4848. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Habela CW, Olsen ML and Sontheimer H: CLC3
is a critical regulator of the cell cycle in normal and malignant
glial cells. J Neurosci. 28:9205–9217. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang GX, Hatton WJ, Wang GL, et al:
Functional effects of novel anti-CLC-3 antibodies on native
volume-sensitive osmolyte and anion channels in cardiac and smooth
muscle cells. Am J Physiol Heart Circ Physiol. 285:H1453–H1463.
2003.PubMed/NCBI
|
|
28
|
Do CW, Lu W, Mitchell CH and Civan MM:
Inhibition of swelling-activated Cl− currents by
functional anti-CLC-3 antibody in native bovine non-pigmented
ciliary epithelial cells. Invest Ophthalmol Vis Sci. 46:948–955.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhou JG, Ren JL, Qiu QY, He H and Guan YY:
Regulation of intracellular CI− concentration through
volume-regulated CLC-3 chloride channels in A10 vascular smooth
muscle cells. J Biol Chem. 280:7301–7308. 2005. View Article : Google Scholar
|
|
30
|
Duran C, Thompson CH, Xiao Q and Hartzell
HC: Chloride channels: often enigmatic, rarely predictable. Annu
Rev Physiol. 72:95–121. 2010. View Article : Google Scholar :
|
|
31
|
Guzman RE, Grieschat M, Fahlke C and
Alekov AK: CLC-3 is an intracellular chloride/proton exchanger with
large voltage-dependent nonlinear capacitance. ACS Chem Neurosci.
4:994–1003. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Scheel O, Zdebik AA, Lourdel S and Jentsch
TJ: Voltage-dependent electrogenic chloride/proton exchange by
endosomal CLC proteins. Nature. 436:424–427. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Picollo A and Pusch M: Chloride/proton
antiporter activity of mammalian CLC proteins CLC-4 and CLC-5.
Nature. 436:420–423. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hara-Chikuma M, Yang B, Sonawane ND,
Sasaki S, Uchida S and Verkman AS: CLC-3 chloride channels
facilitate endosomal acidification and chloride accumulation. J
Biol Chem. 280:1241–1247. 2005. View Article : Google Scholar
|
|
35
|
Rajagopal A and Simon SM: Subcellular
localization and activity of multidrug resistance proteins. Mol
Biol Cell. 14:3389–3399. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Weylandt KH, Nebrig M, Jansen-Rosseck N,
et al: CLC-3 expression enhances etoposide resistance by increasing
acidification of the late endocytic compartment. Mol Cancer Ther.
6:979–986. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Su J, Xu Y, Zhou L, et al: Suppression of
chloride channel 3 expression facilitates sensitivity of human
glioma U251 cells to cisplatin through concomitant inhibition of
Akt and autophagy. Anat Rec (Hoboken). 296:595–603. 2013.
View Article : Google Scholar
|
|
38
|
Xu Y, Zheng H, Kang JS, et al:
5-Nitro-2-(3-phenylpropylamino) benzoic acid induced drug
resistance to cisplatin in human erythroleukemia cell lines. Anat
Rec (Hoboken). 294:945–952. 2011. View Article : Google Scholar
|
|
39
|
Xu B, Mao J, Wang L, et al: CLC-3 chloride
channels are essential for cell proliferation and cell cycle
progression in nasopharyngeal carcinoma cells. Acta Biochim Biophys
Sin (Shanghai). 42:370–380. 2010. View Article : Google Scholar
|
|
40
|
Zhang H, Zhu L, Zuo W, et al: The CLC-3
chloride channel protein is a downstream target of cyclin D1 in
nasopharyngeal carcinoma cells. Int J Biochem Cell Biol.
45:672–683. 2013. View Article : Google Scholar
|
|
41
|
Sontheimer H: An unexpected role for ion
channels in brain tumor metastasis. Exp Biol Med (Maywood).
233:779–791. 2008. View Article : Google Scholar
|
|
42
|
Wang L, Ma W, Zhu L, et al: CLC-3 is a
candidate of the channel proteins mediating acid-activated chloride
currents in nasopharyngeal carcinoma cells. Am J Physiol Cell
Physiol. 303:C14–C23. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Li M, Wu DB and Wang J: Effects of
volume-activated chloride channels on the invasion and migration of
human endometrial cancer cells. Eur J Gynaecol Oncol. 34:60–64.
2013.PubMed/NCBI
|
|
44
|
Wen PY and Kesari S: Malignant gliomas in
adults. N Engl J Med. 359:492–507. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lui VC, Lung SS, Pu JK, Hung KN and Leung
GK: Invasion of human glioma cells is regulated by multiple
chloride channels including CLC-3. Anticancer Res. 30:4515–4524.
2010.PubMed/NCBI
|
|
46
|
Olsen ML, Schade S, Lyons SA, Amaral MD
and Sontheimer H: Expression of voltage-gated chloride channels in
human glioma cells. J Neurosci. 23:5572–5582. 2003.PubMed/NCBI
|
|
47
|
Jantaratnotai N and McLarnon JG: Calcium
dependence of purinergic subtype P2Y1 receptor
modulation of C6 glioma cell migration. Neurosci Lett. 497:80–84.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Montana V and Sontheimer H: Bradykinin
promotes the chemotactic invasion of primary brain tumors. J
Neurosci. 31:4858–4867. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Cuddapah VA and Sontheimer H: Molecular
interaction and functional regulation of CLC-3 by
Ca2+/calmodulin-dependent protein kinase II (CaMKII) in
human malignant glioma. J Biol Chem. 285:11188–11196. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Huang P, Liu J, Di A, et al: Regulation of
human CLC-3 channels by multifunctional
Ca2+/calmodulin-dependent protein kinase. J Biol Chem.
276:20093–20100. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Cuddapah VA, Turner KL, Seifert S and
Sontheimer H: Bradykinin-induced chemotaxis of human gliomas
requires the activation of KCa3.1 and CLC-3. J Neurosci.
33:1427–1440. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Habela CW and Sontheimer H: Cytoplasmic
volume condensation is an integral part of mitosis. Cell Cycle.
6:1613–1620. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Cuddapah VA, Habela CW, Watkins S, Moore
LS, Barclay TT and Sontheimer H: Kinase activation of CLC-3
accelerates cytoplasmic condensation during mitotic cell rounding.
Am J Physiol Cell Physiol. 302:C527–C538. 2012. View Article : Google Scholar :
|
|
54
|
Wang LW, Chen LX and Jacob T: CLC-3
expression in the cell cycle of nasopharyngeal carcinoma cells.
Sheng Li Xue Bao. 56:230–236. 2004.PubMed/NCBI
|
|
55
|
Ye D, Zhang HF, Zhu LY, Wang LW and Chen
LX: CLC-3 siRNA inhibits regulatory volume decrease in
nasopharyngeal carcinoma cells. Nan Fang Yi Ke Da Xue Xue Bao.
31:216–220. 2011.(In Chinese). PubMed/NCBI
|
|
56
|
Yang L, Ye D, Ye W, et al: CLC-3 Is A main
component of background chloride channels activated under isotonic
conditions by autocrine ATP in nasopharyngeal varcinoma cells. J
Cell Physiol. 226:2516–2526. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Zhu L, Yang H, Zuo W, et al: Differential
expression and roles of volume-activated chloride channels in
control of growth of normal and cancerous nasopharyngeal epithelial
cells. Biochem Pharmacol. 83:324–334. 2012. View Article : Google Scholar
|
|
58
|
Raghunand N, Martinez-Zaguilan R, Wright
SH and Gillies RJ: pH and drug resistance. II Turnover of acidic
vesicles and resistance to weakly basic chemotherapeutic drugs.
Biochem Pharmacol. 57:1047–1058. 1999. View Article : Google Scholar
|
|
59
|
Hoffmann EK, Lambert IH and Pedersen SF:
Physiology of cell volume regulation in vertebrates. Physiol Rev.
89:193–277. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Lang F: Mechanisms and significance of
cell volume regulation. J Am Coll Nutr. 26:S613–S623. 2007.
View Article : Google Scholar
|
|
61
|
Sardini A, Amey JS, Weylandt KH, Nobles M,
Valverdez MA and Higgins CF: Cell volume regulation and
swelling-activated chloride channels. Biochim Biophys Acta.
1618:153–162. 2003. View Article : Google Scholar
|
|
62
|
Wondergem R, Gong W, Monen SH, et al:
Blocking swelling-activated chloride current inhibits mouse liver
cell proliferation. J Physiol. 532:661–672. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Nilius B, Prenen J, Kamouchi M, Viana F,
Voets T and Droogmans G: Inhibition by mibefradil, a novel calcium
channel antagonist, of Ca(2+)- and volume-activated Cl−
channels in macrovascular endothelial cells. Br J Pharmacol.
121:547–555. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Liang W, Huang L, Zhao D, et al:
Swelling-activated Cl− currents and intracellular CLC-3
are involved in proliferation of human pulmonary artery smooth
muscle cells. J Hypertens. 32:318–330. 2014. View Article : Google Scholar
|
|
65
|
Shen MR, Droogmans G, Eggermont J, Voets
T, Ellory JC and Nilius B: Differential expression of
volume-regulated anion channels during cell cycle progression of
human cervical cancer cells. J Physiol. 529:385–394. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Duan D, Winter C, Cowley S, Hume JR and
Horowitz B: Molecular identification of a volume-regulated chloride
channel. Nature. 390:417–421. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
67
|
Stobrawa SM, Breiderhoff T, Takamori S,
Engel D, Schweizer M, Zdebik AA, Bösl MR, Ruether K, Jahn H,
Draguhn A, Jahn R and Jentsch TJ: Disruption of CLC-3, a chloride
channel expressed on synaptic vesicles, leads to a loss of the
hippocampus. Neuron. 29:185–196. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Yamamoto-Mizuma S, Wang GX, Liu LL, et al:
Altered properties of volume-sensitive osmolyte and anion channels
(VSOACs) and membrane protein expression in cardiac and smooth
muscle myocytes from Clcn3−/− mice. J Physiol.
557:439–456. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Xiong D, Heyman NS, Airey J, et al:
Cardiac-specific, inducible CLC-3 gene deletion eliminates native
volume-sensitive chloride channels and produces myocardial
hypertrophy in adult mice. J Mol Cell Cardiol. 48:211–219. 2010.
View Article : Google Scholar :
|
|
70
|
Wang GL, Wang XR, Lin MJ, He H, Lan XJ and
Guan YY: Deficiency in CLC-3 chloride channels prevents rat aortic
smooth muscle cell proliferation. Circ Res. 91:E28–E32. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Tang YB, Liu YJ, Zhou JG, Wang GL, Qiu QY
and Guan YY: Silence of CLC-3 chloride channel inhibits cell
proliferation and the cell cycle via G/S phase arrest in rat
basilar arterial smooth muscle cells. Cell Prolif. 41:775–785.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Rouzaire-Dubois B, O’Regan S and Dubois
JM: Cell size-dependent and independent proliferation of rodent
neuroblastoma x glioma cells. J Cell Physiol. 203:243–250. 2005.
View Article : Google Scholar
|
|
73
|
Dubois JM and Rouzaire-Dubois B: The
influence of cell volume changes on tumour cell proliferation. Eur
Biophys J. 33:227–232. 2004. View Article : Google Scholar
|
|
74
|
Van der Wijk T, De Jonge HR and Tilly BC:
Osmotic cell swelling-induced ATP release mediates the activation
of extracellular signal-regulated protein kinase (Erk)-1/2 but not
the activation of osmo-sensitive anion channels. Biochem J.
343:579–586. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Sadoshima J, Qiu Z, Morgan JP and Izumo S:
Tyrosine kinase activation is an immediate and essential step in
hypotonic cell swelling-induced ERK activation and c-fos gene
expression in cardiac myocytes. EMBO J. 15:5535–5546.
1996.PubMed/NCBI
|
|
76
|
Modi PK, Komaravelli N, Singh N and Sharma
P: Interplay between MEK-ERK signaling, cyclin D1 and
cyclin-dependent kinase 5 regulates cell cycle reentry and
apoptosis of neurons. Mol Biol Cell. 23:3722–3730. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Cohen JD, Gard JM, Nagle RB, Dietrich JD,
Monks TJ and Lau SS: ERK crosstalks with 4EBP1 to activate cyclin
D1 translation during quinol-thioether-induced tuberous sclerosis
renal cell carcinoma. Toxicol Sci. 124:75–87. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Ravenhall C, Guida E, Harris T, Koutsoubos
V and Stewart A: The importance of ERK activity in the regulation
of cyclin D1 levels and DNA synthesis in human cultured airway
smooth muscle. Br J Pharmacol. 131:17–28. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Bortner CD, Hughes FM Jr and Cidlowski JA:
A primary role for K+ and Na+ efflux in the
activation of apoptosis. J Biol Chem. 272:32436–32442. 1997.
View Article : Google Scholar
|
|
80
|
Eggermont J, Trouet D, Carton I and Nilius
B: Cellular function and control of volume-regulated anion
channels. Cell Biochem Biophys. 35:263–274. 2001. View Article : Google Scholar
|
|
81
|
Pedersen SF, Hoffmann EK and Novak I: Cell
volume regulation in epithelial physiology and cancer. Front
Physiol. 4:2332013. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Stutzin A and Hoffmann EK:
Swelling-activated ion channels: functional regulation in
cell-swelling, proliferation and apoptosis. Acta Physiol (Oxf).
187:27–42. 2006. View Article : Google Scholar
|
|
83
|
Maeno E, Ishizaki Y, Kanaseki T, Hazama A
and Okada Y: Normotonic cell shrinkage because of disordered volume
regulation is an early prerequisite to apoptosis. Proc Natl Acad
Sci USA. 97:9487–9492. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Okada Y, Shimizu T, Maeno E, Tanabe S,
Wang X and Takahashi N: Volume-sensitive chloride channels involved
in apoptotic volume decrease and cell death. J Membr Biol.
209:21–29. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Qian Y, Du YH, Tang YB, et al: CLC-3
chloride channel prevents apoptosis induced by hydrogen peroxide in
basilar artery smooth muscle cells through mitochondria dependent
pathway. Apoptosis. 16:468–477. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Cheng G, Shao Z, Chaudhari B and Agrawal
DK: Involvement of chloride channels in TGF-beta1-induced apoptosis
of human bronchial epithelial cells. Am J Physiol Lung Cell Mol
Physiol. 293:L1339–1347. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Zhang HN, Zhou JG, Qiu QY, Ren JL and Guan
YY: CLC-3 chloride channel prevents apoptosis induced by
thapsigargin in PC12 cells. Apoptosis. 11:327–336. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Zhang H, Li H, Yang L, et al: The CLC-3
chloride channel associated with microtubules is a target of
paclitaxel in its induced-apoptosis. Sci Rep. 3:26152013.PubMed/NCBI
|
|
89
|
Liu J, Zhang D, Li Y, et al: Discovery of
bufadienolides as a novel class of CLC-3 chloride channel
activators with antitumor activities. J Med Chem. 56:5734–5743.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Lin Y, Fukuchi J, Hiipakka RA, Kokontis JM
and Xiang J: Up-regulation of Bcl-2 is required for the progression
of prostate cancer cells from an androgen-dependent to an
androgen-independent growth stage. Cell Res. 17:531–536. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Yang YD, Cho H, Koo JY, et al: TMEM16A
confers receptor-activated calcium-dependent chloride conductance.
Nature. 455:1210–1215. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Jin NG, Kim JK, Yang DK, et al:
Fundamental role of CLC-3 in volume-sensitive Cl−
channel function and cell volume regulation in AGS cells. Am J
Physiol Gastrointest Liver Physiol. 285:G938–948. 2003.PubMed/NCBI
|
|
93
|
Gomez-Varela D, Zwick-Wallasch E, Knotgen
H, et al: Monoclonal antibody blockade of the human Eag1 potassium
channel function exerts antitumor activity. Cancer Res.
67:7343–7349. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
DeBin JA and Strichartz GR: Chloride
channel inhibition by the venom of the scorpion Leiurus
quinquestriatus. Toxicon. 29:1403–1408. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Deshane J, Garner CC and Sontheimer H:
Chlorotoxin inhibits glioma cell invasion via matrix
metalloproteinase-2. J Biol Chem. 278:4135–4144. 2003. View Article : Google Scholar
|
|
96
|
Qin C, He B, Dai W, et al: The impact of a
chlorotoxin-modified liposome system on receptor MMP-2 and the
receptor-associated protein CLC-3. Biomaterials. 35:5908–5920.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Mamelak AN, Rosenfeld S, Bucholz R, et al:
Phase I single-dose study of intracavitary-administered
iodine-131-TM-601 in adults with recurrent high-grade glioma. J
Clin Oncol. 24:3644–3650. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Newman RA, Yang P, Pawlus AD and Block KI:
Cardiac glycosides as novel cancer therapeutic agents. Mol Interv.
8:36–49. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Abdel-Rahman MA, Ahmed SH and Nabil ZI: In
vitro cardiotoxicity and mechanism of action of the Egyptian green
toad Bufo viridis skin secretions. Toxicol In Vitro. 24:480–485.
2010. View Article : Google Scholar
|
|
100
|
Barrueto F Jr, Kirrane BM, Cotter BW,
Hoffman RS and Nelson LS: Cardioactive steroid poisoning: a
comparison of plant- and animal-derived compounds. J Med Toxicol.
2:152–155. 2006. View Article : Google Scholar
|
|
101
|
Hu K, Zhu L, Liang H, Hu F and Feng J:
Improved antitumor efficacy and reduced toxicity of liposomes
containing bufadienolides. Arch Pharm Res. 34:1487–1494. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Yoshikawa M, Uchida S, Ezaki J, et al:
CLC-3 deficiency leads to phenotypes similar to human neuronal
ceroid lipofuscinosis. Genes Cells. 7:597–605. 2002. View Article : Google Scholar : PubMed/NCBI
|