Introduction
Ovarian cancer is one of the most common
gynecological malignancies and is associated with a poor prognosis.
Historically, it is considered a ‘silent’ cancer since most
patients present with late-stage disease (1,2).
Despite advances in surgery and the development of more effective
chemotherapy, ovarian cancer remains the number one cause of death
from gynecologic cancer. Drug resistance is the predominant cause
of death in late-stage patients. Approximately 30% of patients
whose tumors are platinum-resistant will generally either progress
during primary therapy or shortly thereafter. Moreover, there is no
preferred standard second-line chemotherapy to offer these patients
(3,4). Thus, elucidation of mechanisms and
identification of new therapeutic targets and drugs for ovarian
cancer are critical to reduce the high mortality.
Histone deacetylase inhibitors (HDACis) show promise
as a novel class of anticancer agents in a wide spectrum of tumors
including ovarian cancer (5–7). To
date, at least 14 HDACis are being tested in over 100 clinical
trials and have displayed encouraging therapeutic responses with
surprisingly good safety profiles. The clinical potential of HDACis
has been well documented by the successful development of
vorinostat/SAHA and romidepsin, which have been approved by the
U.S. Food and Drug Administration (5,8,9).
Despite the rapid progress achieved, clinical data have shown that
there is limited efficacy for HDACis as a single agent. Most
current clinical trials are combination studies looking at HDACis
in combination with other agents (6,10,11).
All of these combination trials seek to increase the antitumor
activity of the treatments. Although these combination strategies
follow a rational molecular approach in some cases, in most
instances, they are relatively empirical. Accordingly, synergism in
antitumor efficacy might be accompanied by adverse effects that are
rarely or never seen with HDACis alone such as severe
myelosuppression (11). Therefore,
revealing the molecular mechanisms underlying the low potency of
HDACis is pivotal in determining the optimal application of this
class of therapeutic agents in the treatment of ovarian cancer.
Trichostatin A (TSA) is a natural compound and is
one of the most potent HDACis. In a previous study, we conducted a
functional gene screen approach to identify the key genes
responsible for the tumor-selective action of TSA. LIV1 was
isolated by its marked ability to confer resistance against
TSA-induced apoptosis. Our data preliminarily implied that the
inhibition of TSA-induced apoptosis by knockdown of LIV1 might be
associated with its ability to disrupt intracellular zinc
homeostasis in cervical cancer cells (12). To date, research on LIV1 is very
limited. Only several studies have speculated that LIV1 might be
related to the poor prognosis of breast cancer and could control
epithelial-mesenchymal transition in zebrafish gastrula organizer,
which have not been fully verified (13,14).
Therefore, the effect of LIV1 on ovarian cancer cells is totally
unknown. The present study was designed to explore the effect of
LIV1 on the sensitivity of ovarian cancer cells to TSA and to
provide a theoretical basis for further clinical targeted
therapy.
Materials and methods
Cells and reagents
The human ovarian cancer cell lines A2780 and SKOV3
were purchased from the American Type Culture Collection and
cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing
10% FCS. All cells were cultured at 37°C in a humidified 5%
CO2 atmosphere. HDACis, TSA and SAHA, were purchased
from Sigma and dissolved in DMSO.
Primary ovarian cancer cell culture
Ovarian cancer tissues were obtained from 6 patients
hospitalized in the Beijing Obstetrics and Gynecology Hospital
affiliated to Capital Medical University before any clinical
therapy. All research protocols in the present study were approved
by our Ethics Committee, and all patients gave written informed
consent to enroll in the study. These patients were 17–75 (mean,
47.1±13.5) years of age and were hospitalized between June 2012 and
December 2013; ovarian cancer was confirmed by pathological
diagnosis. Four patients were pathologically classified as serous
adenocarcinoma and 2 as mucoid adenocarcinoma. According to the
Federation International of Gynecology and Obstetrics (FIGO)
staging system, 1 case was classified as FIGO stage I, 2 as FIGO
stage II, 2 as FIGO stage III, and 1 as FIGO stage IV. Cells were
isolated and cultured as previously described (15).
Cell viability assays
Cell viability was determined using an MTT assay. In
brief, 5×103 cells were plated into each well of 96-well
plates at 72 h after the indicated treatments, after which 5 mg/ml
MTT was added and incubated at 37°C for 4 h. Media were then
removed, and 1 ml of DMSO was added to solubilize the MTT-formazan
product. The MTT absorbance was then measured at 570 nm on a
Multiscan JX ver 1.1 (Thermo Labsystems). Results are expressed as
a percentage of the viable cells in the DMSO-treated group. Each
data point is the mean ± SEM of six replicates.
Apoptosis assays
Cells were stained with Annexin V and propidium
iodide (PI) and the percentage of apoptotic cells were determined
by flow cytometry as previously described (15). CellQuest software was used for data
acquisition and analysis.
Real-time PCR
Quantitative PCR was conducted in ABI Prism 7000
using the SYBR-Green PCR Master Mix (Sigma) with the following set
of primers: LIV1, 5′-GGT GAT GGC CTG CAC AAT TTC-3′ and 5-TTA ACG
GTC ATG CCA GCC TTT AGT A-3; 18s RNA, 5′-AGT CCC TGC CCT TTG ACA
CA-3′ and 5′-GAT CCG AGG GCC TCA CTA AAC-3′. 18s RNA was used as
internal control. All primers were designed with the Primer3
software. A melting curve assay was performed to determine the
purity of the amplified product. Contamination with genomic DNA was
not detected in any of the analyzed samples. Each sample was
assayed in triplicate, analysis of the relative gene expression
data used the 2−ΔCT method (15), and the results are expressed as fold
induction compared with the untreated group.
Western blot analysis
Preparation of protein samples and western blotting
were carried out as previously described (15). Antibodies against LIV1 were
purchased from Novus Biologicals. Antibodies against Bcl-2, Bax,
and caspase-3 were purchased from Cell Signaling Technology.
Antibodies against β-actin were purchased from Santa Cruz
Biotechnology.
Colony forming assays
A2780 and SKOV3 cells were stably transfected with
PCEP4-CAT and AS-LIV1/FL-LIV1 and cultured for 24 h. Cells were
then treated with 500 nmol/l TSA or 500 nmol/l apicidine for 24 h
and plated in triplicate in 24-well plates at 50 cells/well. Plates
were subsequently incubated for 14 days in a humidified incubator
at 37°C, and colonies were fixed with 4% paraformaldehyde and
stained with 0.5% crystal violet and counted using a dissecting
microscope (magnification, ×50). Three random fields were counted
for each triplicate of samples, and average values were presented
as the means ± SD.
Results
TSA induces the expression of LIV1 in
ovarian cancer cells
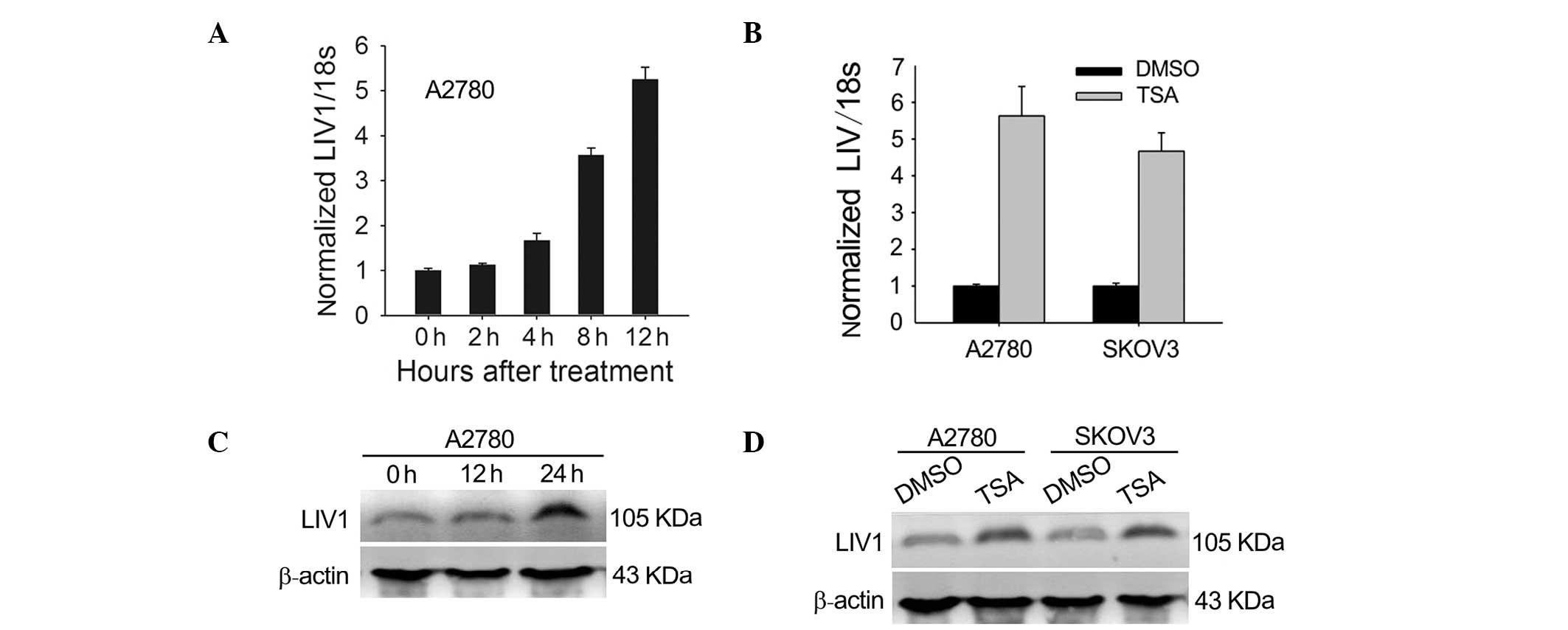
To test whether expression of LIV1 is induced by
TSA, we investigated the effects of TSA on the mRNA and protein
expression of LIV1 in the ovarian cancer cells. A2780 and SKOV3
cells were chosen because they had moderate levels of LIV1. First,
cells were treated with 500 nmol/l TSA for various lengths of time.
As shown in Fig. 1A and B,
transcription of LIV1 was highly induced. At 12 h post-treatment
with TSA, transcription induction reached a maximal level
(5.63±0.80-fold for A2780; 4.67±0.51-fold for SKOV3; P<0.05,
compared with the basal transcriptional level). To address whether
the induction of LIV1 transcription gave rise to the upregulated
level of LIV1 protein, cultured A2780 and SKOV3 cells were treated
with TSA or DMSO and examined for the LIV1 protein using western
blotting. As expected, treatment of TSA significantly enhanced the
protein levels of LIV1 in the ovarian cancer cells (Fig. 1C and D). Next, to determine whether
the TSA-induced expression of LIV1 occurs in primary ovarian cancer
cells, 6 primary tumor samples from patients with ovarian cancer
were treated with 500 nmol/l TSA for different time periods. Again,
TSA significantly induced the expression of LIV1 at a maximal level
(patient 1 for example: 3 h, 15.3±3.24-fold; 6 h, 10.16±2.54-fold;
and 12 h, 8.75±2.93-fold; P<0.05) as early as 3 h after
treatment and the increase lasted up to 12 h in all of the samples
examined (Table I).
 | Table IEffect of TSA on the expression of
LIV1 in the clinical tumor samples. |
Table I
Effect of TSA on the expression of
LIV1 in the clinical tumor samples.
| | | Time course (h) |
|---|
| | |
|
|---|
| Patients | Clinical
diagnosis | Classification | 0 | 3 | 6 | 12 |
|---|
| Patient 1 | Ovarian cancer | Serous | 1 | 15.3±3.24 | 10.16±2.54 | 8.75±2.93 |
| Patient 2 | Ovarian cancer | Mucinous | 1 | 14.5±2.83 | 9.59±2.19 | 5.84±1.45 |
| Patient 3 | Ovarian cancer | Serous | 1 | 13.2±3.56 | 10.96±2.78 | 6.57±1.38 |
| Patient 4 | Ovarian cancer | Serous | 1 | 9.8±1.98 | 8.25±1.33 | 7.18±1.59 |
| Patient 5 | Ovarian cancer | Mucinous | 1 | 14.1±3.72 | 6.41±1.21 | 5.66±1.17 |
| Patient 6 | Ovarian cancer | Serous | 1 | 10.3±2.41 | 8.57±1.65 | 6.92±1.55 |
Knockdown of LIV1 suppresses cell death
induced by TSA in ovarian cancer cells
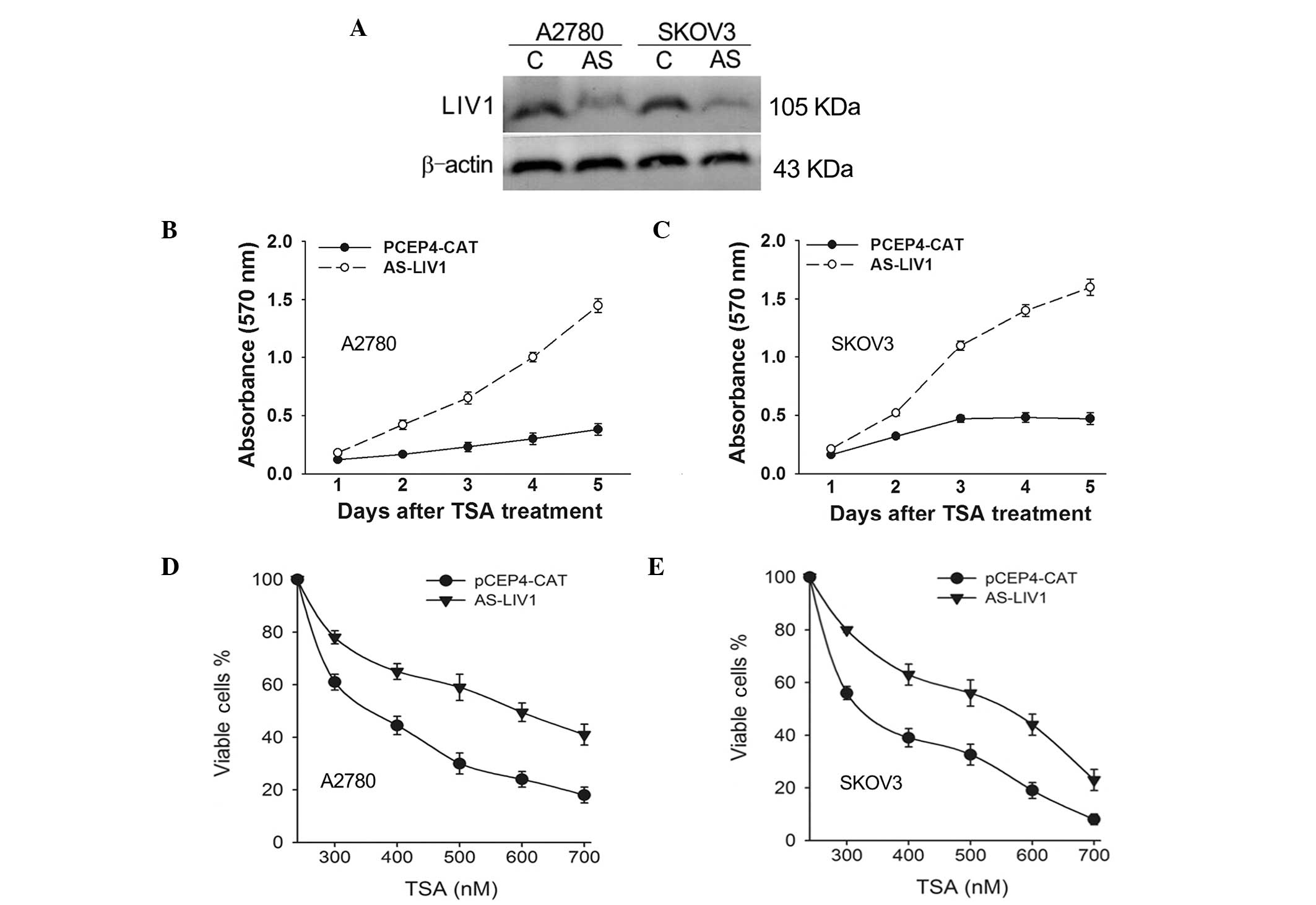
To confirm whether expression of LIV1 significantly
affects TSA-induced cell death in ovarian cancer cells, A2780 and
SKOV3 cells were stably transfected with the LIV1 antisense plasmid
AS-LIV1 or PCEP4-CAT followed by a 5-day treatment with TSA, and
they were then examined for growth inhibition. AS-LIV1 was
confirmed to significantly knockdown the basal and TSA-induced
levels of LIV1 expression (Fig.
2A), and transfection of AS-LIV1 resulted in the resistance of
the cells to TSA treatment (Fig. 2B and
C). Furthermore, we treated the A2780 and SKOV3 cells stably
transfected with AS-LIV1 or PCEP4-CAT with a TSA dose range between
300 and 700 nmol/l for 72 h and measured the cell viability using
the MTT assay. As shown in Fig. 2D and
E, knockdown of LIV1 decreased the TSA-induced killing
efficiency in both the A2780 and SKOV3 cells at every dose, with a
maximum effect observed at a 500 nmol/l concentration, where the
rate of viable cells was increased over 29% in the A2780 and 24% in
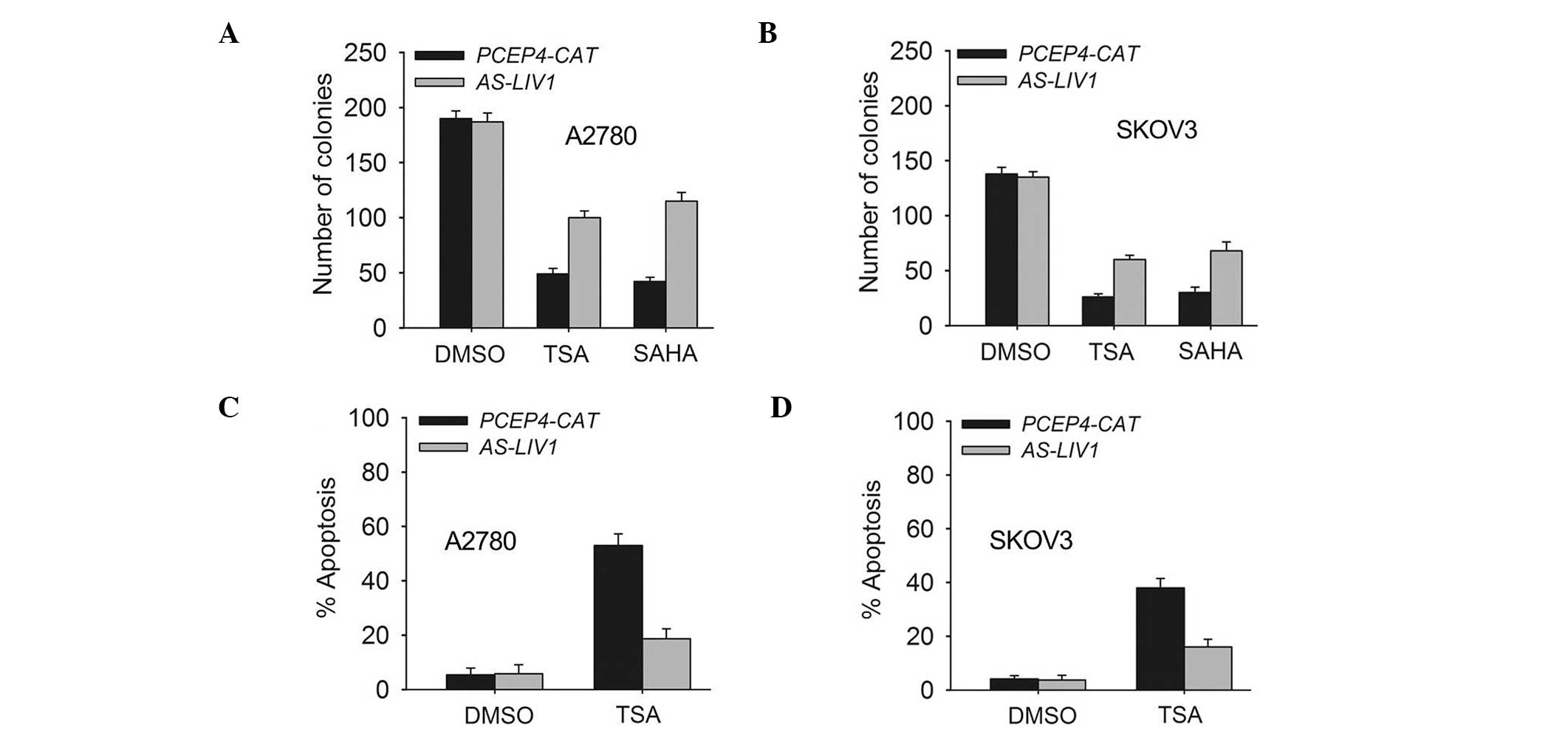
the SKOV3 cells. In addition, knockdown of LIV1 suppressed TSA- or
SAHA (a structurally diverse HDACi)-induced killing and gave rise
to more surviving colonies (Fig. 3A and
B). Accordingly, knockdown of LIV1 made A2780 and SKOV3 cells
resistant to TSA-induced apoptosis (Fig. 3C and D). A2780 and SKOV3 cells
stably transfected with AS-LIV1 were much less sensitive to
TSA-induced apoptosis than cells stably transfected with PCEP4-CAT
(A2780, 18.7±3.6 vs. 49.06±4.3%, P<0.05; SKOV3, 16.28±2.9 vs.
38.13±3.5%, P<0.05).
Upregulation of LIV1 enhances cell death
induced by TSA in ovarian cancer cells
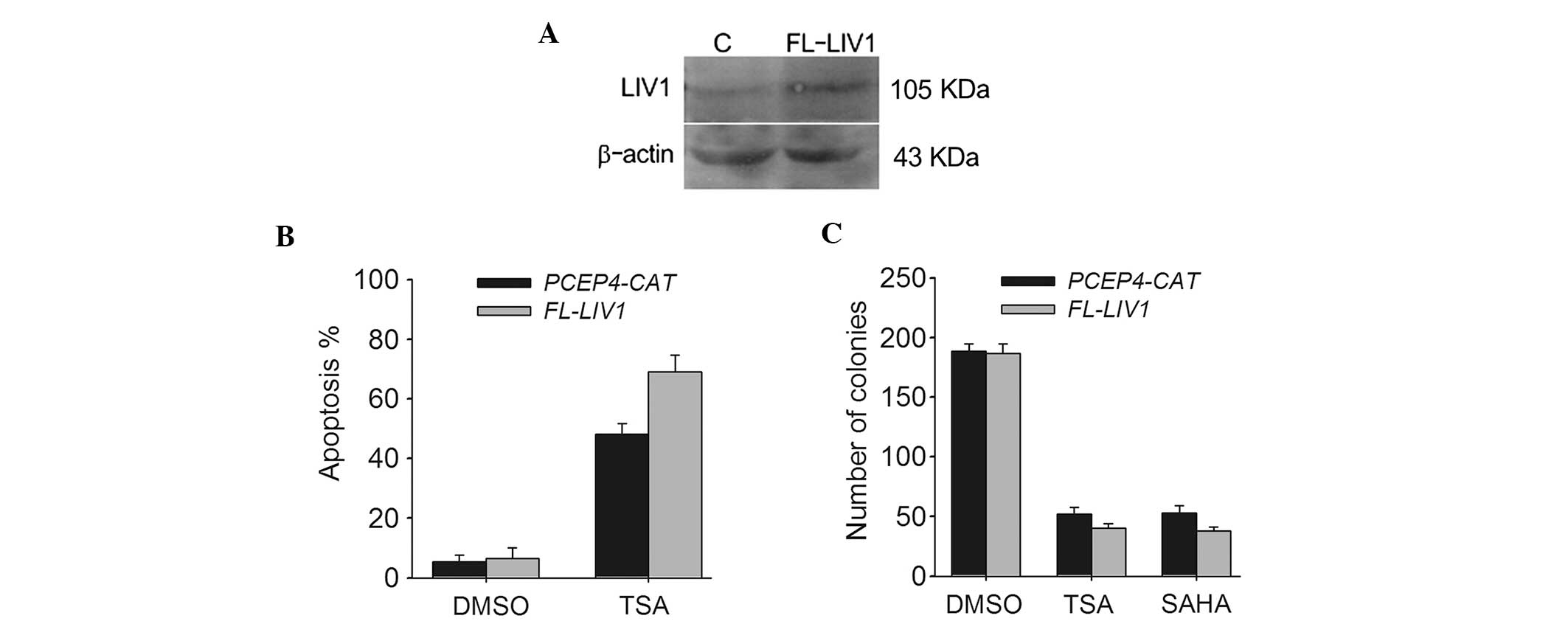
To further verify whether LIV1 is capable of
conferring a significant resistance to TSA-induced cell death in
ovarian cancer cells from a contrasting perspective, we conducted
LIV1 full-length plasmid FL-LIV1, which was confirmed to obviously
increase the level of LIV1 expression (Fig. 4A). A2780 cells stably transfected
with FL-LIV1 were much more sensitive to TSA-induced apoptosis than
cells stably transfected with PCEP4-CAT (69.02±4.5 vs. 48.01±3.7%;
P<0.05) (Fig. 4B). Moreover,
overexpression of LIV1 promoted TSA- and SAHA-induced cell death
and decreased the number of surviving colonies (Fig. 4C). Therefore, it appears that LIV1
modulates the killing efficacy of TSA and may be a critical
regulator of cell growth and death in ovarian cancer cells.
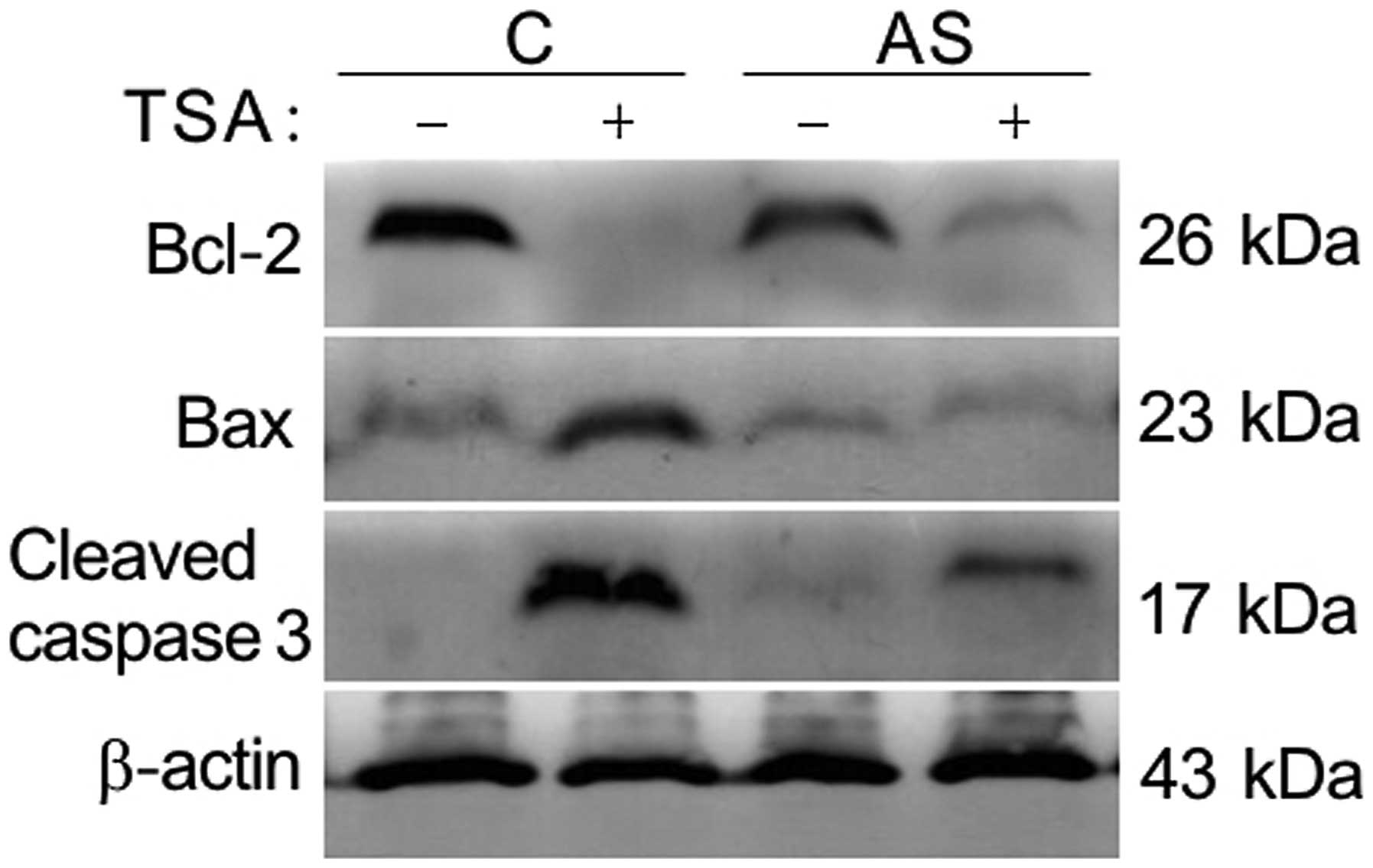
Knockdown of LIV1 protects cells from
TSA-induced apoptosis by affecting Bcl-2 family and activity of
caspase-3
Our previous data suggest that the inhibition of
TSA-induced apoptosis by knockdown of LIV1 might be associated with
its ability to disrupt intracellular zinc homeostasis (12). Zinc was shown to be a critical
regulator of cell growth and death. Previous findings indicate that
the mechanistic actions of zinc are shown through change in caspase
enzyme activities, as well as the direct alteration of apoptotic
regulator expression (16–19). As shown in Fig. 5, TSA induced apoptosis by decreasing
endogenous levels of Bcl-2, enhancing levels of Bax and cleavage of
procaspase-3. In contrast, the TSA-induced alteration mentioned
above could be significantly reversed by LIV-1 knockdown,
indicating that LIV1 plays a critical role rather than a
by-phenomenon in TSA-mediated apoptosis.
Discussion
Most patients with ovarian cancer have progressed to
advanced stages by the first clinical visit, and are not eligible
to be treated with surgery. Thus, these patients can only receive
chemotherapy with poor results. Drug resistance is the primary
cause of death in late-stage patients. The flood of new second line
drugs in recent years has provided many dramatic improvements in
anticancer therapy (20,21). Thus, development of new therapeutic
strategies and the search for novel genes with new mechanisms of
action that lead to drug resistance of ovarian cancer cells have
become the focus of current cancer research.
In our previous research, we conducted a functional
gene screen approach named suppression of mortality by antisense
rescue technique to identify the key genes responsible for the
tumor-selective killing of TSA. LIV1 was identified as a critical
mediator responsible for TSA-induced apoptosis. LIV1 belongs to a
new subfamily of Zrt-, Irt-like protein (ZIP) zinc transporters,
now termed the LIV1 subfamily of ZIP zinc transporters (LZT)
(22). Based on its amino acid
sequence and its cellular location on the plasma membrane, it has
been proposed as a putative zinc transporter involved in
maintaining intracellular zinc homeostasis (23). Previous investigations have
demonstrated that LIV1 expression is associated with small estrogen
receptor-positive tumors of which 92% show lymph node involvement,
and its expression may be both a suitable prognostic marker for
lymph node involvement and metastatic spread in steroid hormone
receptor-positive disease (24). In
breast cancer, high LIV1 protein expression is associated with a
better clinical outcome in patients with breast cancer. In
zebrafish gastrula organizer, LIV1 controls epithelial-mesenchymal
transition. Nevertheless, the biological function of the LIV1 gene
is still not well understood. Our findings presented here highlight
an essential role for LIV1 in drug resistance in ovarian cancer
cells which might extend our understanding of LIV1 in the
regulation of apoptosis in cancer.
The present study confirmed that LIV1 expression is
obviously induced by TSA treatment in ovarian cancer cells.
Knockdown of LIV1 protects ovarian cancer cells from TSA-induced
apoptosis and it is associated with the alteration of activity of
caspase-3 and the Bcl-2 family. It is well-known that the caspase
family is a cysteine protease family, among which the proteolysis
cascade reaction controls the development of cell apoptosis.
Caspase-3 is related to several events during the effector phase of
apoptosis and its activation serves as a common channel for
apoptosis pathways (25,26). This experiment confirmed that
knockdown of LIV1 could significantly decrease activated
casepase-3, eventually leading to irreversible drug resistance.
Furthermore, programmed cell death is a well-orchestrated process
regulated by multiple pro-apoptotic and anti-apoptotic genes,
particularly those of the Bcl-2 gene family. Bcl-2 is an integral
membrane protein located mainly on the outer membrane of
mitochondria. Overexpression of Bcl-2 prevents cells from
undergoing apoptosis in response to a variety of stimuli. In
contrast, Bax promotes apoptosis (27,28).
Our data are also consistent with previous findings that knockdown
of LIV1 increased Bcl-2 expression and decreased Bax expression.
Taken together, our findings have identified LIV1 as a novel target
responsible for sensivity of ovarian cancer cells to TSA. The novel
mechanism proposed here might have important clinical potential.
Given that LIV1 is a novel identified gene with undefined
functions, further animal experiments and clinical studies are
needed to determine their therapeutic effects in vivo, and
further characterization of LIV1 would aid in the development of
more effective protocols.
Acknowledgements
The present study was supported by a grant from the
National Natural Science Foundation of China (no. 81101970); Ph.D.
Programs Foundation of the Ministry of Education of China (no.
20111107120009); and the Scientific Research Common Program of
Beijing Municipal Commission of Education (KM 201210025020).
References
|
1
|
Jemal A, Bray F, Center MM, et al: Global
cancer statistics. CA Cancer J Clin. 61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hennessy BT, Coleman RL and Markman M:
Ovarian cancer. Lancet. 374:1371–1382. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zahedi P, Yoganathan R, Piquette-Miller M
and Allen C: Recent advances in drug delivery strategies for
treatment of ovarian cancer. Expert Opin Drug Deliv. 9:567–583.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Vecchione A, Belletti B, Lovat F, et al: A
microRNA signature defines chemoresistance in ovarian cancer
through modulation of angiogenesis. Proc Natl Acad Sci USA.
110:9845–9850. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
West AC and Johnstone RW: New and emerging
HDAC inhibitors for cancer treatment. J Clin Invest. 2:30–39. 2014.
View Article : Google Scholar
|
|
6
|
Slingerland M, Guchelaar HJ and Gelderblom
H: Histone deacetylase inhibitors: an overview of the clinical
studies in solid tumors. Anticancer Drugs. 25:140–149. 2014.
View Article : Google Scholar
|
|
7
|
Højfeldt JW, Agger K and Helin K: Histone
lysine demethylases as targets for anticancer therapy. Nat Rev Drug
Discov. 12:917–930. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Marchion D and Munster P: Development of
histone deacetylase inhibitors for cancer treatment. Expert Rev
Anticancer Ther. 7:583–598. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Marks PA and Breslow R: Dimethyl sulfoxide
to vorinostat: development of this histone deacetylase inhibitor as
an anticancer drug. Nat Biotechnol. 25:84–90. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rasheed WK, Johnstone RW and Prince HM:
Histone deacetylase inhibitors in cancer therapy. Expert Opin
Investig Drugs. 16:659–678. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fakih MG, Pendyala L, Fetterly G, et al: A
phase I, pharmacokinetic and pharmacodynamic study on vorinostat in
combination with 5-fluorouracil, leucovorin, and oxaliplatin in
patients with refractory colorectal cancer. Clin Cancer Res.
15:3189–3195. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ma XL, Ma QF, Liu J, et al: Identification
of LIV1, a putative zinc transporter gene responsible for
HDACi-induced apoptosis, using a functional gene screen approach.
Mol Cancer Ther. 8:3108–3116. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yamashita S, Miyagi C, Fukada T, et al:
Zinc transporter LIV-1 controls epithelial-mesenchymal transition
in zebrafish gastrula organizer. Nature. 429:298–302. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kasper G, Weiser AA, Rump A, et al:
Expression levels of the putative zinc transporter LIV-1 are
associated with a better outcome of breast cancer patients. Int J
Cancer. 117:961–973. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ma XL, Liu J, Wu J, et al: Synergistic
killing effect between vorinostat and target of CD146 in malignant
cells. Clin Cancer Res. 16:5165–5176. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sankavaram K, Chong L, Bruno RS and Freake
HC: Zinc status alters growth and oxidative stress responses in rat
hepatoma cells. Nutr Cancer. 66:104–116. 2014. View Article : Google Scholar
|
|
17
|
Nygaard SB, Larsen A, Knuhtsen A, et al:
Effects of zinc supplementation and zinc chelation on in vitro
β-cell function in INS-1E cells. BMC Res Notes. 7:842014.
View Article : Google Scholar
|
|
18
|
Zhang X, Zhao Y, Chu Q, et al: Zinc
modulates high glucose-induced apoptosis by suppressing oxidative
stress in renal tubular epithelial cells. Biol Trace Elem Res.
158:259–267. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chimienti F, Aouffen M, Favier A and Seve
M: Zinc homeostasis-regulating proteins: new drug targets for
triggering cell fate. Curr Drug Targets. 4:323–338. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jayson GC, Kohn EC, Kitchener HC and
Ledermann JA: Ovarian cancer. Lancet. 13:62146–62147. 2014.
|
|
21
|
Marcus CS, Maxwell GL, Darcy KM, et al:
Current approaches and challenges in managing and monitoring
treatment response in ovarian cancer. J Cancer. 5:25–30. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Taylor KM and Nicholson RI: The LZT
proteins; the LIV-1 subfamily of zinc transporters. Biochim Biophys
Acta. 1611:16–30. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Taylor KM: LIV-1 breast cancer protein
belongs to a new family of histidine-rich membrane proteins with
potential to control intracellular Zn2+ homeostasis.
IUBMB Life. 49:294–253. 2000. View Article : Google Scholar
|
|
24
|
Taylor KM, Morgan HE, Johnson A, et al:
Structure-function analysis of LIV-1, the breast cancer-associated
protein that belongs to a new subfamily of zinc transporters.
Biochem J. 375:51–59. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hensley P, Mishra M and Kyprianou N:
Targeting caspases in cancer therapeutics. Biol Chem. 394:831–843.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Maghsoudi N, Zakeri Z and Lockshin RA:
Programmed cell death and apoptosis-where it came from and where it
is going: from Elie Metchnikoff to the control of caspases. Exp
Oncol. 34:146–152. 2012.PubMed/NCBI
|
|
27
|
Ola MS, Nawaz M and Ahsan H: Role of Bcl-2
family proteins and caspases in the regulation of apoptosis. Mol
Cell Biochem. 351:41–58. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Thomas S, Quinn BA, Das SK, et al:
Targeting the Bcl-2 family for cancer therapy. Expert Opin Ther
Targets. 17:61–75. 2013. View Article : Google Scholar
|