Introduction
The essential characteristic of tumors is rapid
growth; therefore, tumors need to generate a large number of new
blood vessels to provide nutrients. Nonetheless, compared with
normal tissue, tumor angiogenesis is disarranged and functions
poorly (1). The rapid tumor growth
and the physiological characteristics of the tumor vasculature mean
that the rate of formation of blood vessels providing the energy
substances is unable to catch up with the rate of increase in tumor
volume, such that many tumors exist in a low glucose environment.
The concentration of glucose in colon and stomach tumor tissue is
only 0.12 and 0.4 mM, respectively (2). Tumors can survive in harsh
environments, such as poor glucose and oxygen depletion (3). In the case of glucose deficiency,
tumors in vivo adjust their own state to adapt to the
environment and obtain more nutrition. By contrast, lack of glucose
in cultured tumor cells in vitro does not support the
survival of tumor cells. Therefore, to explore how tumors survive
glucose depletion in vivo is important.
Cancer cells preferentially use the glycolysis
pathways for energy generation, even in the presence of oxygen, the
so called ‘aerobic glycolysis’, as first proposed by Warburg
(4). Glycolysis is far less
efficient than oxidative phosphorylation in terms of ATP
generation; therefore, cancer cells exhibit abnormally high
glycolytic rates to maintain energy homeostasis (5). Such dysregulated metabolism in cancer
cells also leads to the accumulation of the metabolic product of
glycolysis, lactic acid, in solid tumors. Many measurements have
been made to determine the level of tumor lactate and significant
variations have been found, with the average ranging from 7 to 10
mM/g and a maximum of up to 25.9 mM/g (6,7). In
contrast to tumor hypoxia, tumor glycolysis and lactate biology
have received little scientific attention for many years. However,
findings concerning the overexpression of glycolysis-related genes
in 70% of all human cancers worldwide and the exploitation of
increased glucose uptake of cancer cells for tumor diagnostics by
positron emission tomography (PET) with
18F-fluorodeoxyglucose (FDG), have contributed to the
topic experiencing a renaissance. This may lead to improvement of
cancer diagnosis and therapeutic follow-up in a clinical
setting.
As an important part of the tumor microenvironment,
studies of lactate were justified for the following reasons.
Extracellular lactate inhibits the differentiation of monocytes to
dendritic cells (DCs) and inactivates cytokine release from DCs
(8) and cytotoxic T cells (9), the key players in antitumoral
response. The addition of exogenous lactate led to a
concentration-dependent increase in random migration of various
cancer cell lines (10). Lactate
concentrations are positively correlated with radioresistance
(11). This reprogramming is
necessary for the growth and survival of tumors in stress
conditions (12).
In the present study, we found that lactate could
rescue cancer cells from glucose starvation-induced death.
Furthermore, we explored the mechanism of the role of lactic acid
in the process of tumor adaptation to glucose deficiency. We found
that lactate rescues cancer cells from glucose starvation-induced
cell death by regulating the Akt/mammalian target of rapamycin
(mTOR)/B-cell lymphoma 2 (Bcl-2) signaling pathway. These data
suggest that lactate is an important determinant of the sensitivity
of tumors to glucose starvation, and reducing lactate or inhibiting
the Akt/mTOR/Bcl-2 signaling pathway may influence the response of
cancers to glucose starvation.
Materials and methods
Cell lines and cell culture
A549, H1299, PC3, DU145 and U87-MG cell lines were
purchased from the Cell Bank of the Type Culture Collection of the
Chinese Academy of Sciences. A549 and U87-MG were cultured in
complete Dulbecco’s modified Eagle’s medium (DMEM, cat. no.
12430-054), with 10% fetal bovine serum (cat. no. 10100-147) (both
from Gibco, USA), 100 U/ml penicillin and 100 μg/ml streptomycin.
DU145 and H1299 were cultured in RPMI-1640 medium (cat. no.
11875-093; Gibco) supplemented as above. PC3 cells were cultured in
F-12 medium (cat. no. 21700-075; Gibco) supplemented as above. The
glucose concentration in the medium was 25 mM, unless otherwise
stated. For the glucose-starvation experiment, we mixed no-glucose
DMEM (cat. no. 11966-025) or RPMI-1640 (cat. no. 11879-020) (both
from Gibco) with the complete medium mentioned above in a certain
proportion to make the glucose concentration 5 mM. We added sodium
L-lactate (cat. no. 71718) or L-lactic acid (cat. no. L1750) (both
from Sigma-Aldrich, USA) or HCl into the medium to create different
culture environments with different lactate concentrations and
pHs.
Cell viability assay
Cells (104/well) were seeded in 24-well
plates in complete medium with 25 mM glucose, 24 h before the
experiment. The following day we changed the medium to one
containing no glucose. Meanwhile, we added a different
concentration of lactate into the medium at the start of the
experiment. From day 2 to 12, we counted the live cells every 2
days; 3 wells/day for each culture environment.
Transfection
A small interfering RNA (siRNA) targeting Bcl-2
(cat. no. GS596; Qiagen, Germany) was transfected into cells to
block its function. We used Lipofectamine 2000 (cat. no. 11668019;
Invitrogen, USA) as the transfection reagent and a negative control
siRNA (cat. no. SI03650318; Qiagen). Forty-eight hours after
transfection, we collected the mRNA and protein of the transfected
cells. To assess knockdown efficiency, we analyzed the mRNA level
using real-time PCR (LightCycler 480; Roche, Switzerland) and the
level of protein by western blotting (Mini-PROTEAN®
165–8004; Bio-Rad, USA). Both values were normalized to the
expression of β-actin. Experiments were performed between 24 and 72
h after transfection.
Analysis of cell metabolism using a
Seahorse Bioscience XF24 instrument
The oxygen consumption rate (OCR) measurements of
cells were performed using a Seahorse Bioscience XF24 instrument
(Seahorse Bioscience, Billerica, MD, USA). Before running the
experiment, the growth medium was removed and the cells were washed
with PBS containing Ca2+/Mg2+ (pH 7.4), which
was then aspirated and replaced with 700 ml of reduced serum (RS)
buffer [CaCl2 (1.8 mM), MgCl2 (0.6 mM),
KH2PO4 (0.5 mM), KCl (5.33 mM),
Na2HPO4 (0.5 mM), NaCl (130 mM), glucose (5.6
mM)], glutamax, minimum essential medium (MEM) amino acid solution,
MEM nonessential amino acids, MEM vitamin solution,
penicillin/streptomycin, 1% bovine serum albumin (BSA, factor V
fatty acid free), 1% FBS and insulin (100 nM). All components,
except FBS and insulin, were combined before filter sterilization.
Following the addition of FBS and insulin (usually 24–48 h
pre-experiment), the RS buffer was warmed to 37°C and the pH
adjusted to 7.4. The template for testing was set up as follows.
Measurements were performed every 5 min, repeating metabolic
measurements 3–4 times per condition for statistical analyses. The
5-min cycle included a 2-min mix period, a 1-min wait and a 2-min
measuring time. After measurements of baseline activity, oligomycin
(an ATP coupler) was injected, rates were measured, carbonylcyanide
p-trifluoromethoxyphenylhydrazone (FCCP) (an electron
transport chain accelerator) was injected, and finally rotenone and
antimycin (mitochondrial inhibitors) were injected, followed by a
final rate measurement.
Measurement of extracellular lactate
levels
Cells were seeded onto plates and allowed to grow in
medium with 25 mM glucose for attachment overnight. The next day,
we replaced the medium with fresh medium with 0 mM glucose at time
zero. From time zero, every 24 h, we collected the medium samples
and counted the number of cells in the same plate. The
concentrations of lactate in the medium samples were measured with
lactate reagent (cat. no. P0000024; CMA Microdialysis, Sweden). The
results were normalized to the number of the cells in each
sample.
Measurement of glucose starvation-induced
apoptosis
Cells were placed in 6-well plates and incubated for
24 h before the experiment. Cells in the control group were
cultured in complete medium containing 25 mM glucose. Cells in the
treated group were cultured in complete medium containing 0 mM
glucose with different lactate contents. After 48 h of treatment,
cells were trypsinized, centrifuged and resuspended in binding
buffer with Annexin V-FITC and propidium iodide (PI) from the Dead
Cell Apoptosis Kit #2 (cat. no. V13241; Life Technologies, USA).
Stained cells were incubated for 15 min at room temperature in the
dark. Flow cytometry was used to analyze the stained cells,
measuring the fluorescence emission at 530 and 575 nm using 488 nm
excitation.
Western blotting and antibodies
Treated cells were lysed using RIPA lysis buffer
with 1% phenylmethanesulfonyl fluoride (PMSF). Cell lysates were
separated through a 10% SDS gel and blotted onto nitrocellulose
membranes. The membranes were blocked with 5% non-fat dry milk in
PBST at room temperature for 1 h and incubated separately with
primary antibodies against Akt (cat. no. 4685), phospho-Akt
(Thr308, cat. no. 13038) (both from Cell Signaling Technology,
USA), phospho-Akt (Ser473, cat. no. ab66138), PTEN (cat. no.
ab32199), Mcl-1 (cat. no. ab114016), Bcl-2 (cat. no. ab117115),
Bcl-xL (cat. no. ab32370), phospho-mTOR (Ser2448, cat. no. ab84400)
(all from Abcam, UK) or β-actin (cat. no. A1978; Sigma-Aldrich)
overnight at 4°C. The next day, the membranes were washed 3 times
with PBST and incubated with secondary antibodies for 1 h at room
temperature. After washing the membranes 3 times with PBST, the
membranes were scanned using an Odyssey Infrared Imaging System
(Li-COR Biosciences, USA).
Reagents
LY294002, perifosine and rapamycin were purchased
from Selleck Chemicals, USA (cat. nos. S1105, S1037 and S1039).
Insulin and resveratrol were purchased from Sigma-Aldrich (cat. no.
R5010). Insulin-like growth factor-1 (IGF-1) was purchased from
Invitrogen (cat. no. PHG0071).
Statistical analysis
Statistical analysis was performed using GraphPad
Prism 5.0 and Instat 3.1 packages (GraphPad Software, Inc., San
Diego, CA, USA). The results are expressed as the means ± standard
error (SE). Statistical differences between the groups were
compared using t-tests. P-values <0.05 were considered to
indicate statistically significant results.
Results
Lactate rescues cancer cells from glucose
starvation-induced cell death
To determine the effect of lactate on glucose
starvation, we exposed A549 cells to different culture conditions:
complete DMEM containing different sodium lactate concentrations
and without glucose. We found that in glucose-free conditions,
adding sodium lactate into the medium effectively prolonged the
survival times of the A549 cell line (Fig. 1A). At low doses, cell proliferation
and survival time increased with the increment of sodium lactate
concentration. The survival time of A549 cells in the presence of
20 mM sodium lactate was longer than that of any other
concentrations of sodium lactate. Indeed, A549 cells grown with
sodium lactate in glucose-free conditions survived for more than 2
weeks; even 1 month later there were still viable cells. At sodium
lactate concentrations >20 mM, cell proliferation and survival
time decreased. Without sodium lactate, in A549 cells onset of cell
death started in 1–2 days and all the cells died after 4 days.
However, high concentrations of sodium lactate (such as 80 mM)
caused immediate death of A549 cells (Fig. 1B).
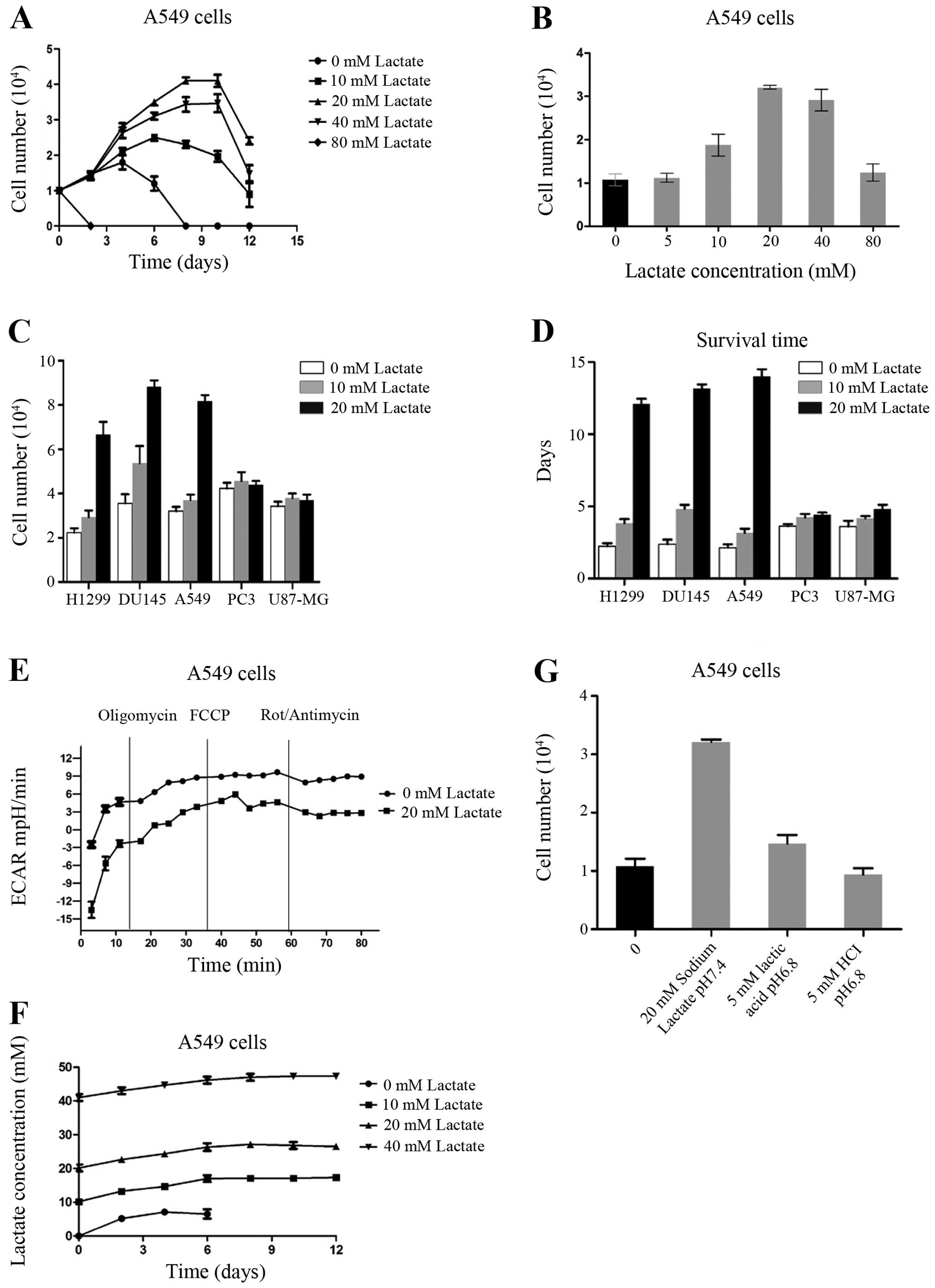 | Figure 1Lactate promotes cancer cell
resistance to glucose starvation. (A) Cell growth curves of A549
cells, which were cultured in glucose-free medium with different
concentrations of lactate. (B) The numbers of A549 cells were
counted 3 days after culture in glucose-free medium with different
concentrations of lactate. (C) The numbers of H1299, DU145, A549,
PC3 and U87-MG cells counted 3 days after culture in glucose-free
medium with different concentrations of lactate. (D) Survival of
H1299, DU145, A549, PC3 and U87-MG cells after culture in
glucose-free medium with different concentrations of lactate. (E)
The oxygen consumption rate (OCR) of A549 cells with or without
lactate was measured at baseline and continuously throughout the
experimental period and in the presence of the indicated drugs:
oligomycin (1 μg/ml), carbonylcyanide
p-trifluoromethoxyphenylhydrazone (FCCP) (1 μM), rotenone (1
μM) plus antimycin A (1 μM). (F) The lactate concentration in the
medium was detected under the same experimental conditions as in A.
(G) The numbers of A549 cells were counted 3 days after culture in
different environments: no glucose, no glucose + 20 mM sodium
lactate (pH 7.4), no glucose + 5 mM lactic acid (pH 6.8), and no
glucose + 5 mM hydrochloric acid (pH 6.8). Data are mean ± standart
error (SE), n=3. |
We also confirmed the above phenomena in other types
of tumor cell lines. We cultured four human cancer cell lines
derived from the brain (U87-MG), prostate (PC3 and DU145), and lung
(H1299) in no-glucose cultivation conditions with various lactate
concentrations. The addition of sodium lactate (20 mM) effectively
prolonged the survival time of all 5 types of tumor cells. We then
compared the proliferation and survival time between all 5 types of
cancer cells in the presence of sodium lactate. We found that
DU145, A549 and H1299 cell proliferation activity was stronger than
PC3 and U87-MG cells (Fig. 1C), and
their survival times were longer than those of the PC3 and U87-MG
cell lines (Fig. 1D).
Some studies have reported that tumor cells have the
ability to take up lactate and utilize it as an energy source via
oxidative phosphorylation. Determination of dissolved oxygen in the
culture of cells monitored in the Seahorse Bioscience XF24
instrument showed that, upon addition of lactate, oxidative
phosphorylation levels did not change significantly (Fig. 1E). In addition, we examined the
lactate concentrations in the cell culture; the extracellular
lactate concentrations did not change significantly over the course
of the experiment (Fig. 1F). These
results suggest that under deprivation of glucose, lactate was not
used as a substrate for energy metabolism.
Cell secretion of lactic acid would affect the value
of pH in the extracellular environment. To confirm whether the
acidic environment plays a role in tumor cell survival in a
glucose-free environment, we cultured A549 cells separately in the
following 4 types of environment: no glucose, no glucose + 20 mM
sodium lactate (pH 7.4), no glucose + 20 mM lactic acid (pH 6.8),
no glucose + 20 mM hydrochloric acid (pH 6.8). After 72 h of
culture, we found that adding hydrochloric acid to the tumor cells
in glucose-free environment had no significant effect on survival,
adding lactic acid had some effect, and adding lactate had the most
significant effect (Fig. 1G). We
concluded that tumor cell survival in a glucose-free environment
was mainly a function of lactate itself, and had little to do with
the acidic environment.
Effect of activation of Akt signaling on
the role of lactate in tumor cell survival in glucose-free
conditions
In the above experiments, we found that sodium
lactate had different effects on the survival of the different cell
lines in glucose-free conditions. Accordingly, we divided the five
cancer cell lines into 2 groups: a lactate-sensitive group,
including DU145, A549 and H1299; and a lactate-insensitive group,
including PC3 and U87-MG. To explore why lactate had different
effects on the survival of the 2 groups of cells in glucose-free
conditions, we compared the genetic background of the 2 groups of
cells. We found that the 2 lactate insensitive cell lines, PC3 and
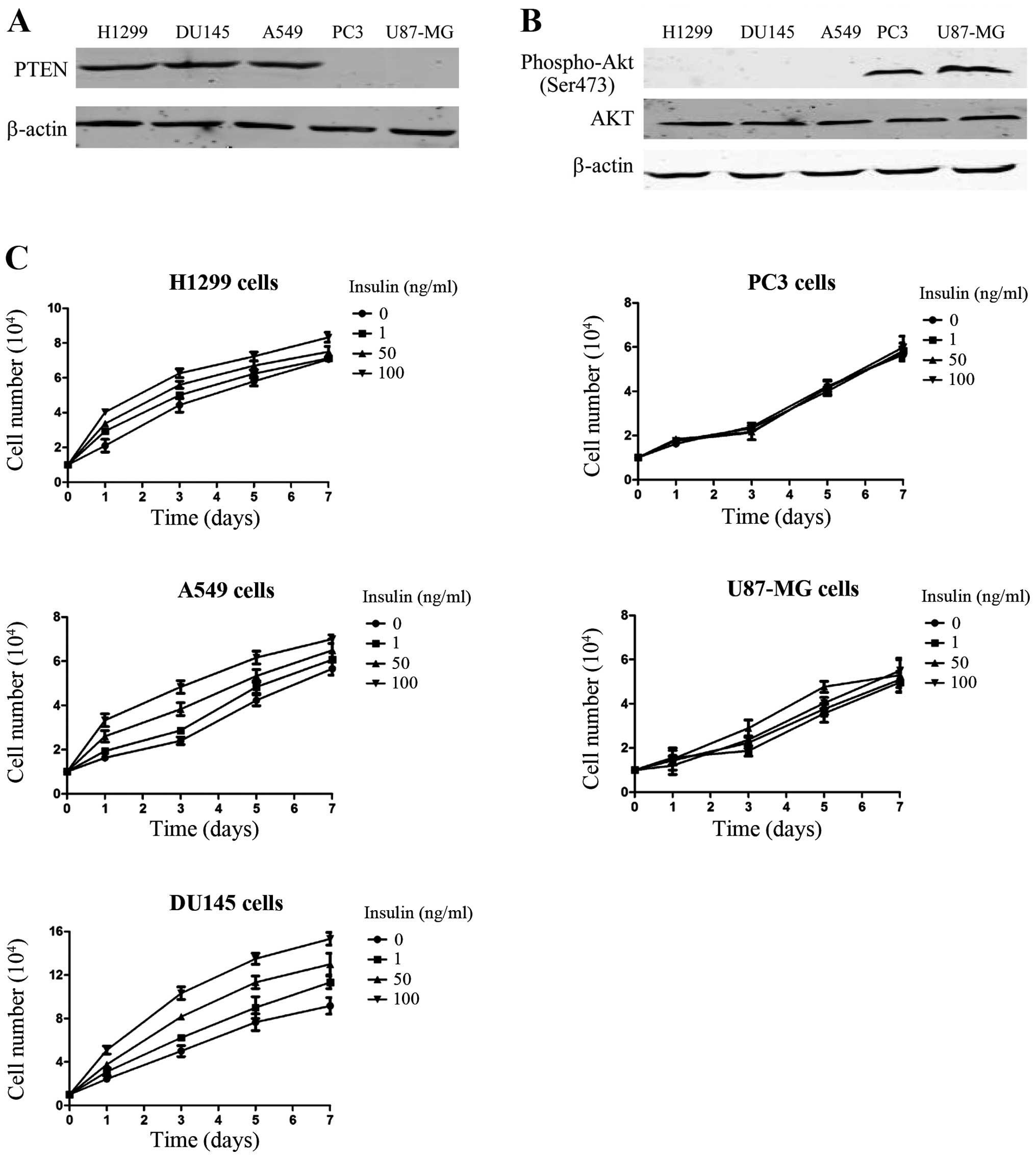
U87-MG, had lost PTEN activity (phosphatase and tensin homolog
deleted on chromosome 10) (Fig.
2A). PTEN is a tumor-suppressor gene with phosphatase activity,
which is involved in the negative regulation of the Akt signaling
pathway, where it blocks the activation of Akt and its downstream
effector molecules (13). The Akt
signaling pathway plays an important role in cell proliferation and
survival (14). Therefore, to
confirm the existence of activation of the Akt signaling pathway in
PC3 and U87-MG cell lines with PTEN deletion, we examined the
phosphorylation status of Akt in the 2 cell lines (Fig. 2B). The results showed that, compared
with the lactate-sensitive group, Akt phosphorylation in the
lactate-insensitive group increased, which would result in Akt
signaling pathway activation. We also compared the reactions of the
2 groups of cells to insulin, an activator of the Akt signaling
pathway (Fig. 2C). Insulin caused a
dose-dependent increase in cell numbers in the cells of the
lactate-sensitive group. By contrast, the growth of the cells in
the lactate-insensitive group was not affected by insulin. The
above results suggest that the state of the Akt signaling pathway
was a key factor that distinguished the role of lactate in cancer
cell proliferation and survival time in glucose-free
conditions.
Lactate induces Akt phosphorylation
through PI3K
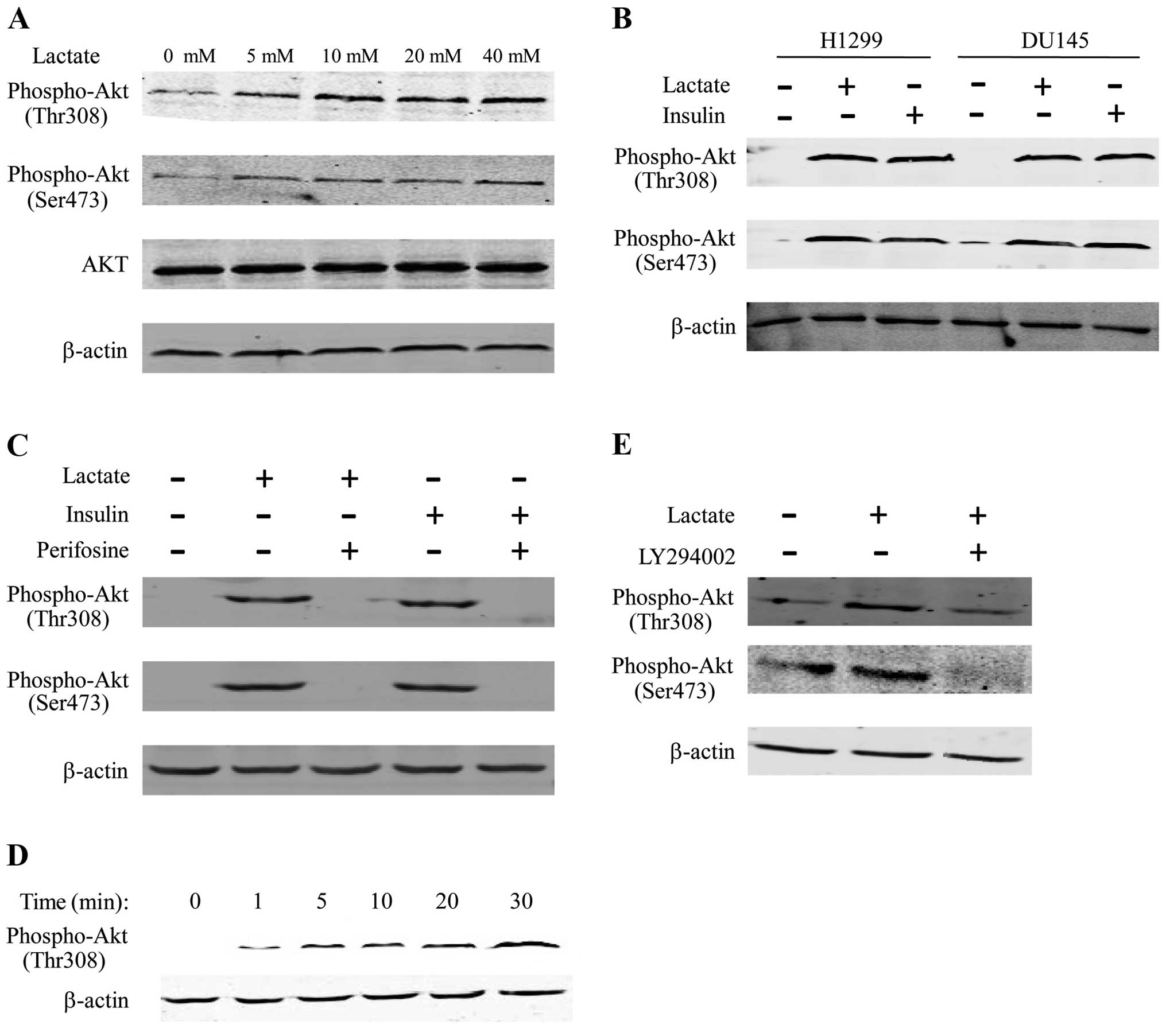
To analyze Akt signaling induced by lactate, A549
cells were cultured overnight in DMEM without serum, and the cells
were stimulated with sodium lactate for 30 min. Lysates of these
cells were analyzed by western blotting, and Akt activation was
measured by detection of Thr308 and Ser473 phosphorylated forms of
Akt. We used insulin to stimulate the serum-deprived cells as a
positive control for the activation of Akt. Under this environment,
we observed that Akt Thr308 and Ser473 phosphorylation increased in
the presence of lactate to a similar extent to that obtained with
insulin (Fig. 3A and C). We then
used perifosine (an inhibitor of Akt) to suppress the activation of
Akt. We found that Akt Thr308 and Ser473 phosphorylation by lactate
or insulin were suppressed by perifosine (Fig. 3C). We obtained the same result when
we used different cell lines, such as H1299 and DU145, indicating
that the differences in Akt phosphorylation were not specific to
the A549 cell line (Fig. 3B). Time
course experiments showed that Akt phosphorylation at Thr308 and
Ser473 phosphorylation sites were both rapid (1–5 min after
incubation with lactate), with maximum levels being achieved
between 10 and 30 min (Fig. 3D). To
estimate whether lactate induced Akt phosphorylation downstream of
PI3K, we used LY294002 (an inhibitor of PI3K). Akt Thr308 and
Ser473 phosphorylation in the presence of lactate were
substantially reduced in the presence of LY294002 (Fig. 3E). These results suggest that
lactate induced the activation of Akt significantly and rapidly
through Thr308 and Ser473 phosphorylation mediated by PI3K.
Lactate activates the Akt/mTOR/Bcl-2
signaling pathway to modulate the apoptotic response of cancer
cells to glucose starvation
Akt plays an important role in cell apoptosis and
survival in response to extracellular stimuli, such as insulin or
growth factors. Our previous experiments showed that the state of
the PI3K/Akt signaling pathway was a key factor to distinguish the
effect of lactate on cancer cell proliferation and survival in
glucose-free conditions, where lactate induces the activation of
Akt by Thr308 and Ser473 phosphorylation. We hypothesized that
lactate helps tumor cells to survive by activating Akt in
glucose-free conditions. To test the hypothesis, we cultured A549
cells with perifosine, an Akt inhibitor. We found that treatment
with perifosine inhibited the cell survival induced by lactate in
glucose-free conditions. In addition, treatment with an Akt
activator, IGF-1, increased A549 cell survival in glucose-free
conditions, but less effectively compared with lactate (Fig. 4A). These results showed that the
lactate increased tumor cell survival in glucose free conditions
through Akt activation.
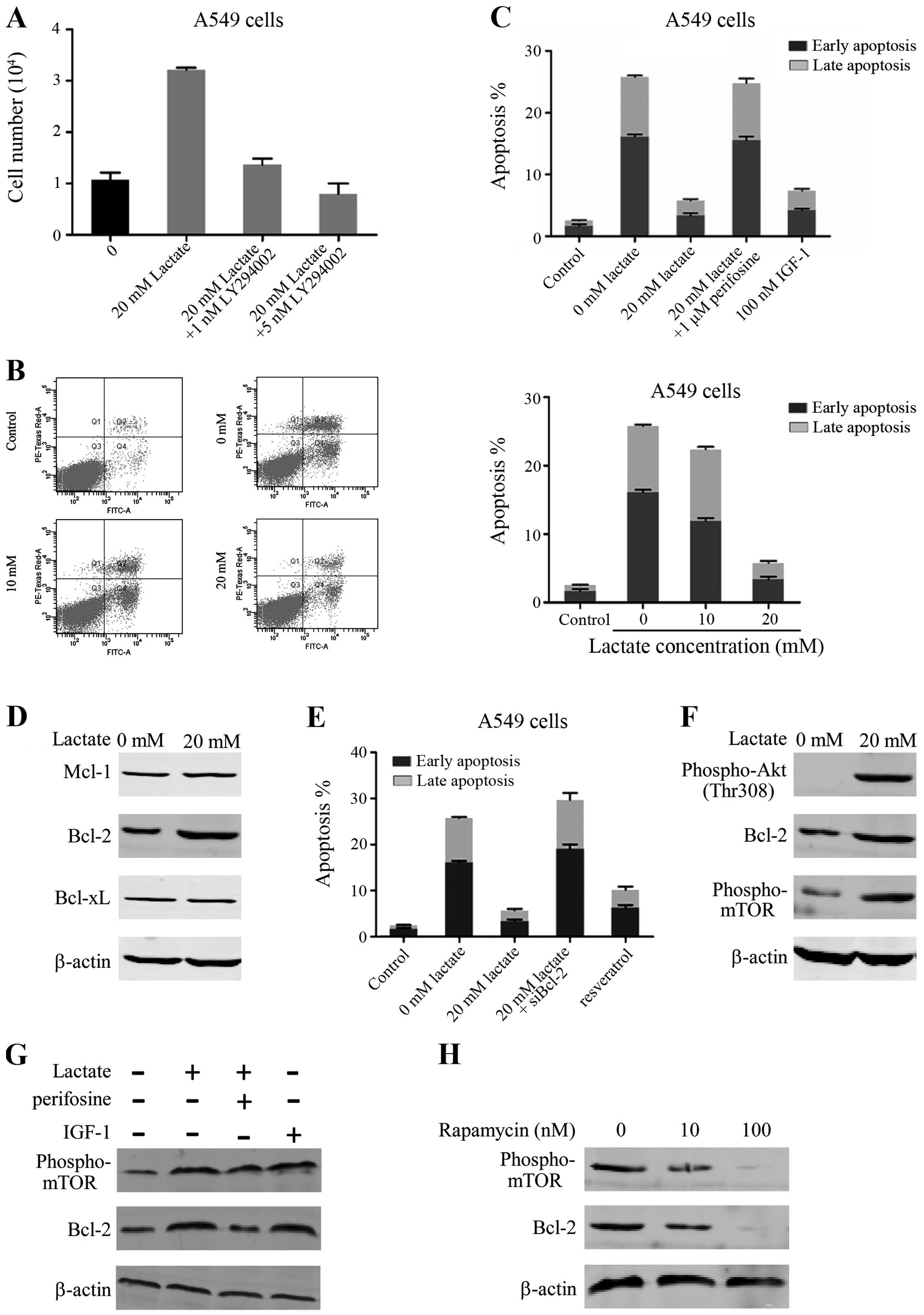 | Figure 4Reduced apoptosis by lactate during
glucose starvation is mediated through the activation of the
PI3K/Akt/mammalian target of rapamycin (mTOR)/B-cell lymphoma
(Bcl-2) signaling pathway. (A) The numbers of A549 cells were
counted 3 days after culture in glucose-free medium treated with
lactate (20 mM) or lactate (20 mM) + LY294002 (1 nM) or lactate (20
mM) + LY294002 (5 nM). (B) A549 cells were cultured in 25 mM
glucose medium or no glucose medium with lactate 48 h before
staining with Annexin V-FITC and propidium iodide (PI). Flow
cytometry to measure the fluorescence intensity at 530 and 575 nm,
using 488 nm excitation, allowed us to calculate the early and late
apoptosis rates of the cells. (C) The early and late apoptosis
rates of A549 cells were calculated after they were cultured in 25
mM glucose medium, no glucose medium with lactate (20 mM), lactate
(20 mM) + perifosine (1 μM) or IGF-1 (100 nM) for 48 h. (D) Protein
expression of Mcl-1, Bcl-2 and Bcl-xL in the A549 cell line treated
with or without lactate (20 mM) in glucose-free medium was examined
by western blotting. (E) The early and late apoptosis rates of A549
cells were calculated after they were cultured in 25 mM glucose
medium, no glucose medium with lactate (20 mM) + control small
interfering RNA (siRNA), lactate (20 mM) + siBcl-2 or resveratrol
(100 μM) for 48 h. (F) Protein expression of phospho-Thr308 Akt,
Bcl-2 and phospho-Ser2448 mTOR in the A549 cell line treated with
or without lactate (20 mM) in glucose-free medium was examined by
western blotting. (G) Protein expression of phospho-Ser2448 mTOR
and Bcl-2 in the A549 cell line treated with lactate (20 mM),
lactate (20 mM) + perifosine (1 μM), or IGF-1 (100 nM) was examined
by western blotting. (H) Protein expression of phospho-Ser2448 mTOR
and Bcl-2 in the A549 cell line treated with rapamycin was examined
by western blotting. β-actin was used as the normalization
control. |
Many studies have demonstrated that under the
metabolic stress of no glucose, tumor cells undergo significant
apoptosis. Thus, we investigated metabolic stress resistance to
apoptosis in tumor cells incubated with lactate (Fig. 4B). We cultured A549 cells in high
glucose (25 mM glucose), glucose-free and glucose-free with added
lactate conditions. Twenty-four hours later, cells were
trypsinized, centrifuged and resuspended in binding buffer with
Annexin V-FITC and PI. The stained cells were analyzed by flow
cytometry. We found that compared with high glucose conditions,
there were many apoptotic cells in the glucose-free environment.
There was an ~10.1- fold (P<0.001) increase in early apoptosis
and an ~12-fold (P<0.001) increase in late apoptosis in the
population of cells without glucose compared to the cells with high
glucose. The apoptosis rate in the culture with added lactate
culture was greatly reduced, and was close to that under the high
glucose cultivation. The above results showed that under
glucose-free conditions, a large number of A549 underwent
apoptosis, and the addition of lactate could prevent this
apoptosis, the mechanism of which will be further explored
below.
We checked the apoptosis rate of A549 cells and then
added perifosine or IGF-1. We found that treatment with perifosine
prevented the decrease in apoptosis induced by lactate in
glucose-free conditions, and that IGF-1 reduced the cell apoptosis
caused by no glucose (Fig. 4C). We
concluded that lactate prevented apoptosis in glucose-free
conditions through Akt activation.
Several studies have pointed out that the
anti-apoptotic Bcl-2 family is pivotal for cell survival under
metabolic stress. We found lactate can reduce tumor cell apoptosis
caused by the lack of glucose. We, therefore, examined whether
lactate affected the expression levels of anti-apoptotic Bcl-2
family proteins such as Mcl-1, Bcl-2 and Bcl-xL. A549 cells were
cultured with added lactate in glucose-free conditions. After 48 h,
lysates of these cells were analyzed by western blotting. Treatment
with lactate increased Bcl-2 levels, while Mcl-1 or Bcl-xL levels
were not affected by treatment with lactate (Fig. 4D). To confirm whether Bcl-2 plays a
major role in cell survival, we used a Bcl-2 targeting siRNA to
lower its protein expression. The results showed that treatment
with siBcl-2 prevented resistance to apoptosis induced by lactate
in glucose-free conditions. Furthermore, we observed that
resveratrol, a Bcl-2 activator, reduced the apoptosis of A549 cells
in glucose-free conditions, similarly to lactate (Fig. 4E). These findings revealed that the
upregulation of Bcl-2 by lactate is important for tumor cell
resistance to apoptosis in glucose-free conditions.
Akt plays a core role in promoting cell survival,
through activation of anti-apoptotic substances. Therefore, we
aimed to confirm whether Bcl-2 upregulation is mediated via
activation of the Akt/mTOR signaling pathway upon treatment with
lactate in glucose-free conditions. First, we found that in A549
cells treated with lactate, Akt was activated and the expression of
the Bcl-2 protein was increased, accompanied by mTOR activation
(Fig. 4F). To demonstrate that the
increase in the expression levels of Bcl-2 and mTOR were dependent
on the activation of Akt, we added perifosine and found that the
expression levels of Bcl-2 and mTOR were restored to the original
levels. Furthermore, the expression levels of Bcl-2 and mTOR were
upregulated in the presence of IGF-1 (Fig. 4G). Addition of rapamycin, an mTOR
inhibitor, blocked the increased expression of Bcl-2 (Fig. 4H). These results demonstrated that
Akt activation and mTOR upregulation by lactate in glucose-free
conditions led to the upregulation of Bcl-2.
Taking all the above results into consideration, we
conclude that lactate, through activation of Akt by phosphorylation
mediated by PI3K, activates mTOR and further increases the
expression of anti-apoptotic protein Bcl-2, to help tumor cells
resist apoptosis caused by glucose starvation.
Discussion
Reprogramming energy metabolism is a hallmark of
cancer (5). Warburg (4) first observed that even in cases with
an adequate oxygen supply, tumors still preferred to utilize
glucose via glycolysis. Compared with oxidative phosphorylation,
glycolysis is a low efficiency method of energy production;
therefore, compared with normal tissue, tumors often require more
glucose. The clinical diagnosis of cancer by PET with a
radiolabeled analog of glucose (FDG) as a reporter, a widely used
method, is possible as tumor cells have increased glucose uptake
(15). These changes in tumor
metabolic patterns, increased glucose uptake and glycolysis as the
main production method, eventually lead to the accumulation of
lactate, the end product of glycolysis, in tumors. Several studies
have reported increased lactate levels within tumors, which
reflects the high metastasis rate and poor prognosis in human
cervical cancers (16), human head
and neck cancers (17), human
rectal adenocarcinomas (18), human
hepatocellular carcinoma (19) and
non-small cell lung cancers (20).
Some studies have suggested that lactate could be used as an energy
source by oxidative phosphorylation to generate ATP (21,22).
Lactate can also be used as a signaling molecule in tumor cells
(23). Lactate can produce the
promotion of VEGF in wound healing (24), and lactate is also sufficient to
instigate signals for angiogenesis (25).
In solid tumors, along with the rapid growth of the
tumor, the development of blood vessels within is incomplete, which
leads to certain areas of the tumor to suffer glucose deficit. In
blood-rich regions, aerobic glycolysis consumes a large amount of
glucose to produce lactate, whereas the lactate in the blood-poor
regions of tumor cells plays an important role. In the present
study, in the absence of glucose, added lactate in culture
significantly prolonged the survival of A549 cells in a
concentration-dependent manner. This result was consistent with
those of the Wu et al (26).
Subsequent experiments in different cell lines confirmed the role
of lactate. Some studies reported that lactate could be used as an
energy substrate to produce ATP by tumor cells through oxidative
phosphorylation; we determined whether under no glucose conditions,
lactate could be used as an energy substrate. The results showed no
significant changes in oxygen consumption and or any lactate
consumption. Thus, under glucose-free conditions, lactate is not an
energy substrate. We also ruled out an acidic environment in
glucose-free conditions as having any influence on maintaining
tumor cell growth.
Notably, lactate had different effects on different
cell lines, allowing cell lines to be divided into sensitive and
insensitive lactate groups. We compared the genetic backgrounds of
the five cell lines and found that the cell lines insensitive to
lactate lacked PTEN function. Insulin-mediated stimulation of
growth confirmed the activation of the Akt signaling pathway in the
insensitive cell lines. Therefore, to further explore the mechanism
of lactate, we focused on the Akt signaling pathway. Akt signaling
pathway is an important signaling pathway in tumor cell survival
and development. It is activated by upstream signaling molecules,
such as growth factors, and is then further regulated by downstream
molecules to participate in the occurrence and development of
tumors (14). The activation of the
PI3K/Akt pathway could help tumor resistance under dietary
deficient conditions (27). Western
blotting showed that lactate activated Akt via the rapid
phosphorylation at Thr308 and Ser473 mediated by PI3K. Lactate can
be used as a signaling molecule in the regulation of certain
signaling pathways. For example, lactate was found to upregulate
the transcription of 673 genes in L6 cells and was further involved
in mitochondrial biogenesis (28).
In tumors, lactate, in the presence of oxygen, stimulated the
expression of HIF-1α and upregulated various hypoxia-inducible
dependent genes (29). Lactate
could increase TLR4 signaling and NF-κB pathway-mediated gene
transcription in macrophages (30).
Lactate increased the level of TGF-β2 in glioma (31). Interestingly, further experiments
demonstrated that lactate helped tumor cell survival in
glucose-free conditions through activation of Akt by
phosphorylation.
Apoptosis tends to occur when cells are under
metabolic stress because of lack of glucose. We found significant
apoptosis of A549 cells under glucose-free environment conditions,
and the addition of lactate prevented this apoptosis. The Bcl-2
family of anti-apoptotic proteins are key regulators of cell
apoptosis (32). We found that
lactate increased the expression of Bcl-2, and downregulation of
Bcl-2 protein expression using an siRNA reduced the resistance to
apoptosis induced by lactate in glucose-free conditions.
The PI3K/Akt signaling pathway plays a key role in
inhibiting apoptosis, thereby promoting cell proliferation. The
activation of Akt can act directly on apoptosis-related proteins to
regulate apoptosis. Activation of Akt induces phosphorylation of
caspase-9 at the Ser196 site, which inhibits apoptosis (33). The PI3K/Akt signaling pathway can
directly or indirectly affect the functions of transcription
factors to regulate cell survival. Akt can inhibit the enzyme IκBα
that phosphorylates NF-κB, whereas unphosphorylated NF-κB in the
nucleus regulates anti-apoptotic gene transcription (34). Akt can prevent the mitochondrial
release of cytochrome c and apoptosis-inducing factors,
contributing to apoptosis resistance (35). In the present study, we confirmed
that lactate acts via the PI3K/Akt pathway to regulate Bcl-2,
inhibiting apoptosis caused by the lack of glucose.
In conclusion, lactate helps tumor cells to resist
apoptosis caused by glucose starvation. Lactate, through PI3K,
activates Akt by phosphorylation, which activates mTOR and further
increases the expression of anti-apoptotic protein Bcl-2. This
study indicates that treatments targeting lactate could more
effectively inhibit the survival of tumors.
Acknowledgements
The present study was supported by research grants
from the National Basic Research Program of China (973 Program,
2012CB932600), Significant New Drug Creation Five-Year Plan Special
Science and Technology Major (2012ZX09506001-005), the National
Natural Science Foundation of China (nos. 30830038, 81071180 and
30970842), the Key Project of Science and Technology Commission of
Shanghai Municipality (no. 10JC1410000), and the Shanghai Leading
Academic Discipline Project (project no. S30203).
References
|
1
|
Vaupel P: Tumor microenvironmental
physiology and its implications for radiation oncology. Semin
Radiat Oncol. 14:198–206. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hirayama A, Kami K, Sugimoto M, Sugawara
M, Tok N, Onozuka H, Kinoshita T, Saito N, Ochiai A, Tomita M,
Esumi H and Soga T: Quantitative metabolome profiling of colon and
stomach cancer microenvironment by capillary electrophoresis
time-of-flight mass spectrometry. Cancer Res. 69:4918–4925. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hahnfeldt P, Panigrahy D, Folkman J and
Hlatky L: Tumor development under angiogenic signaling: a dynamical
theory of tumor growth, treatment response, and postvascular
dormancy. Cancer Res. 59:4770–4775. 1999.PubMed/NCBI
|
|
4
|
Warburg O: On respiratory impairment in
cancer cells. Science. 124:269–270. 1956.PubMed/NCBI
|
|
5
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Quennet V, Yaromina A, Zips D, Rosner A,
Walenta S, Baumann M and Mueller-Klieser W: Tumor lactate content
predicts for response to fractionated irradiation of human squamous
cell carcinomas in nude mice. Radiother Oncol. 81:130–135. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Brizel DM, Schroeder T, Scher RL, Walenta
S, Clough RW, Dewhirst MW and Mueller-Klieser W: Elevated tumor
lactate concentrations predict for an increased risk of metastases
in head-and-neck cancer. Int J Radiat Oncol Biol Phys. 51:349–353.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gottfried E, Kunz-Schughart LA, Ebner S,
Mueller-Klieser W, Hoves S, Andreesen R, Mackensen A and Kreutz M:
Tumor-derived lactic acid modulates dendritic cell activation and
antigen expression. Blood. 107:2013–2021. 2006. View Article : Google Scholar
|
|
9
|
Fischer K, Hoffmann P, Voelkl S,
Meidenbauer N, Ammer J, Edinger M, Gottfried E, Schwarz S, Rothe G,
Hoves S, Renner K, Timischl B, Mackensen A, Kunz-Schughart L,
Andreesen R, Krause SW and Kreutz M: Inhibitory effect of tumor
cell-derived lactic acid on human T cells. Blood. 109:3812–3819.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Goetze K, Walenta S, Ksiazkiewicz M,
Kunz-Schughart LA and Mueller-Klieser W: Lactate enhances motility
of tumor cells and inhibits monocyte migration and cytokine
release. Int J Oncol. 39:453–463. 2011.PubMed/NCBI
|
|
11
|
Sattler UG, Meyer SS, Quennet V, Hoerner
C, Knoerzer H, Fabian C, Yaromina A, Zips D, Walenta S, Baumann M
and Mueller-Klieser W: Glycolytic metabolism and tumour response to
fractionated irradiation. Radiother Oncol. 94:102–109. 2010.
View Article : Google Scholar
|
|
12
|
Le A, Cooper CR, Gouw AM, Dinavahi R,
Maitra A, Deck LM, Royer RE, Vander Jagt DL, Semenza GL and Dang
CV: Inhibition of lactate dehydrogenase A induces oxidative stress
and inhibits tumor progression. Proc Natl Acad Sci USA.
107:2037–2042. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sharrard RM and Maitland NJ: Regulation of
protein kinase B activity by PTEN and SHIP2 in human
prostate-derived cell lines. Cell Signal. 19:129–138. 2007.
View Article : Google Scholar
|
|
14
|
Osaki M, Oshimura M and Ito H: PI3K-Akt
pathway: its functions and alterations in human cancer. Apoptosis.
9:667–676. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gambhir SS, Czernin J, Schwimmer J,
Silverman DH, Coleman RE and Phelps ME: A tabulated summary of the
FDG PET literature. J Nucl Med. 42(Suppl 5): 1S–93S.
2001.PubMed/NCBI
|
|
16
|
Walenta S, Wetterling M, Lehrke M,
Schwickert G, Sundfør K, Rofstad EK and Mueller-Klieser W: High
lactate levels predict likelihood of metastases, tumor recurrence,
and restricted patient survival in human cervical cancers. Cancer
Res. 60:916–921. 2000.PubMed/NCBI
|
|
17
|
Brizel DM, Schroeder T, Scher RL, Walenta
S, Clough RW, Dewhirst MW and Mueller-Klieser W: Elevated tumor
lactate concentrations predict for an increased risk of metastases
in head-and-neck cancer. Int J Radiat Oncol Biol Phys. 51:349–353.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Walenta S, Chau TV, Schroeder T, Lehr HA,
Kunz-Schughart LA, Fuerst A and Mueller-Klieser W: Metabolic
classification of human rectal adenocarcinomas: a novel guideline
for clinical oncologists? J Cancer Res Clin Oncol. 129:321–326.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sheng SL, Liu JJ, Dai YH, Sun XG, Xiong XP
and Huang G: Knockdown of lactate dehydrogenase A suppresses tumor
growth and metastasis of human hepatocellular carcinoma. FEBS J.
279:3898–3910. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yokota H, Guo J, Matoba M, Higashi K,
Tonami H and Nagao Y: Lactate, choline, and creatine levels
measured by vitro 1H-MRS as prognostic parameters in patients with
non-small-cell lung cancer. J Magn Reson Imaging. 25:992–999. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Semenza GL: Tumor metabolism: cancer cells
give and take lactate. J Clin Invest. 118:3835–3837.
2008.PubMed/NCBI
|
|
22
|
Bonuccelli G, Tsirigos A, Whitaker-Menezes
D, Pavlides S, Pestell RG, Chiavarina B, Frank PG, Flomenberg N,
Howell A, Martinez-Outschoorn UE, Sotgia F and Lisanti MP: Ketones
and lactate ‘fuel’ tumor growth and metastasis: Evidence that
epithelial cancer cells use oxidative mitochondrial metabolism.
Cell Cycle. 9:3506–3514. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Vegran F, Boidot R, Michiels C, Sonveaux P
and Feron O: Lactate influx through the endothelial cell
monocarboxylate transporter MCT1 supports an NF-κB/IL-8 pathway
that drives tumor angiogenesis. Cancer Res. 71:2550–2560. 2011.
View Article : Google Scholar
|
|
24
|
Trabold O, Wagner S, Wicke C, Scheuenstuhl
H, Hussain MZ, Rosen N, Seremetiev A, Becker HD and Hunt TK:
Lactate and oxygen constitute a fundamental regulatory mechanism in
wound healing. Wound Repair Regen. 11:504–509. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hunt TK, Aslam R, Hussain Z and Beckert S:
Lactate, with oxygen, incites angiogenesis. Adv Exp Med Biol.
614:73–80. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wu H, Ding Z, Hu D, Sun F, Dai C, Xie J
and Hu X: Central role of lactic acidosis in cancer cell resistance
to glucose deprivation-induced cell death. J Pathol. 227:189–199.
2012. View Article : Google Scholar
|
|
27
|
Kalaany NY and Sabatini DM: Tumours with
PI3K activation are resistant to dietary restriction. Nature.
458:725–731. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hashimoto T, Hussien R, Oommen S, Gohil K
and Brooks GA: Lactate sensitive transcription factor network in L6
cells: activation of MCT1 and mitochondrial biogenesis. FASEB J.
21:2602–2612. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lu H, Forbes RA and Verma A:
Hypoxia-inducible factor 1 activation by aerobic glycolysis
implicates the Warburg effect in carcinogenesis. J Biol Chem.
277:23111–23115. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Samuvel DJ, Sundararaj KP, Nareika A,
Lopes-Virella MF and Huang Y: Lactate boosts TLR4 signaling and
NF-kappaB pathwaymediated gene transcription in macrophages via
monocarboxylate transporters and MD-2 up-regulation. J Immunol.
182:2476–2484. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Baumann F, Leukel P, Doerfelt A, Beier CP,
Dettmer K, Oefner PJ, Kastenberger M, Kreutz M, Nickl-Jockschat T,
Bogdahn U, Bosserhoff AK and Hau P: Lactate promotes glioma
migration by TGF-beta2-dependent regulation of matrix
metalloproteinase-2. Neuro Oncol. 11:368–380. 2009. View Article : Google Scholar :
|
|
32
|
Cory S and Adams JM: The Bcl2 family:
regulators of the cellular life-or-death switch. Nat Rev Cancer.
2:647–656. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Song G, Ouyang GL and Bao SD: The
activation of Akt/PKB signaling pathway and cell survival. J Cell
Mol Med. 9:59–71. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jeong SJ, Pise-Masison CA, Radonovich MF,
Park HU and Brady JN: Activated Akt regulates NF-kappaB activation,
p53 inhibition and cell survival in HTLV-1-transformed cells.
Oncogene. 24:6719–6728. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kim EC, Yun BS, Ryoo IJ, Min JK, Won MH,
Lee KS, Kim YM, Yoo I and Kwon YG: Complestatin prevents apoptotic
cell death: inhibition of a mitochondrial caspase pathway through
AKT/PKB activation. Biochem Biophys Res Commun. 313:193–204. 2004.
View Article : Google Scholar
|