Introduction
Glioblastoma multiforme (GBM) is one of the most
common gliomas, and is extremely lethal in all central nervous
system tumors (1). GBM infiltrates
into normal brain tissues where a tumor and the normal brain tissue
have no clear demarcation, due to glioma cells having a high
mobility and possessing strong invasive properties (2,3). Thus,
it is difficult to completely remove the tumor through surgical
resection. Additionally, GBM has been proven to resist radio- and
chemotherapy (4). Although patients
undergo the most aggressive regimens of debulking surgeries,
radiotherapy together with adjuvant chemotherapy results in a
median survival of ~14 months (5).
At present, there are no effective methods to prevent a relapse of
the tumor by residual neoplastic cells following surgery and
radiotherapy.
Temozolomide (TMZ) is a class of alkylating agent
approved by the Food and Drug Administration (FDA). TMZ is widely
used as a standard-of-care during clinical treatment. However, it
only results in a slight increase of overall survival of GBM
patients. Furthermore, most patients are resistant to TMZ in the
clinic (6).
O6-methylguanine produced by DNA methyl transferase
mainly mediates the cytotoxicity of TMZ and triggers cell
cycle-dependent DNA damage, ensuring cell death. Thus,
O6-methylguanine-DNA methyl transferase (MGMT) limits
the therapeutic effect of TMZ by removing
O6-methylguanine (7,8).
However, findings of previous GBM cell line studies showed that the
activity of MGMT was not entirely consistent with the resistance of
GBM to TMZ, i.e., even in MGMT-silenced GBM cells, the effect of
TMZ was limited (8). The exact cell
death pathway induced by TMZ and the molecular mechanisms affecting
the efficacy of TMZ in GBM cells remain to be determined.
The glutamate/cystine antiporter system
xc− is an obligate sodium-independent amino
acid antiporter, comprising 12-pass transmembrane transporter
protein xCT (SLC7A11) which is connected to the 4F2 cell surface
antigen 4F2hc (CD98/SLC3A2) by a disulfide bridge (9,10).
System xc− transports extracellular cystine
into cells in exchange for intracellular glutamate at a ratio of
1:1, and maintaining intracellular cysteine pools is important
(10). Cysteine is a crucial
material in glutathione (GSH) synthesis, which is indispensable for
maintaining intracellular redox balance and drug metabolism
(11,12). xCT expression is mediated by the
oxidative stress-response transcription factor NF-E2 related to
factor 2 (Nrf2) and activation of transcription factor 4 (ATF4)
(13). xCT is expressed in many
types of malignancies and is associated with tumor growth and
metastasis. It is also associated with resistance to chemotherapy
and poor survival (13–17). Accordingly, xCT has been considered
as a potential therapeutic target (13,14,16,17).
Sulfasalazine (SASP) is a sulfa drug used for inflammatory bowel
diseases and rheumatoid arthritis treatment and is a widely
recognized xCT-specific inhibitor (18). Although it has been suggested that
the inhibitory effect of SASP on xCT can suppress GBM cell growth,
the combination of SASP combined with TMZ for GBM treatment in the
clinic has yielded controversial results (19,20).
Erastin (ERA) is a voltage-dependent anion channels (VDAC)-binding
small molecule that is selectively lethal to some cancer cells
(21). Compared to SASP, ERA exerts
a stronger inhibitory effect on xCT (22). In addition, ERA is able to inhibit
the activity of certain GSH-related enzymes, such as glutathione
peroxidase 4 (GPx4), resulting in more lethal oxidative damage to
cells (23). ERA can cause a unique
form of cell death on iron-dependent tumor cells as compared to
conventional apoptosis, necrosis and autophagy (22).
In many types of cells cysteine is derived from an
imported xCT process, however, there is also a transsulfuration
pathway in which methionine is transferred to cysteine via
catalysis by cystathionine β-synthase (CSE) and
cystathionine-γ-lyase (CTH) (24,25).
In brain carcinoma cells such as glioma cells and astrocytes, most
cysteine originates from the reduction process of cystine imported
by xCT under normal conditions. However, when xCT is blocked or GSH
is decreased, the transsulfuration pathway is activated, which
insures the cysteine supply for GSH synthesis in a compensatory
manner (26,27). Furthermore, CTH activity may limit
cysteine synthesis via the methionine transsulfuration pathway
(28,29). However, whether transsulfuration and
CTH are involved in TMZ resistance remains to be elucidated.
In this study, we demonstrated that GBM cells were
sensitive to TMZ with a downregulation of the GSH level. TMZ
enhanced xCT expression in an Nrf2- and ATF4-dependent manner.
Moreover, it activated the transsulfuration pathway in GBM cells by
enhancing CTH activity. Erastin restrained xCT and CTH resulting in
a marked increase of TMZ cytotoxicity, which may be beneficial to
GBM therapy.
Materials and methods
Materials
Temozolomide (TMZ, 3,
4-dihydro-3-methyl-4-oxoimidazo [5, 1-d]-1, 2, 3,
5-tetrazine-8-carboxamide), deferoxamine mesylate (DFO), erastin
(ERA), N-Acetyl-L-cysteine (NAC), sulfasalazine (SASP),
propargylglycine (PPG), as well as Dulbecco’s modified Eagle’s
medium (DMEM) and bovine serum albumin (BSA) were obtained from
Sigma-Aldrich (St. Louis, MO, USA). Dimethylsulfoxide (DMSO) was
obtained from Wako Pure Chemical (Osaka, Japan). CellROX Orange
reagent was purchased from Life Technologies (Tokyo, Japan).
Anti-xCT antibody (ab37185) was obtained from Abcam (Cambridge, MA,
USA). Anti-Nrf2 antibody (sc-722), anti-ATF4 antibody (sc-200) and
anti-LaminB antibody (sc-56144) were purchased from Santa Cruz
Biotechnology, Inc. (Dallas, TX, USA).
Cell culture
The human A172, U87-MG and T98G malignant
glioblastoma multiforme cells were purchased from the American
Tissue Culture Collection (Rockville, MD, USA). GBM-N6 and GBM-N15,
isolated from a grade IV human glioblastoma, were a generous gift
from Dr Dongcheng Wang at Shandong University.
The cells were maintained in DMEM containing 10%
fetal bovine serum (Gibco, Grand Island, NY, USA) with 100 U/ml
penicillin and 100 μg/ml streptomycin (Gibco). The cells were
cultured at 37°C with 5% CO2 and saturated humidity.
Cloning of CTH cDNA and stable
transfection
The overexpression vector of CTH was constructed by
using the primer 5′-CGT CCC AGC ATG CAG AAG AA-3′ and 5′-CAG TTA
TTC AGA AGG TCT GGC CC-3′. The constructs containing CTH cDNA were
then subcloned into the pIRES2-EGFP expression vector (Clontech) as
previously reported (30). For
stable transfection, GBM-N15 cells were transfected with linearized
constructs by using FuGENE transfection reagent (Roche Applied
Science) according to the manufacturer’s instructions. After 48 h
of transfection, the cells were seeded in 35-mm dish at a density
of 5×104 cells/dish. The following day, the culture
media was refreshed containing 500 μg/ml G418 (Life Technologies)
for antibiotic selection. After five weeks of culturing the
selected antibiotics, survival-transfected cells were collected.
Stable-transfected cells were used within 15 passages as previously
reported (30).
Determination of intracellular reactive
oxygen species (ROS)
U87, T98G and GBM-N15 cells were plated in 12-well
plates at a density of 5.0×105 cells/well and cultured
overnight. The cells were treated with DMSO or 400 μM TMZ for 3 h.
In the experiment in which TMZ and ERA or SASP were used as
co-treatment methods, 5.0×105 cells/well of GBM-N15
cells were plated in 12-well plates. After 24 h, the culture medium
was refreshed with 400 μM TMZ and 5 μM ERA or 0.3 mM SASP. After
3-h incubation, the culture medium was replaced with fresh culture
medium containing 5 μM CellRox Orange Reagent, and the cells were
incubated at 37°C for an additional 30 min. After washing with PBS
twice, the Tali Image-based Cytometer (Life Technologies) was used
to detect the stained cells.
RNA preparation and RT-qPCR
Total RNA from GBM-N15 cells was isolated by using
TRIzol reagent (Life Technologies, Carlsbad, CA, USA) according to
the manufacturer’s instructions. cDNAs were synthesized using the
Transcriptor First Strand cDNA Synthesis kit (Roche, Shanghai,
China). The quantitative RT-PCR analyses were performed using SYBR
Premix Ex Taq II (Takara Bio) and the CFX Real-time PCR Detection
System (Bio-Rad, Hercules, CA, USA). The primers used for RT-qPCR
were: human xCT, forward: 5′-CCA TGA ACG GTG GTG TGT T-3′
and reverse: 5′-GAC CCT CTC GAG ACG CAA C-3′); human Nrf2,
forward: 5′-ACT CCC AGG TTG CCC AC-3′ and reverse: 5′-GTA GCC GAA
GAA ACC TCA TTG TC -3′); human ATF4, forward: 5′-TGA AGG AGT
TCG ACT TGG ATG CC-3′ and reverse: 5′-CAG AAG GTC ATC TGG CAT GGT
TTC-3′); human CTH, forward: 5′-GCC CAG TTC CGT GAA TCT
AA-3′ and reverse: 5′-CAT GCT GAA GAG TGC CCT TA-3′); and human
Cyclophilin A, forward: 5′-ATG CTG GAC CCA ACA CAA AT-3′ and
reverse 5′-TCT TTC ACT TTG CCA AAC ACC-3′). Cyclophilin A
was used as an internal control.
Protein extraction and immunoblot
analysis
The cells were washed three times with PBS, lysed by
using the buffer of 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% Triton
X-100, 1 mM EDTA, and sonicated to shear the DNA. Protein
concentrations were determined using the bicinchoninic acid assay
kit (Pierce, Rockford, IL, USA) according to the manufacturer’s
instructions. 2-Mercaptoethanol (1%) and bromophenolblue (0.01%)
were added into each sample. Protein (10 μg) per lane was separated
by SDS-PAGE and then transferred onto PVDF membranes (Millipore,
Billerica, MA, USA). The membranes were blocked with 5% BSA-PBS
with 0.1% Tween-20 (PBST). For protein visualization, the membranes
were blotted with primary antibodies against Nrf2, ATF4, xCT and
LaminB. Peroxidase-conjugated anti-rabbit IgG was applied as the
secondary antibody. Protein bands were detected using ImmunoStar
chemiluminescent reagent (Wako Pure Chemical).
siRNA transfection
After 24 h of seeding in 6-well plates, GBM-N15
cells were transfected with siRNA by using Lipofectamine RNAiMAX
(Life Technologies). Human CTH siRNA (sc-78973), human xCT siRNA
(sc-76933), human Nrf2 (sc-37030) and control siRNA (sc-37007) were
purchased from Santa Cruz Biotechnology, Inc.; and human ATF4 siRNA
was synthesized as the following sequence: forward: 5′-GCC UAG GUC
UCU UAG AUG ATT-3′ and reverse: 5′-UCA UCU AAG AGA CCU AGG CTT-3′.
After another 24 h of incubation, the transfected cells were
treated with TMZ for the indicated times. Subsequently, immunoblot
analysis, cell viability determination and intracellular GSH
detection were performed.
Cell viability analysis
Cell viability was evaluated by using the Cell
Counting kit-8 (DojinDo, Kumamoto, Japan) according to the
manufacturer’s instructions. Briefly, A172, U87, T98G, GBM-N6 and
GBM-N15 cells were plated in 96-well plates at a density of
5.0×104 cells/well. On the following day, the cells were
treated with TMZ of a concentration from 50 to 800 μM. After 48 h
of incubation, the cell viability was detected. For the
TMZ-inducible cell death in xCT knockdown GBM-N15 cells experiment,
the cells were transfected with control or xCT siRNA as described
above. After 24 h of transfection, the cells were seeded in 96-well
plates at a density of 5.0×104 cells/well and incubated
overnight. The following day, 50–600 μM TMZ were added to the
transfected cells and incubated for an additional 48 h. In TMZ and
SASP (or ERA) co-treatment experiments, the GBM-N15 cells were
seeded in 96-well plates at a density of 5.0×104
cells/well. After 24 h, the culture media were replaced with the
media containing increased doses of TMZ in the presence and absence
of 0.3 mM SASP or 5 μM ERA. After 48 h, the cell viability was
analyzed. For the experiment in which desferrioxamine (DFO)
decreased ERA-processed TMZ cytotoxicity, the cells were seeded as
described above. The following day, the medium was replaced with 50
or 100 μM TMZ and 5 μM ERA in the presence or absence of 100 μM
DFO.
Intracellular cysteine and GSH
analysis
U87, T98G and GBM-N15 cells were plated in 6-well
plates at a density of 6.0×105 cells/well and cultured
overnight. The following day, DMSO or 400 μM TMZ was introduced for
a 24-h incubation. For the TMZ-induced GSH levels experiment, xCT
was knocked down with siRNA as described above. The following day,
the cells were incubated in the presence or absence of 400 μM TMZ.
After 24 h, the cells were subjected to GSH analysis. For the TMZ
and SASP (or ERA) co-treatment experiment, the GBM-N15 cells were
plated in 6-well plates as described above. The following day, 400
μM TMZ with 0.3 mM SASP or 5 μM ERA were added into the medium, and
the cells were incubated for 24 h. To determine amino acid
deprivation, the cells were plated as mentioned above. The
following day, the culture medium was replaced with a certain amino
acid deprivation culture medium (methionine, cystine or
cystathionine) as shown in Fig. 5A.
Following incubation for 30 min at 37°C, 400 μM TMZ was introduced.
After 24 h of treatment, intracellular cysteine and GSH levels were
measured as previously described (31).
Determination of CTH activity
GBM-N15 cells were plated on 6-well plates at a
density of 5.0×105 cells/well. After 24 h, the medium
was refreshed with SASP, ERA or PPG. After 30-min incubation at
37°C, 400 μM TMZ was introduced for the next 24-h treatment.
Subsequently, intracellular CTH activity was determined as
previously described (27).
Statistical analysis
Data were presented as the means ± SEM of at least
three independent experiments. One-way ANOVA with the Bonferroni
post-hoc test was used to determine significant differences between
the means. The difference between the means was considered
significant at p<0.05.
Results
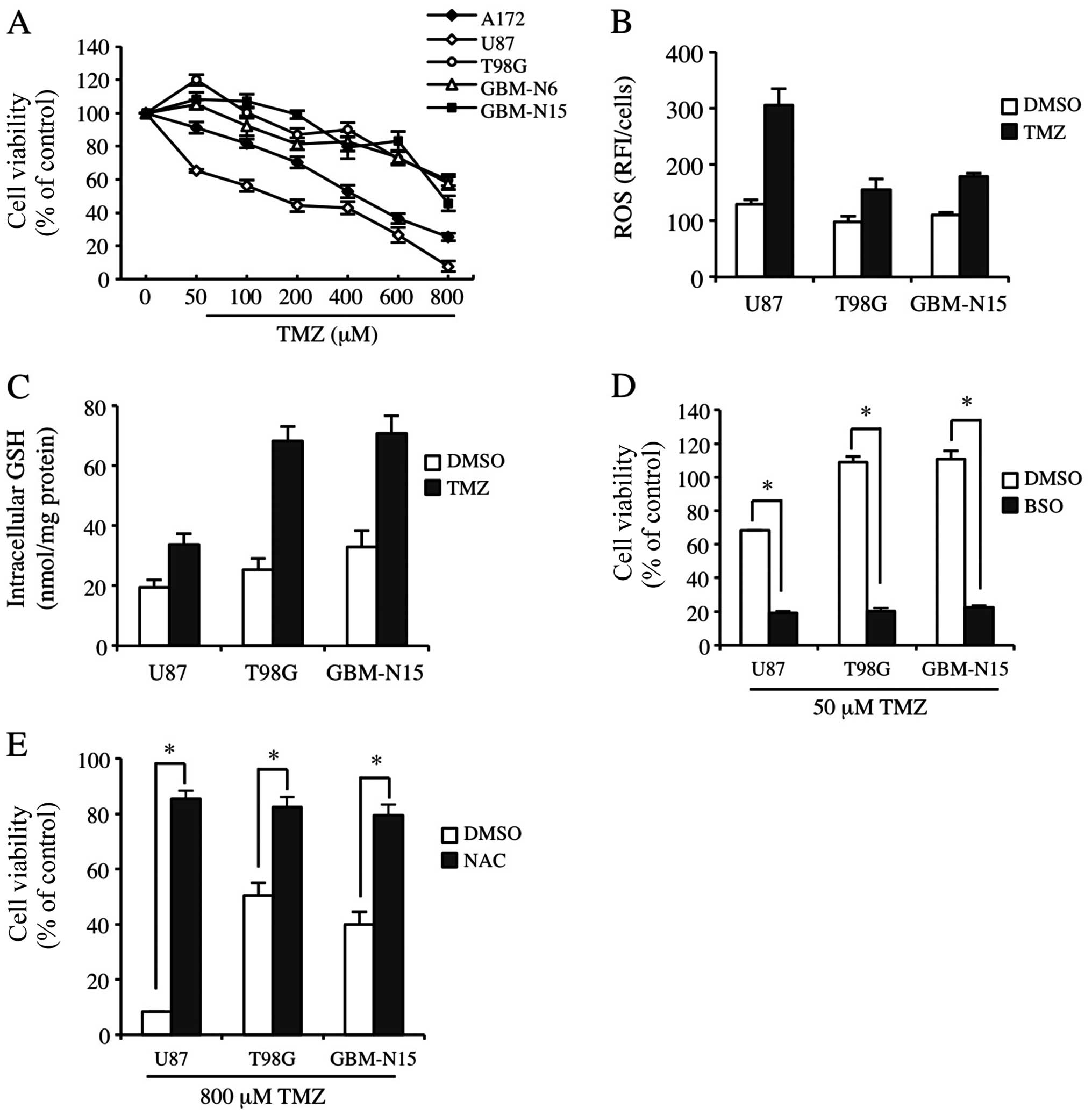
ROS and GSH levels are closely associates
with the sensitivity of GBM cells to TMZ treatment
To determine GBM cell line sensitivity to TMZ, we
identified three frequently used GBM cell lines in the laboratory:
A172, U87 and T98G cells. GBM-N6 and GBM-N15GBM cell lines were
also identified in glioma patients in stage IV. As shown in
Fig. 1A, U87 and A172 cells were
moderately sensitive to TMZ; however, T98G, GBM-N6 and GBM-N15
cells were significantly resistant to TMZ. The three cell lines
were screened and U87, T98G and GBM-N15 cells were used to detect
constitutive and TMZ-inducible ROS levels. The ROS level in U87
cells was significantly increased by TMZ treatment, while there
were no significant changes for the ROS level in T98G and GBM-N15
cells (Fig. 1B). Correspondingly,
the GSH level was only slightly enhanced by TMZ treatment in U87
cells, but was much more significantly elevated in T98G and GBM-N15
cells, suggesting that the cells with a low GSH level and
consequently a high ROS level were sensitive to TMZ (Fig. 1C). To confirm the association
between resistance to TMZ and the GSH level in the cells, buthione
sulfoximine (BSO), an inhibitor in GSH synthesis, was introduced in
the experiment. The results showed that BSO almost completely
abrogated the resistance of GBM cells to TMZ (Fig. 1D). We also analyzed the effect of
NAC on TMZ-inducible cytotoxicity, which is an antioxidant and
functions as a source of cysteine and GSH (32). As shown in Fig. 1E, NAC abolished the cytotoxicity of
TMZ to GBM cells. These results indicated that the intracellular
ROS and GSH levels are closely associated with the resistance of
GBM cells to TMZ treatment.
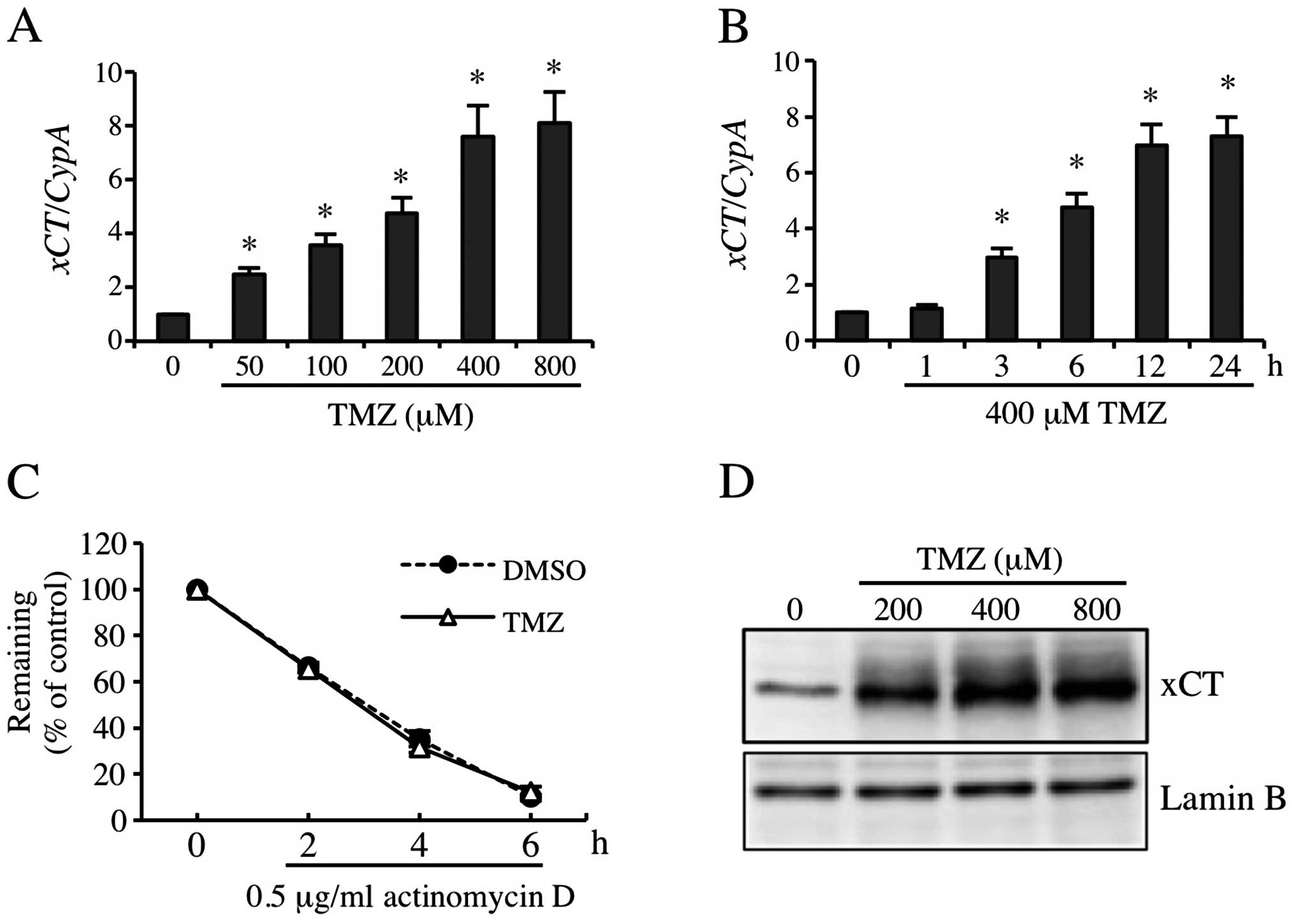
TMZ markedly induced xCT expression in
GBM-N15 cells
Results of previous studies indicated that GSH and
ROS levels were associated with xCT expression. Moreover, the
pharmacologic inhibitor for xCT enhanced the cytotoxicity of TMZ
(13,16,20,33).
To examine the contribution of xCT to resistance to TMZ, we
detected TMZ-inducible xCT expression. The RT-qPCR results showed
that 50, 100, 200, 400 and 800 μM TMZ increased xCT mRNA
expression by 2.5-, 3.6-, 4.8-, 7.6- and 8.1-fold, respectively
(Fig. 2A), and the induced
xCT mRNA expression increased significantly from 6 to 24 h
(Fig. 2B). Consistent with this
result, the xCT protein level was also increased significantly by
TMZ treatment (Fig. 2D). To
identifiy the stability of xCT mRNA induced by TMZ, 0.5
μg/ml of actinomycin D was used to sever the gene transcription. As
shown in Fig. 2C, TMZ did not
change the stability of xCT mRNA significantly. These
observations suggested that TMZ markedly induced the expression of
xCT at transcription level.
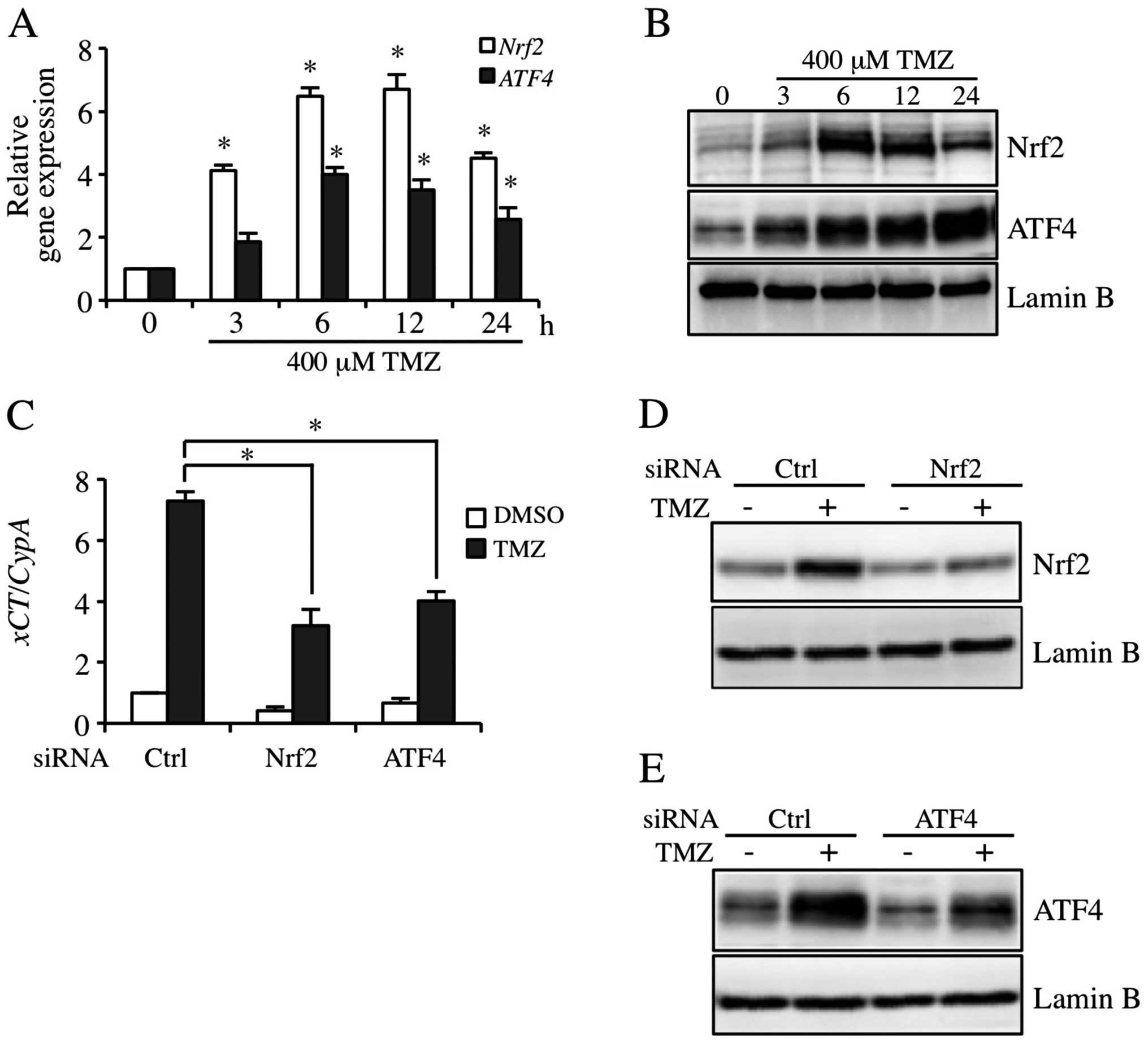
TMZ induces xCT expression via Nrf2 and
ATF4 activation pathway
Nrf2 and ATF4 are involved in the regulation of xCT
expression in human bladder carcinoma cells (13). To investigate the contribution to
the expression of Nrf2 and ATF4 in TMZ-inducible xCT, Nrf2 and ATF4
expression levels were detected in GBM-N15 cells. The results
showed that 400 μM TMZ markedly induced the expression of Nrf2 and
ATF4 at mRNA and protein levels, and the inductions peaked from 6
to 12 h (Fig. 3A and B). As shown
in Fig. 3C, the constitutive- and
TMZ-triggered expression of xCT mRNA was decreased in Nrf2
and ATF4 knockdown cells. In addition, TMZ-inducible Nrf2 and ATF4
protein expression was decreased individually by siRNA targeting
Nrf2 and ATF4 (Fig. 3D and E).
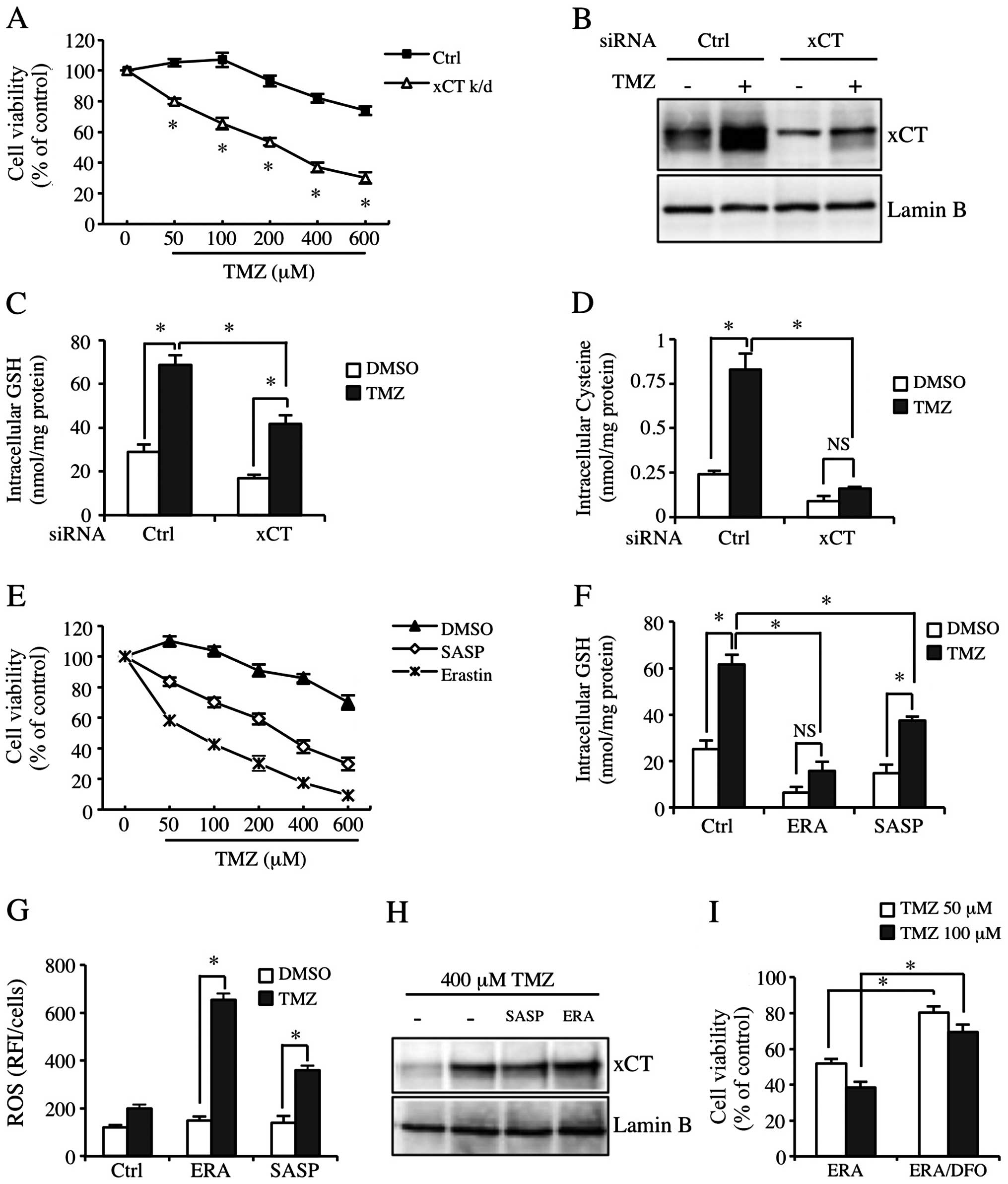
xCT inhibition significantly sensitizes
GBM-N15 cells to TMZ
In order to study the role of xCT in the resistance
to TMZ, we inhibited the function of xCT by applying xCT siRNA or
pharmacologic xCT inhibitors. We confirmed that transfection with
xCT siRNA effectively reduced constitutive and TMZ-inducible xCT
expression (Fig. 4B). Fig. 4A shows that the sensitivity of GBM
cells to TMZ was significantly increased in the xCT knockdown
GBM-N15 cells. The cytotoxicity of TMZ was also increased at a
physiological concentration (50 μM) (Fig. 4A). Since xCT was crucial for
maintaining cysteine pool and GSH synthesis in cells, we detected
intracellular cysteine and GSH levels. The results showed that the
basic level and TMZ-inducible cysteine level in xCT knockdown cells
was decreased significantly (Fig.
4D). Although the TMZ-inducible GSH level was inhibited by the
effect of xCT knockdown, the cysteine level was not significantly
reduced as compared to the control group (Fig. 4C). In addition, we used the
pharmacologic inhibitors of xCT, SASP and ERA to block the
properties of xCT in another experiment. Similarly, SASP and ERA
significantly decreased the TMZ-inducible GSH level and the
resistance to TMZ (Fig. 4E and F).
However, the regulation of ERA on TMZ-inducible cytotoxicity and
intracellular GSH level in the cells was much stronger than the
regulation of xCT siRNA and SASP, and it almost completely
abolished the increase of TMZ-inducible GSH level (Fig. 4E and F, middle lane). Consistent
with this result, SASP and ERA enhanced the TMZ-inducible ROS level
in GBM-N15 cells, with the effect of ERA being much more
significant than that of SASP (Fig.
4G). Additionally, SASP and ERA did not affect TMZ-inducible
xCT expression (Fig. 4H).
Additionally, the increase of sensitivity to TMZ by applied ERA was
partially eliminated by an Fe (III) chelator known as
desferrioxamine (DFO) (Fig. 4I).
These data indicated TMZ-induced cell death partially resulted from
oxidative stress. The TMZ-inducible xCT upregulation increased the
cysteine and GSH levels in GBM-N15 cells, which inhibited oxidative
stress and reduced the effect of TMZ.
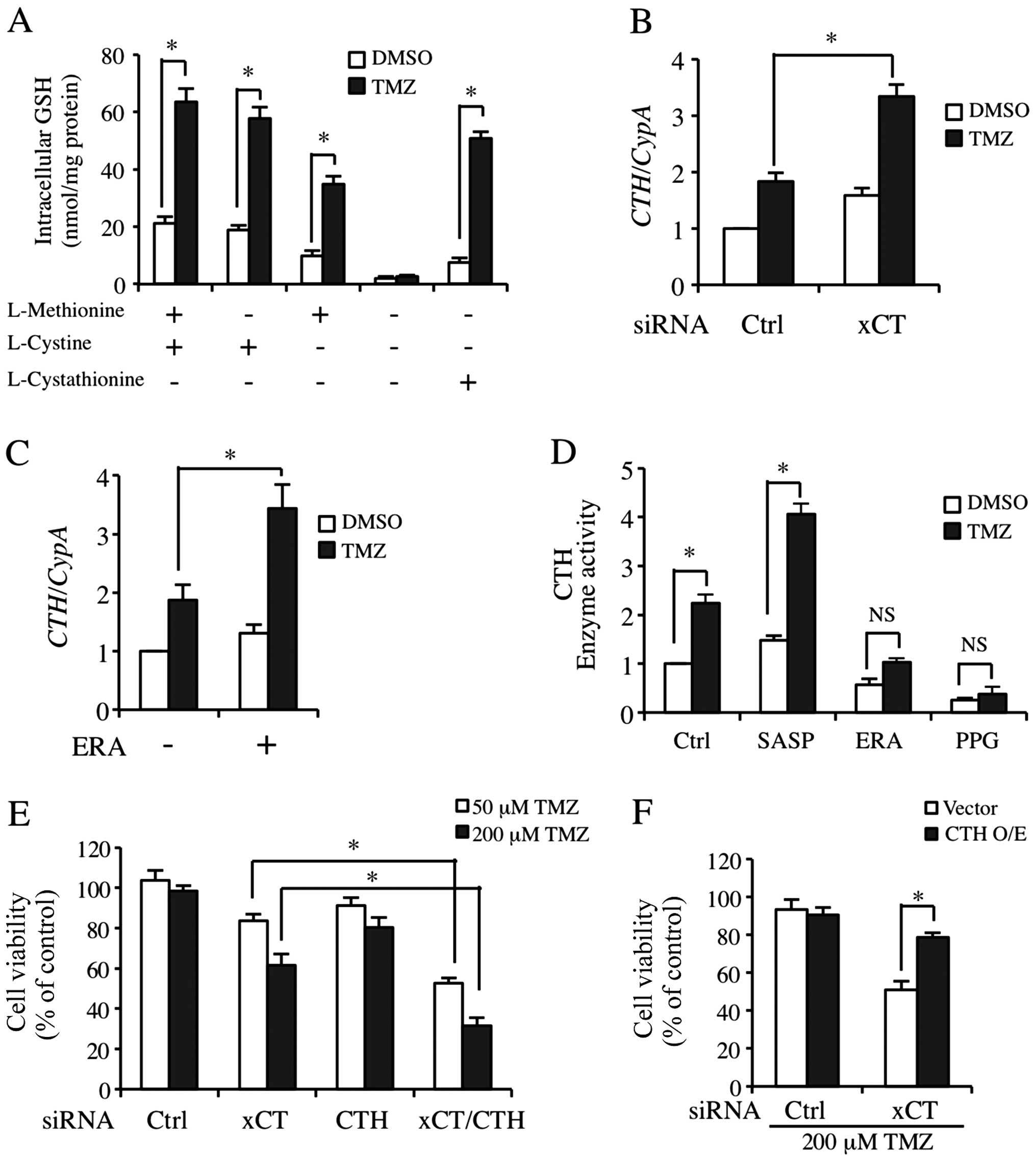
Transsulfuration pathway supports the
necessary cysteine for GSH synthesis as a compensatory pathway when
xCT expression was inhibited
Fig. 4C and D shows
that although the cysteine level was markedly decreased, GSH
synthesis was only partly affected. Kandil et al stated that
cystathionine-γ-lyase (CTH), the key regulatory enzyme in
transsulfuration pathway is activated to maintain GSH synthesis
when xCT was blocked (34). We
found that the TMZ-inducible GSH level was only partially
downregulated due to cysteine deprivation in the media (Fig. 5A). However, GSH level was markedly
decreased in the basic and TMZ-processed cells when methionine and
cysteine were deprived (Fig. 5A,
lane 3). Moreover, cystathionine almost completely acquired the
role of methionine and cysteine (Fig.
5A, lane 5). We then detected CTH mRNA regulation by TMZ
treatment in the cells transfected with ctrl or xCT siRNA. As shown
in Fig. 5B, TMZ significantly
upregulated the expression of CTH mRNA in xCT knockdown
cells. CTH expression was also increased slightly in
xCT-silenced cells without TMZ treatment in a compensatory manner
(Fig. 5B). In another experiment,
we detected the effect of TMZ on CTH expression and its
enzyme activities when xCT was inhibited by pharmacological
inhibitors. Of note, CTH enzyme activity that was processed by TMZ
and SASP co-treatment was markedly increased (Fig. 5D, Lane 2). By contrast, ERA almost
completely abolished the increase of TMZ-inducible CTH enzyme
activity (Fig. 5D, Lane 3). PPG as
an inhibitor to the transsulfuration pathway was the positive
control (Fig. 5D, Lane 4). We also
found that ERA did not decrease the basic and TMZ-inducible
CTH mRNA expression (Fig.
5C). In order to clarify that CTH was involved in the
resistance to TMZ in GBM-N15 cells, we applied siRNA transfection
targeting xCT and CTH independently or in combination. CTH
knockdown alone was not able to decrease resistance to TMZ.
However, when CTH and xCT were knocked down simultaneously,
TMZ-inducible cyto toxicity was significantly increased under
either a physiological (50 μM) or a high (200 μM) concentration of
TMZ (Fig. 5E). Similarly, the
overexpression of CTH suppressed the increase of TMZ-triggered
cytotoxicity in xCT knockdown cells (Fig. 5F).
Discussion
In clinical therapy for GBM patients, new combined
treatments and drugs are currently under the Phase 1 or 2 clinic
trials (35,36); however, TMZ as a first-line
treatment is indispensable. Wide resistance to TMZ affects its
application, and the mechanism of resistance remain to be
elucidated (6–8). Therefore, elucidation of the mechanism
involved in the resistance to TMZ is crucial for improving the
effect as an anticancer drug. Previous studies have reported that
the cytotoxicity of TMZ was mainly mediated by
O6-methylguanine, and a satisfactory result of TMZ
treatment required functional DNA mismatch repair (MMR) and low
MGMT levels as preconditions (37,38).
However, recent studies have found that resistance to TMZ was
associated with various factors. For instance, miR-128 and miR-149
regulate the invasion and chemosensitivity of GBM cells to TMZ by
targeting Rap1B-mediated cytoskeletal and associated molecular
alterations (39). Another example
is Galectin-1 (Gal1) which regulates resistance to TMZ by targeting
the unfolded protein response to endoplasmic reticulum stress
(ERS). Moreover, the cell protective autophagy has been reported to
contribute to TMZ-induced cell death (40). The production of ROS and the
disruption of AKT/mTOR signal have been demonstrated to contribute
to the TMZ resistance (41). In
this study, we focused on the contribution of TMZ-inducible ROS
upregulation and the maintainable cysteine pool and GSH synthesis
by xCT transporting and transsulfuration pathway in the resistance
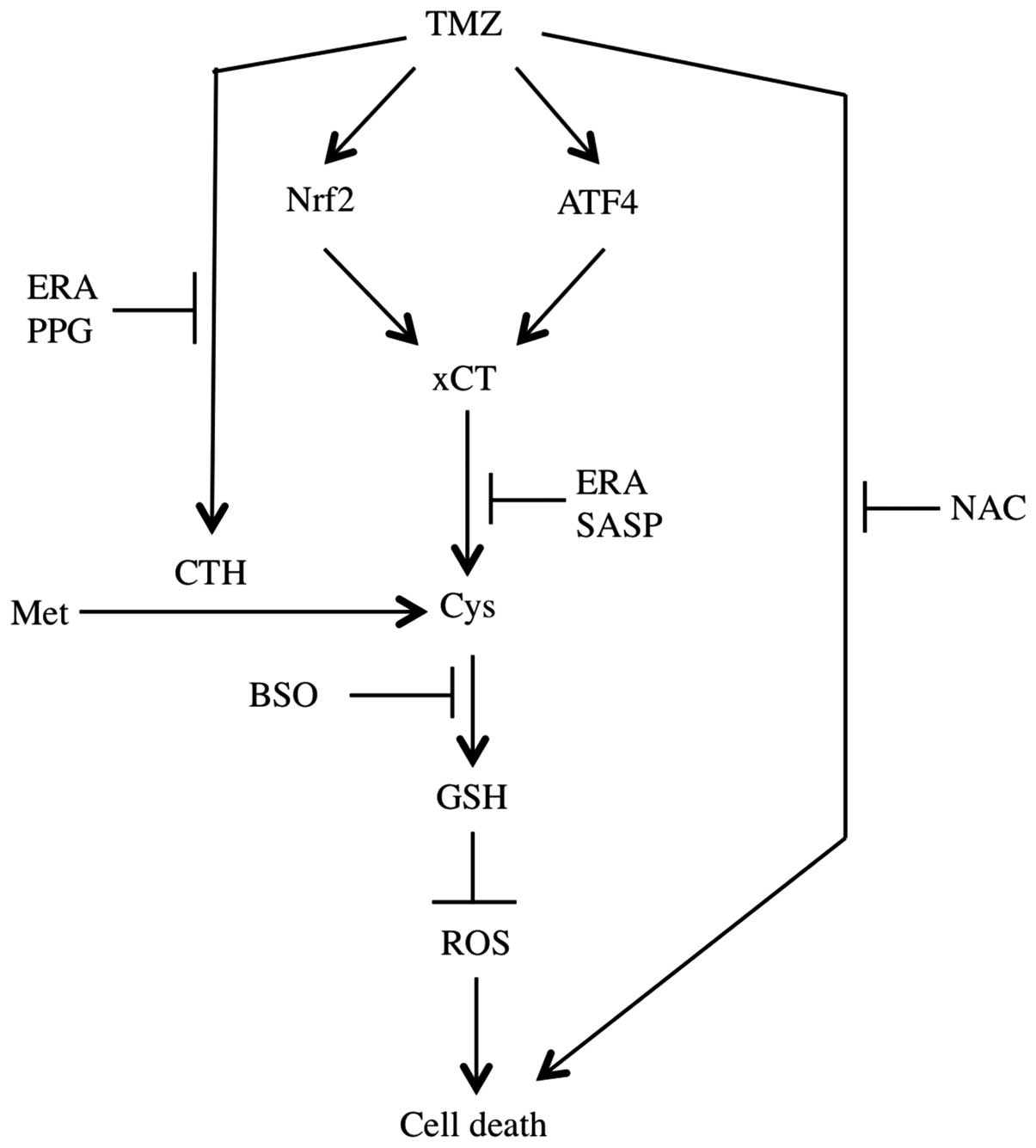
to TMZ (Fig. 6).
xCT as a transporter is closely associated with GSH
synthesis and ROS accumulation. Additionally, xCT is a potential
target of cancer treatment (13,16).
In many different types of tumors, pharmacological inhibition of
xCT function by SASP exerts an inhibitory effect on tumor cell
growth, and decreases the invasion of tumors, such as lymphoma,
hepatocellular carcinoma, prostate cancer, bladder carcinoma and,
glioma (13,16,34,42,43).
Furthermore, it has been proven that the effect of chemo- and
radiotherapy may be improved with inhibition on the function of
xCT. xCT is an independent predictive factor of poor prognosis and
associated with tumor invasion in GBM patients. The findings in the
present study coincide with the results of Ye et al
(13), xCT is strongly induced by
TMZ in an Nrf2- and ATF4-dependent manner.
Following the knockdown by xCT siRNA or
pharmacological inhibition by SASP and ERA, the ROS level increased
while the GSH synthesis level decreased. This process regulates the
sensitivity of GBM cells to TMZ treatment. Consequently, xCT is an
important factor in the resistance of GBM cells to TMZ. However,
the cysteine level in the cells decreased significantly when xCT
was knocked down by siRNA, although a marked reduction in the GSH
level was not observed. A similar result was confirmed in the
co-processing experiment by SASP and TMZ. We found that when we
deprived methionine and cysteine in media independently or
together, methionine was important to GSH synthesis when cysteine
was deprived.
Additionally, cystathionine almost completely
replaced cysteine and methionine. These results suggest that
another GSH synthesis pathway besides that of xCT exists, such as
the transsulfuration pathway. TMZ strongly induces the expression
of CTH mRNA and enhances the related enzyme activity, which is
critical in the transsulfuration pathway, especially when xCT is
inhibited. TMZ has been found to significantly induce the
expression of Nrf2 and ATF4. Nrf2 and ATF4 have been found to
increase GSH production via multiple mechanisms (44). However, loss of ATF4 impairs GSH
production by inhibiting the expression of CTH (45). However, the relevance between
TMZ-induced increase of Nrf2 and ATF4 expression and the CTH enzyme
regulation, and whether TMZ induced other related enzymes such as
CBS (cystathionine β-synthase) in the transsulfuration pathway for
GSH synthesis remain to be clarified and the experiments
confirmed.
SASP as an xCT pharmacological inhibitor is a class
of sulfa drugs that is approved by the Food and Drug Administration
(FDA). SASP has been applied to Chron disease therapy in the clinic
for a long time. ERA as small molecular compounds is another xCT
pharmacological inhibitor that has been recently identified
(22). Although the two inhibitors
can effectively block cysteine uptake as well as GSH synthesis,
there are other synthesis pathways besides xCT, the GSH synthesis
has no absolute reliability on xCT. ERA significantly inhibits CTH
activity and affects synthesis resulting from the xCT and
transsulfuration pathways, thus the effect of TMZ is significantly
elevated. However, ERA is limited with regard to the amelioration
of TMZ cytotoxicity by applied SASP, because a low cysteine level
in cells enables CTH activity with a co-processing treatment by
SASP and TMZ. This may be one reason for the controversy regarding
whether SASP has a therapeutic effect for GBM. Of note, the cell
death caused by applying TMZ and ERA together may be partially
rescued by DFO. This type of cell death is to some degree an Fe
(III)-dependent process, described as ferroptosis in a recent study
(22). ERA highly preferentially
selected to injure RAS-mutant tumors (22). In the present study, the results did
not show that there is a RAS-mutant in GBM cells. However, the
continuously activated RAS was involved in the dialog between tumor
and immune system (46). ERA was
involved in the regulation of ferroptotic cancer cell death by
inhibiting the activity of GPx4 (glutathione peroxidase 4)
(23). Moreover, GPx4 is highly
associated with tumor growth, and is a significant risk factor for
breast cancer when GPx4 is continuously activated (47,48).
However, whether ERA ameliorated GBM cell resistance to TMZ by
inhibiting GPx4 activity and increasing the injury of oxidative
stress for cells requires further investigation.
The present study shows that TMZ-inducible xCT
upregulation and CTH activation were involved in the resistance of
GBM cells to TMZ. In addition, ERA was able to block xCT and reduce
CTH activity simultaneously. As a result, the cytotoxicity of TMZ
was significantly increased. The combined treatment of TMZ and ERA
may therefore greatly benefit GBM therapy.
Acknowledgements
This study was financially supported by grants from
the Natural Science Foundation of China (nos. 81372484, 81172197,
81272564 and 81171131).
Abbreviations:
|
GBM
|
glioblastoma multiforme
|
|
FDA
|
Food and Drug Administration
|
|
TMZ
|
temozolomide
|
|
MGMT
|
O6-methylguanine-DNA methyl
transferase
|
|
Nrf2
|
NF-E2 related factor 2
|
|
ATF4
|
activating transcription factor 4
|
|
SASP
|
sulfasalazine
|
|
ERA
|
erastin
|
|
CTH
|
cystathionine γ-lyase
|
|
CBS
|
Cystathionine β-synthase
|
|
DFO
|
deferoxamine mesylate
|
|
PPG
|
propargylglycine
|
|
GSH
|
glutathione
|
|
ROS
|
reactive oxygen species
|
References
|
1
|
Friedman HS, Kerby T and Calvert H:
Temozolomide and treatment of malignant glioma. Clin Cancer Res.
6:2585–2597. 2000.PubMed/NCBI
|
|
2
|
Gunther W, Pawlak E, Damasceno R, Arnold H
and Terzis AJ: Temozolomide induces apoptosis and senescence in
glioma cells cultured as multicellular spheroids. Br J Cancer.
88:463–469. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Knauth M, Wirtz CR, Tronnier VM, Aras N,
Kunze S and Sartor K: Intraoperative MR imaging increases the
extent of tumor resection in patients with high-grade gliomas. AJNR
Am J Neuroradiol. 20:1642–1646. 1999.PubMed/NCBI
|
|
4
|
Nakada M, Nakada S, Demuth T, Tran NL,
Hoelzinger DB and Berens ME: Molecular targets of glioma invasion.
Cell Mol Life Sci. 64:458–478. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Stupp R, Mason WP, van den Bent MJ, et al:
Radiotherapy plus concomitant and adjuvant temozolomide for
glioblastoma. N Engl J Med. 352:987–996. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Burnet NG, Jefferies SJ, Benson RJ, Hunt
DP and Treasure FP: Years of life lost (YLL) from cancer is an
important measure of population burden - and should be considered
when allocating research funds. Br J Cancer. 92:241–245.
2005.PubMed/NCBI
|
|
7
|
Bocangel DB, Finkelstein S, Schold SC,
Bhakat KK, Mitra S and Kokkinakis DM: Multifaceted resistance of
gliomas to temozolomide. Clin Cancer Res. 8:2725–2734.
2002.PubMed/NCBI
|
|
8
|
Hegi ME, Diserens A, Gorlia T, et al: MGMT
gene silencing and benefit from temozolomide in glioblastoma. New
Engl J Med. 352:997–1003. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Conrad M and Sato H: The oxidative
stress-inducible cystine/glutamate antiporter, system x (c) (−):
cystine supplier and beyond. Amino Acids. 42:231–246. 2012.
View Article : Google Scholar
|
|
10
|
Sato H, Tamba M, Ishii T and Bannai S:
Cloning and expression of a plasma membrane cystine/glutamate
exchange transporter composed of two distinct proteins. J Biol
Chem. 274:11455–11458. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Okuno S, Sato H, Kuriyama-Matsumura K, et
al: Role of cystine transport in intracellular glutathione level
and cisplatin resistance in human ovarian cancer cell lines. Br J
Cancer. 88:951–956. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lu SC: Glutathione synthesis. Biochim
Biophys Acta. 1830:3143–3153. 2013. View Article : Google Scholar :
|
|
13
|
Ye P, Mimura J, Okada T, et al: Nrf2- and
ATF4-dependent up-regulation of xCT modulates the sensitivity of
T24 bladder carcinoma cells to proteasome inhibition. Mol Cell
Biol. 34:3421–3434. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Toyoda M, Kaira K, Ohshima Y, et al:
Prognostic significance of amino-acid transporter expression (LAT1,
ASCT2, and xCT) in surgically resected tongue cancer. Br J Cancer.
110:2506–2513. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yoshikawa M, Tsuchihashi K, Ishimoto T, et
al: xCT inhibition depletes CD44v-expressing tumor cells that are
resistant to EGFR-targeted therapy in head and neck squamous cell
carcinoma. Cancer Res. 73:1855–1866. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Guo W, Zhao Y, Zhang Z, et al: Disruption
of xCT inhibits cell growth via the ROS/autophagy pathway in
hepatocellular carcinoma. Cancer Lett. 312:55–61. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen RS, Song YM, Zhou ZY, et al:
Disruption of xCT inhibits cancer cell metastasis via the
caveolin-1/beta-catenin pathway. Oncogene. 28:599–609. 2009.
View Article : Google Scholar
|
|
18
|
Gout PW, Buckley AR, Simms CR and
Bruchovsky N: Sulfasalazine, a potent suppressor of lymphoma growth
by inhibition of the x(c)- cystine transporter: a new action for an
old drug. Leukemia. 15:1633–1640. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Robe PA, Martin DH, Nguyen-Khac MT, et al:
Early termination of ISRCTN45828668, a phase 1/2 prospective,
randomized study of sulfasalazine for the treatment of progressing
malignant gliomas in adults. BMC Cancer. 9:3722009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Takeuchi S, Wada K, Nagatani K, Otani N,
Osada H and Nawashiro H: Sulfasalazine and temozolomide with
radiation therapy for newly diagnosed glioblastoma. Neurol India.
62:42–47. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yagoda N, von Rechenberg M, Zaganjor E, et
al: RAS-RAF-MEK-dependent oxidative cell death involving
voltage-dependent anion channels. Nature. 447:864–868. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dixon SJ, Lemberg KM, Lamprecht MR, et al:
Ferroptosis: an iron-dependent form of nonapoptotic cell death.
Cell. 149:1060–1072. PubMed/NCBI
|
|
23
|
Yang WS, SriRamaratnam R, Welsch ME, et
al: Regulation of ferroptotic cancer cell death by GPX4. Cell.
156:317–331. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Giordana L, Mantilla BS, Santana M, Silber
AM and Nowicki C: Cystathionine γ-lyase, an enzyme related to the
reverse transsulfuration pathway, is functional in Leishmania spp.
J Eukaryot Microbiol. 61:204–213. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Romero I, Téllez J, Yamanaka LE, Steindel
M, Romanha AJ and Grisard EC: Transsulfuration is an active pathway
for cysteine biosynthesis in Trypanosoma rangeli. Parasit Vectors.
7:1972014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
McBean GJ: The transsulfuration pathway: a
source of cysteine for glutathione in astrocytes. Amino Acids.
42:199–205. 2012. View Article : Google Scholar
|
|
27
|
Manna P and Jain SK: Vitamin D
up-regulates glucose transporter 4 (GLUT4) translocation and
glucose utilization mediated by cystathionine-gamma-lyase (CSE)
activation and H2S formation in 3T3L1 adipocytes. J Biol Chem.
287:42324–42332. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rao AM, Drake MR and Stipanuk MH: Role of
the transsulfuration pathway and of gamma-cystathionase activity in
the formation of cysteine and sulfate from methionine in rat
hepatocytes. J Nutr. 120:837–845. 1990.PubMed/NCBI
|
|
29
|
Liu G, Casqueiro J, Bañuelos O, Cardoza
RE, Gutiérrez S and Martín JF: Targeted inactivation of the mecB
gene, encoding cystathionine-gamma-lyase, shows that the reverse
transsulfuration pathway is required for high-level cephalosporin
biosynthesis in Acremonium chrysogenum C10 but not for methionine
induction of the cephalosporin genes. J Bacteriol. 183:1765–1772.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yang G, Cao K, Wu L and Wang R:
Cystathionine gamma-lyase overexpression inhibits cell
proliferation via a H2S-dependent modulation of ERK1/2
phosphorylation and p21Cip/WAK-1. J Biol Chem. 279:49199–49205.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sasaki H, Sato H, Kuriyama-Matsumura K, et
al: Electrophile response element-mediated induction of the
cystine/glutamate exchange transporter gene expression. J Biol
Chem. 277:44765–44771. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wu G, Fang YZ, Yang S, Lupton JR and
Turner ND: Glutathione metabolism and its implications for health.
J Nutr. 134:489–492. 2004.PubMed/NCBI
|
|
33
|
Chung WJ and Sontheimer H: Sulfasalazine
inhibits the growth of primary brain tumors independent of nuclear
factor-kappaB. J Neurochem. 110:182–193. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kandil S, Brennan L and McBean GJ:
Glutathione depletion causes a JNK and p38MAPK-mediated increase in
expression of cystathionine-gamma-lyase and upregulation of the
transsulfuration pathway in C6 glioma cells. Neurochem Int.
56:611–619. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hottinger AF, Aissa AB, Espeli V, et al:
Phase I study of sorafenib combined with radiation therapy and
temozolomide as first-line treatment of high-grade glioma. Br J
Cancer. 110:2655–2661. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bartels U, Wolff J, Gore L, et al: Phase 2
study of safety and efficacy of nimotuzumab in pediatric patients
with progressive diffuse intrinsic pontine glioma. Neuro Oncol.
16:1554–1559. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Margison GP and Santibanez-Koref MF:
O6-alkylguanine-DNA alkyltransferase: role in carcinogenesis and
chemotherapy. Bioessays. 24:255–266. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhang J, Stevens MF and Bradshaw TD:
Temozolomide: mechanisms of action, repair and resistance. Curr Mol
Pharmacol. 5:102–114. 2012. View Article : Google Scholar
|
|
39
|
She X, Yu Z, Cui Y, et al: miR-128 and
miR-149 enhance the chemosensitivity of temozolomide by
Rap1B-mediated cytoskeletal remodeling in glioblastoma. Oncol Rep.
32:957–964. 2014.PubMed/NCBI
|
|
40
|
Lin CJ, Lee CC, Shih YL, et al:
Resveratrol enhances the therapeutic effect of temozolomide against
malignant glioma in vitro and in vivo by inhibiting autophagy. Free
Radic Biol Med. 52:377–391. 2012. View Article : Google Scholar
|
|
41
|
Yin H, Zhou Y, Wen C, et al: Curcumin
sensitizes glioblastoma to temozolomide by simultaneously
generating ROS and disrupting AKT/mTOR signaling. Oncol Rep.
32:1610–1616. 2014.PubMed/NCBI
|
|
42
|
Dai L, Cao Y, Chen Y, Parsons C and Qin Z:
Targeting xCT, a cystine-glutamate transporter induces apoptosis
and tumor regression for KSHV/HIV-associated lymphoma. J Hematol
Oncol. 7:302014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Doxsee DW, Gout PW, Kurita T, et al:
Sulfasalazine-induced cystine starvation: potential use for
prostate cancer therapy. Prostate. 67:162–171. 2007. View Article : Google Scholar
|
|
44
|
Ehren JL and Maher P: Concurrent
regulation of the transcription factors Nrf2 and ATF4 mediates the
enhancement of glutathione levels by the flavonoid fisetin. Biochem
Pharmacol. 85:1816–1826. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Dickhout JG, Carlisle RE, Jerome DE, et
al: Integrated stress response modulates cellular redox state via
induction of cystathionine gamma-lyase: cross-talk between
integrated stress response and thiol metabolism. J Biol Chem.
287:7603–7614. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Yang BC, Wang YS, Liu HS and Lin SJ: Ras
signaling is involved in the expression of Fas-L in glioma. Lab
Invest. 80:529–537. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Bermano G, Smyth E, Goua M, Heys SD and
Wahle KW: Impaired expression of glutathione peroxidase-4 gene in
peripheral blood mononuclear cells: a biomarker of increased breast
cancer risk. Cancer Biomark. 7:39–46. 2010.PubMed/NCBI
|
|
48
|
Schneider M, Wortmann M, Mandal PK, et al:
Absence of Absence of glutathione peroxidase 4 affects tumor
angiogenesis through increased 12/15-lipoxygenase activity.
Neoplasia. 12:254–263. 2010.PubMed/NCBI
|