Introduction
Colorectal cancer (CRC) is one of the world’s most
common type of malignancy, with more than 1.2 million cases
occurring every year. The prognosis of CRC is poor due to frequent
metastasis and tumor recurrence. Worldwide, approximately one
million new cases of CRC occur annually, amounting to 492,000
related deaths (1). With many
changes in the diet and lifestyle of individuals, CRC has become
the third most common type of digestive tumor in China, and the
number of new cases is increasing each year (2). The overall incidence is identical in
men and women, with the risk beginning at age 40 and increasing
with age. Thus, CRC ranks among the most frequent causes of
cancer-related deaths in China. Despite improvements in surgery,
radiotherapy and chemotherapy, unfortunately, the prognosis for CRC
has not improved over the past decades (3), with an overall 5-year survival rate of
approximately 40–50%. Therefore, novel diagnostic tools and
treatments need to be developed.
Phosphatidylinositol-3-kinase (PI3K) is a lipid
kinase and is responsible for the phosphorylation of position 3 of
the inositol ring of PI (4,5)P2. This generates the
second-messenger PI (3–5) P3, which is required for fundamental
cellular functions including transcription, translation,
proliferation, growth and survival. Abnormal activation of PI3K
signaling not only leads to neoplastic transformation of normal
cells, but also correlates with tumor cell migration, adhesion,
angiogenesis and degradation of the extracellular matrix (4). Phosphatase and tensin homolog deleted
on chromosome ten (PTEN) is a tumor-suppressor gene located
on human chromosome 10q23.3. PTEN is both a lipid phosphatase and a
protein phosphatase. As a lipid phosphatase, PTEN classically
converts phosphatidylinositol-3,4,5-trisphosphate (PIP3) to
phosphatidylinositol-4,5-bisphosphate (PIP2) in the cytoplasm,
thereby directly antagonizing the activity of PI3 kinase (PI3K)
(5). Inactivation of PTEN results
in constitutive activation of the PI3K/AKT pathway and therefore
increases protein synthesis, proliferation, migration and survival
(6). It has been reported that
deletion and mutation of PTEN are involved in the pathogenesis of
cancer by activating the PI3K/Akt signaling pathway (7).
Forkhead box O (FoxO) transcription factors belong
to the Forkhead family of proteins, a family characterized by a
conserved DNA binding domain termed the Forkhead box (Fox). These
transcription factors are downstream targets of the
serine/threonine protein kinase B (PKB)/Akt (8). Phosphorylation of FoxOs by Akt
inhibits the transcription function of FoxOs and contributes to
cell survival, growth and proliferation (9). FoxOs can promote cell growth
inhibition and/or apoptosis signaling by inducing expression of
multiple pro-apoptotic members of the Bcl2 family, stimulating
expression of death receptor ligands, or by enhancing levels of
various cyclin-dependent kinase inhibitors (CDKIs) (10). Coupled with their ability to
cross-talk with p53, FoxOs represent an important class of tumor
suppressors in a variety of cancers.
At present, the relationship between biological
changes resulting from changes in PTEN expression and the FoxO
transcription factor is unknown. In this study, both the expression
and significance of PTEN function in the progression of CRCs were
investigated. CRC cell lines LoVo and SW480 overexpressing PTEN
were constructed to investigate the effects of PTEN expression on
CRC cell proliferation, cell cycle and cell apoptosis. Proteins
involved in the PI3K signaling pathway including Akt, FoxO
transcription factors and proteins associated with the cell cycle
were also investigated to elucidate the molecular mechanisms behind
tumor proliferation and apoptosis.
Materials and methods
Specimens
Thirty-nine cases of colorectal adenomas and
hyperplastic polyp specimens were collected from August 2007 to
September 2009 at Nanfang Hospital, Southern Medical University,
Guangzhou, China. Among these 39 cases, 22 males and 17 females
were enrolled, ranging in age from 20 to 75 years with an average
age of 46.3. Forty-five cases of colorectal cancer (CRC) and
adjacent normal tissues (at least 10 cm away from the tumor) were
obtained simultaneously from the patients at Nanfang Hospital.
Among these 45 cases, 27 males and 18 females were enrolled,
ranging in age from 32 to 83 years with an average age of 56. All
of these cases were pathologically confirmed post-surgery.
According to the criteria of the International Colorectal Cancer
Collaborative Group, 6 cases were categorized as colorectal cancer
Dukes’ A stage. Thirteen cases, 20 cases, and 6 cases belonged to
Dukes’ B stage, Dukes’ C stage, and Dukes’ D stage,
respectively.
Cell lines
CRC cell lines SW480 and LoVo were purchased from
the American Tissue Culture Collection (ATCC) and were provided by
the Shanghai Institute of Cell Biology, Chinese Academy of
Sciences. LoVo cells were derived from a left supraclavicular lymph
node metastases of a 56-year-old male colon adenocarcinoma patient.
SW480 cells were derived from the primary tumor site of a
51-year-old male patient with Duke’s B stage colon cancer.
Cell transfection
The plasmid pc-DNA3.1 (+) containing a CMV promoter
and a neomycin selection marker was purchased from Invitrogen.
Plasmid pc-DNA3.1-PTEN was a kind gift from Dr Guiqin Hou (Cellular
Biology Laboratory of Zhengzhou University). LoVo and SW480 cells
were transfected with pc-DNA3.1-PTEN or the control plasmid
pcDNA3.1 (Gibco Invitrogen, Grand Island, NY, USA) using the
Lipofectamine 2000™ transfection reagent (Invitrogen). For stable
cell population selection, at 24 h post-transfection, cells were
re-plated in RPMI-1640 (Gibco BRL, Rockville, MD, USA) with 10%
(vol/vol) fetal calf serum (FCS) and 1000 μg/ml G418 (Sigma, St.
Louis, MO, USA). G418-resistant clones were selected and expanded.
The mRNA and protein levels of PTEN in these cells were analyzed by
RT-PCR and Western blot analysis. Of note, cells transfected with
the control plasmid pc-DNA3.1 were cultured under the same
conditions.
Isolation of total RNA and RT-PCR
Total RNA was isolated using Trizol reagent
(Invitrogen Life Tech) according to the manufacturer’s
instructions. Total RNA (3 μg) was reverse transcribed into
single-stranded cDNA using RevertAid First Strand cDNA Synthesis
kit (Fermentas). cDNA was amplified with forward (F) and reverse
(R) primers by PCR as described below. The primer sequences for
GAPDH were 5′-CGGG AAGCTTGTCATCAATGG-3′ (F) and 5′-GGCAGTGAT
GGCATGGACTG-3′ (R). The primer sequences for PETN were
5′-GGAAAGGGACGAACTGGTGT-3′ (F) and 5′-CAG GTAACGGCTGAGGGAAC-3′ (R).
Primers were used at a final concentration of 1 μM. Reaction
mixture was first denatured at 94°C for 5 min. The PCR was
performed for 30 cycles of 94°C for 45 sec, 58°C for 45 seconds,
and 72°C for 45 sec, followed by 72°C for 10 min. PCR products were
analyzed using an agarose gel and visualized by ethidium bromide
staining.
Western blotting
Nuclear and cytoplasmic protein was extracted using
a Nuclear and Cytoplasmic Protein Extraction kit (Thermo
Scientific, USA). Cells were harvested and lysed in RIPA buffer
(RIPA kit, Shanghai Shenneng Bocai Biotechnology Co., Shanghai,
China). The supernatant was collected by centrifugation for 20 min
at 4°C. The extracted proteins (20 μg) were resolved on a 10%
SDS-PAGE gel, and transferred to polyvinylidene difluoride
Immobilon membranes. The membranes were blocked with 5% non-fat
milk and incubated with the appropriate primary antibody. After
washing, the membranes were stained with the secondary antibody.
Protein bands were visualized by chemiluminescent detection (ECL)
(Amersham Biosciences).
Immunohistochemical staining to detect
PTEN expression in CRC, adenomas and normal tissues
Paraffin-embedded tissues were sectioned (4-μm) and
placed onto glass slides. The slides were dehydrated and antigen
was retrieved in 1% citric acid solution under microwave
conditions. The slides were blocked with serum, followed by
treatment with a catalase inhibitor for 10 min at room temperature.
The slides were then incubated with the PTEN antibody (1:100) in a
humidified chamber at 4°C for 12 h. Slides were washed 3 times with
PBS and stained with a biotin-labeled secondary antibody for 40
min, followed by a streptavidin peroxidase solution for 30 min at
room temperature after 3 washes with PBS. The slides were developed
using DAB staining solution for 5 min followed by hematoxylin for
10 sec.
The intensity of staining was determined as
described previously (43). No staining, light yellow staining,
yellow staining, and brown staining were marked as 0, 1, 2 and 3
points, respectively. The number of positive cells was scored as ≤,
25%; 2, 26–50%; 3, 51–75%; and 4, ≥75%. Thetotalscores were
obtained by multiplying the score of the color staining and the
point score for the percentage of positive staining. A score of 0
was considered negative. A total score from 1 to 3 points was
considered weakly positive. A total score from 4 to 5 points was
considered moderately positive. More than 5 points was considered
strongly positive.
Measurement of cell proliferation using
CCK-8
LoVo, SW480, LoVo pc-DNA3.1, LoVo PTEN, SW480
pc-DNA3.1 and SW480 PTEN cells were seeded in a 96-well plate at
4000 cells/well and cultured in complete RPMI-1640 medium. CCK-8
was added at 4, 24, 48, 72 and 96 h. The absorbance at 450 and 650
nm was measured.
Fluorescence-activated cell sorting
(FACS)
Cells were released using 0.25% trypsin without EDTA
and fixed with 70% ethanol at −20°C or 12 h. The cells were then
washed and incubated with RNase for 30 min at 37°C. After PI
staining, the cell cycle distribution was assessed using flow
cytometry.
For apoptosis analysis, the released LoVo, SW480,
LoVo pc-DNA3.1, LoVo PTEN, SW480pc-DNA3.1 and SW480 PTEN cells were
stained with Annexin V-FITC and propidium iodide for 15 min at room
temperature. Apoptotic cells were quantified using FACS.
Statistical analysis
All data are presented as the mean ± standard
deviation (SD). Statistical analysis was performed using SPSS13.0
software. A Kruskal-Wallis test was used to analyze the
semi-quantitative immunohistochemistry data. Data between two
groups were compared using a t-test. One-way ANOVA was used to
compare multiple groups. A value of p<0.05 was considered to
indicate a statistically significant result.
Results
PTEN expression is decreased during human
CRC progression and metastasis
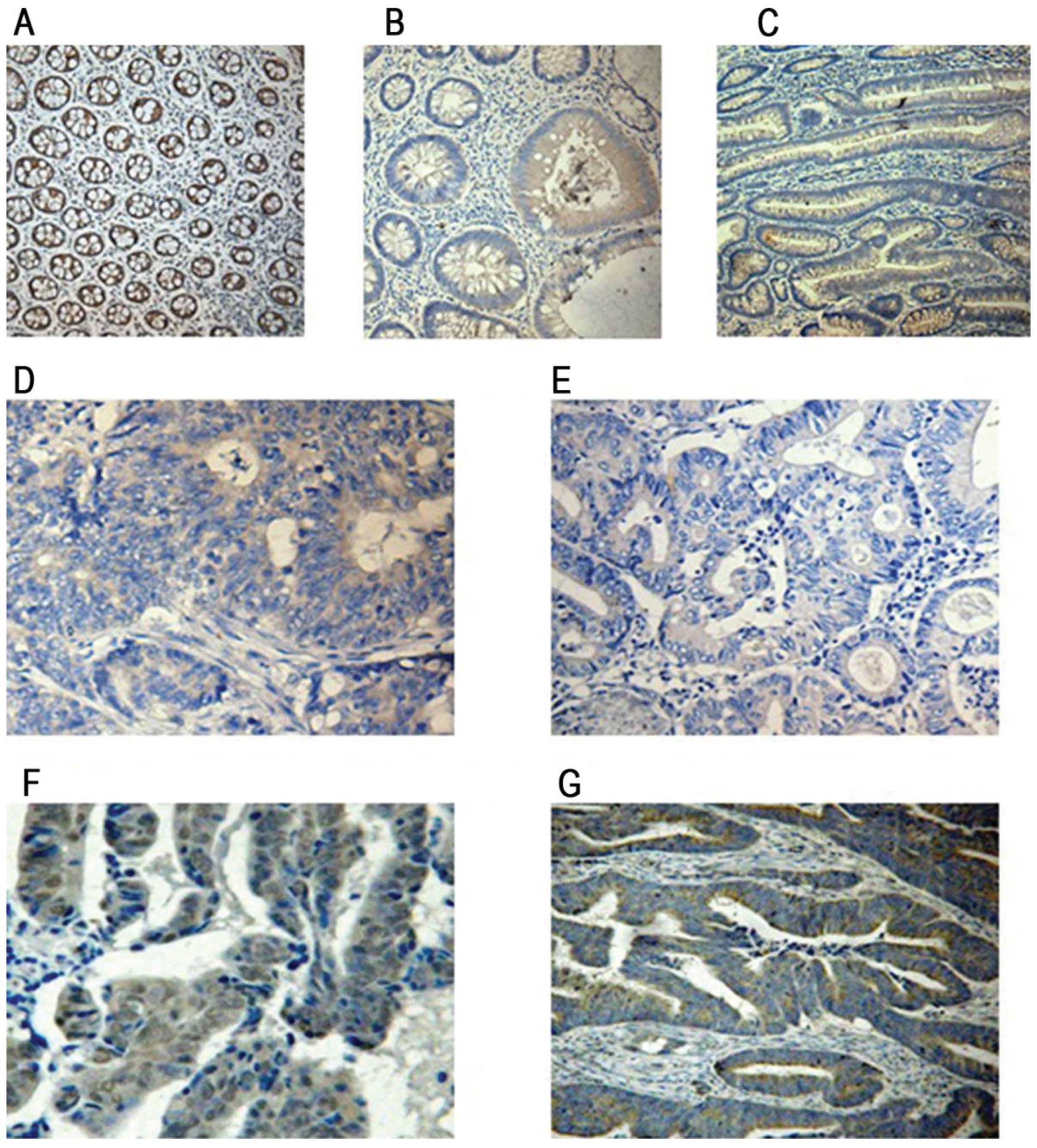
Immunohistochemical staining indicated that PTEN
expression was mainly localized in the cytoplasm of colonic
epithelial cells. In the normal colorectal mucosa, PTEN staining
was negative, weakly positive, positive and strongly positive in
0.00, 2.63, 18.4 and 78.9% of the cells, respectively (Fig. 1A). In colon hyperplastic polyps,
PTEN staining was negative, weakly positive, positive and strongly
positive in 0.00, 5.26, 10.5 and 84.29% of the cells, respectively
(Fig. 1B). In colorectal adenomas,
PTEN staining was negative, weakly positive, positive and strongly
positive in 25, 5, 10 and 60% of the cells, respectively (Fig. 1C). In the CRC tissues, PTEN staining
was negative, weakly positive, positive and strongly positive in
48.89, 28.89, 17.78 and 4.44% of the cells, respectively (Fig. 1D–G). As shown in Table I, a significant difference in PTEN
expression was found between normal colorectal mucosa, colorectal
hyperplastic polyps, colorectal adenomas, and CRC (P<0.05,
Kruskal-Wallis test). PTEN expression levels were gradually
decreased from normal colorectal mucosa to colorectal hyperplastic
polyps to colorectal adenoma and to CRC. PTEN staining was
negative, weakly positive, positive and strongly positive in 33.3,
16.67, 33.3 and 16.67% of the cells, respectively, in Dukes’ A
colorectal cancer. PTEN staining was negative, weakly positive,
positive and strongly positive in 38.46, 38.46, 15.38 and 7.69% of
the cells, respectively in Dukes’ B colorectal cancer. PTEN
staining was negative, weakly positive, positive and strongly
positive in 55, 30, 15 and 0.00% of the cells, respectively in
Dukes’ C colorectal cancer. PTEN staining was negative, weakly
positive, positive and strongly positive in 66.67, 16.67, 16.67 and
0.00% of the cells, respectively, in Dukes’ D colorectal cancer.
Although PTEN expression levels differed in Dukes’ stage colorectal
cancer, no significant difference was found (P=0.336) due to the
small number of samples collected (Table I). It is difficult to collect Dukes’
A and Dukes’ D colorectal cancers. Therefore, no significant
difference in PTEN expression was drawn among CRCs of different
grades of differentiation (P=0.052) (Table I).
 | Table IRelationship between PTEN expression
and clinical pathology features in the patients with colorectal
carcinoma. |
Table I
Relationship between PTEN expression
and clinical pathology features in the patients with colorectal
carcinoma.
| Features | N | PTEN
expression | Mean rank | P-value |
|---|
|
|---|
| 0 | 1+ | 2+ | 3+ |
|---|
|
Differentiation |
| Low | 18 | 12 | 5 | 1 | 0 | 17.92 | 0.052 |
| Moderate | 10 | 5 | 2 | 2 | 1 | 23.90 | |
| Well | 17 | 5 | 6 | 5 | 1 | 27.85 | |
| Tissue type |
| Normal mucosa | 38 | 0 | 1 | 7 | 30 | 83.72 | 0.000 |
| Polyp | 19 | 0 | 1 | 2 | 16 | 85.34 | |
| Adenoma | 20 | 5 | 1 | 2 | 12 | 66.08 | |
| Primary tumor | 45 | 22 | 13 | 8 | 2 | 30.63 | |
| Dukes’ stage |
| A | 6 | 2 | 1 | 2 | 1 | 29.25 | 0.366 |
| B | 13 | 5 | 5 | 2 | 1 | 25.08 | |
| C | 20 | 11 | 6 | 3 | 0 | 20.95 | |
| D | 6 | 4 | 1 | 1 | 0 | 19.08 | |
Upregulation of PTEN inhibits the
proliferation of colon cancer cells
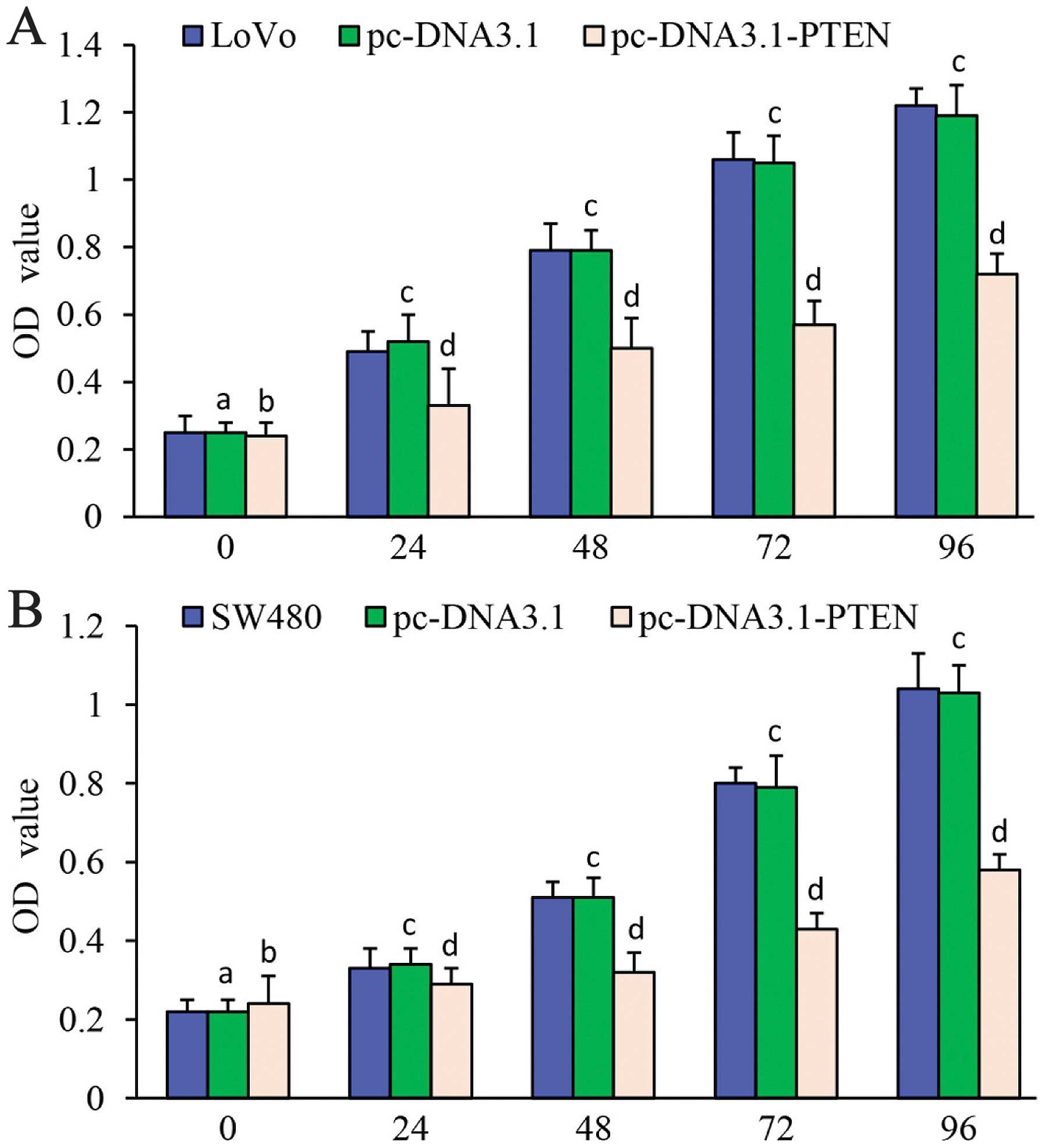
Cell proliferation was detected using CCK-8.
Proliferation of LoVo cells transfected with pc-DNA3.1-PTEN was
significantly decreased at 24, 48, 72 and 96 h compared with the
LoVo cells alone or LoVo cells transfected with pc-DNA3.1.
Proliferation rates of the LoVo cells alone and the LoVo cells
transfected with pc-DNA3.1 were not significantly different;
(P>0.05) (Fig. 2A).
Proliferation of the SW480 cells transfected with pc-DNA3.1-PTEN
was significantly decreased at 24, 48, 72 and 96 h compared with
the SW480 cells alone and the SW480 cells transfected with
pc-DNA3.1 (24 h: F=7.092, P=0.002; 48 h: F=88.731, P<0.001; 72
h: F=205.396, P<0.001; 96 h: F=202.248, P<0.001).
Proliferation rates of the SW480 cells alone and the SW480 cells
transfected with pc-DNA3.1 were not significantly different;
(P>0.05) (Fig. 2B). These data
revealed that upregulation of PTEN expression significantly
inhibited the proliferation of the malignant cells.
Upregulation of PTEN blocks cell cycle
progression
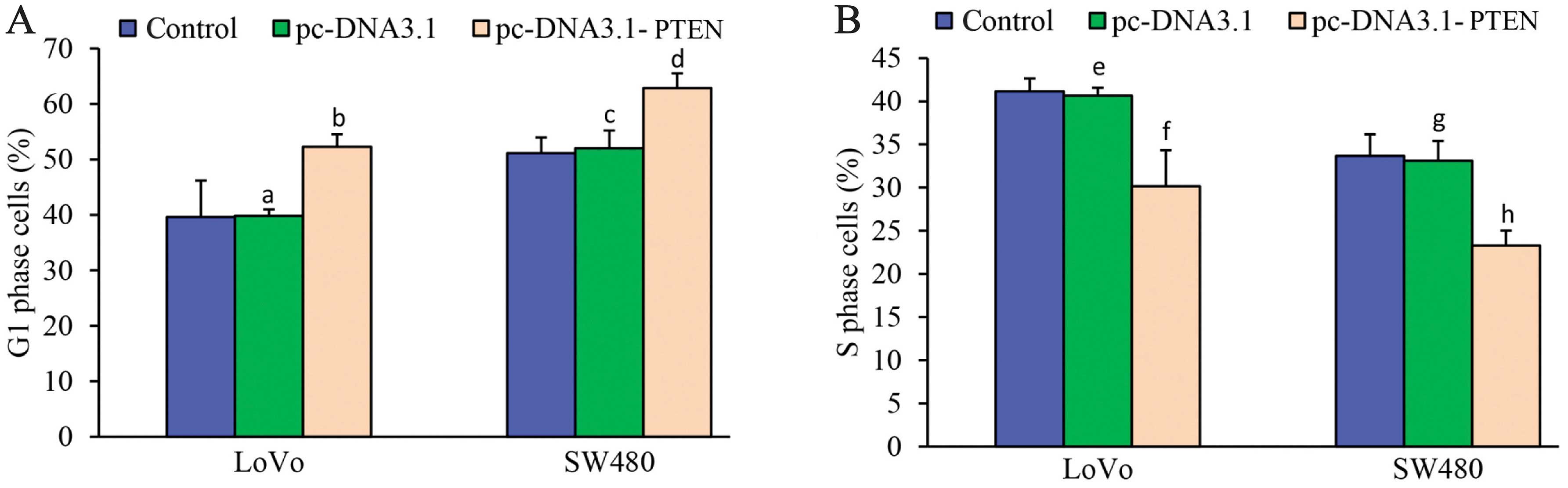
Compared to the LoVo cells and the LoVo cells
transfected with pc-DNA3.1, G1 phase arrest was significantly
increased in the LoVo cells transfected with pc-DNA3.1-PTEN
(F=15.695, P<0.001) (Fig. 3A).
However, the number of cells observed in the S phase was
significantly decreased in the LoVo cells transfected with
pc-DNA3.1-PTEN (F=28.116, P<0.001) (Fig. 3B). No differences were observed in
the cell cycle between the LoVo cells and the LoVo cells
transfected with pc-DNA3.1 (P>0.05) (Fig. 3). Similarly, the number of cells
observed in the G1 phase was significantly increased (F=25.185,
P<0.001) (Fig. 3A), whereas the
number of S phase cells was significantly decreased (F=35.199,
P<0.001) (Fig. 3B) in the SW480
cells transfected with pc-DNA3.1-PTEN compared to the SW480 and
SW480 cells transfected with pc-DNA3.1. No significant difference
was found in the cell cycle between the SW480 cells and the SW480
cells transfected with pc-DNA3.1 (P>0.05) (Fig. 3A and B). These data revealed that
upregulation of PTEN blocked G1 phase entry into the S phase, which
further inhibited the proliferation of malignant cells.
Upregulation of PTEN increases
5-FU-induced apoptosis in colon cancer cells
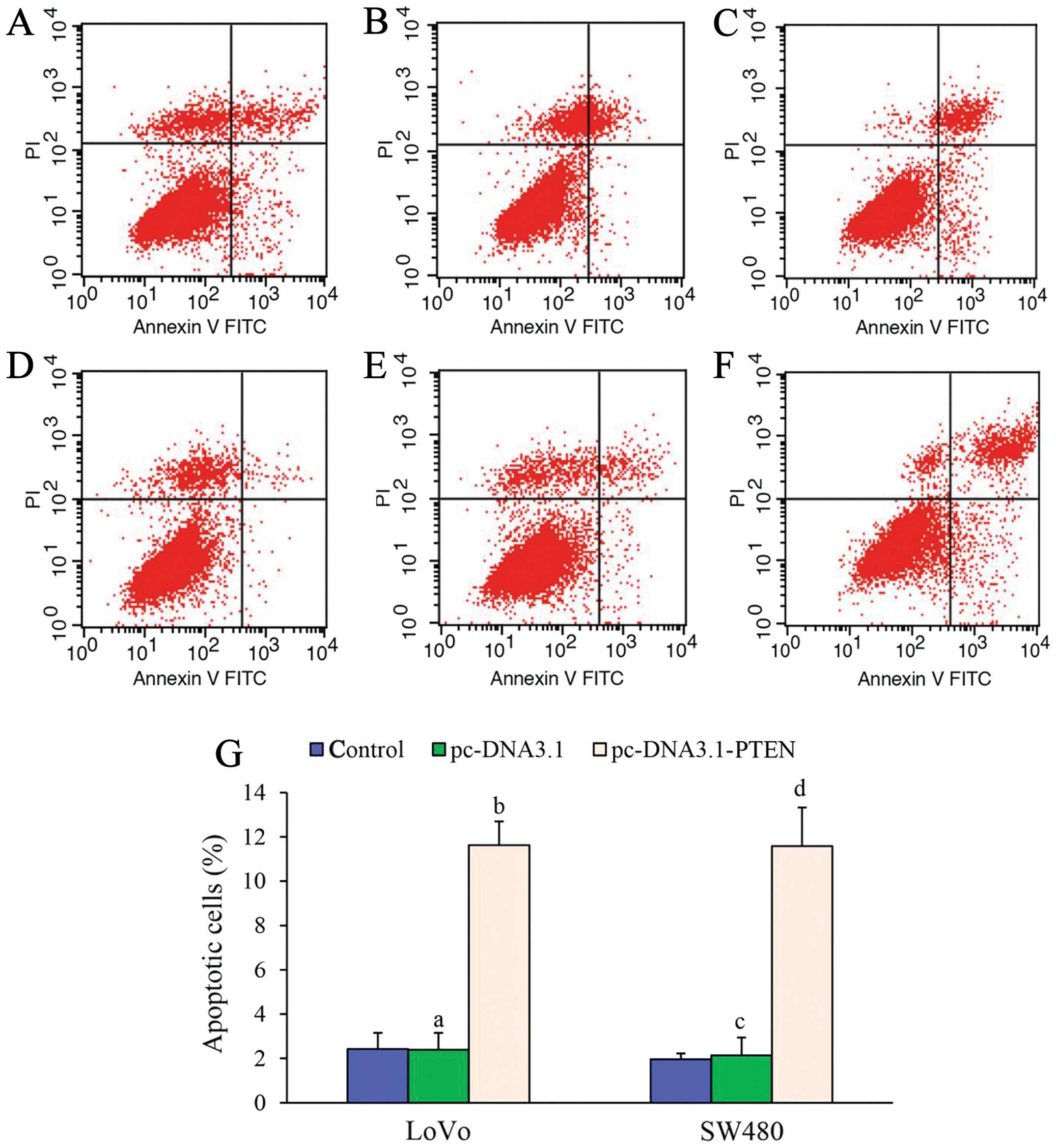
Apoptosis was not observed in the LoVo cells or the
SW480 cells at 48 h without 5-FU treatment. As shown in Fig. 4, after cells were treated with 0.01
μmol/ml or 0.03 μmol/ml 5-FU, a significant increase in apoptosis
was observed in both the LoVo cells and the SW480 cells transfected
with pc-DNA3.1-PTEN compared to the cells alone or cells
transfected with pc-DNA3.1. At least a 5-fold increase in apoptosis
was found in both the LoVo cells and the SW480 cells transfected
with pc-DNA3.1-PTEN compared to the control cells (LoVo: F=189.955,
P<0.001; SW480: F=121.719, P<0.001) (Fig. 4). These results suggest that
upregulation of PTEN could increase apoptosis in colon cancer cells
treated with chemotherapeutic drugs, such as 5-FU.
Effects of upregulation of PTEN on the
PI3K/AKT signaling pathway
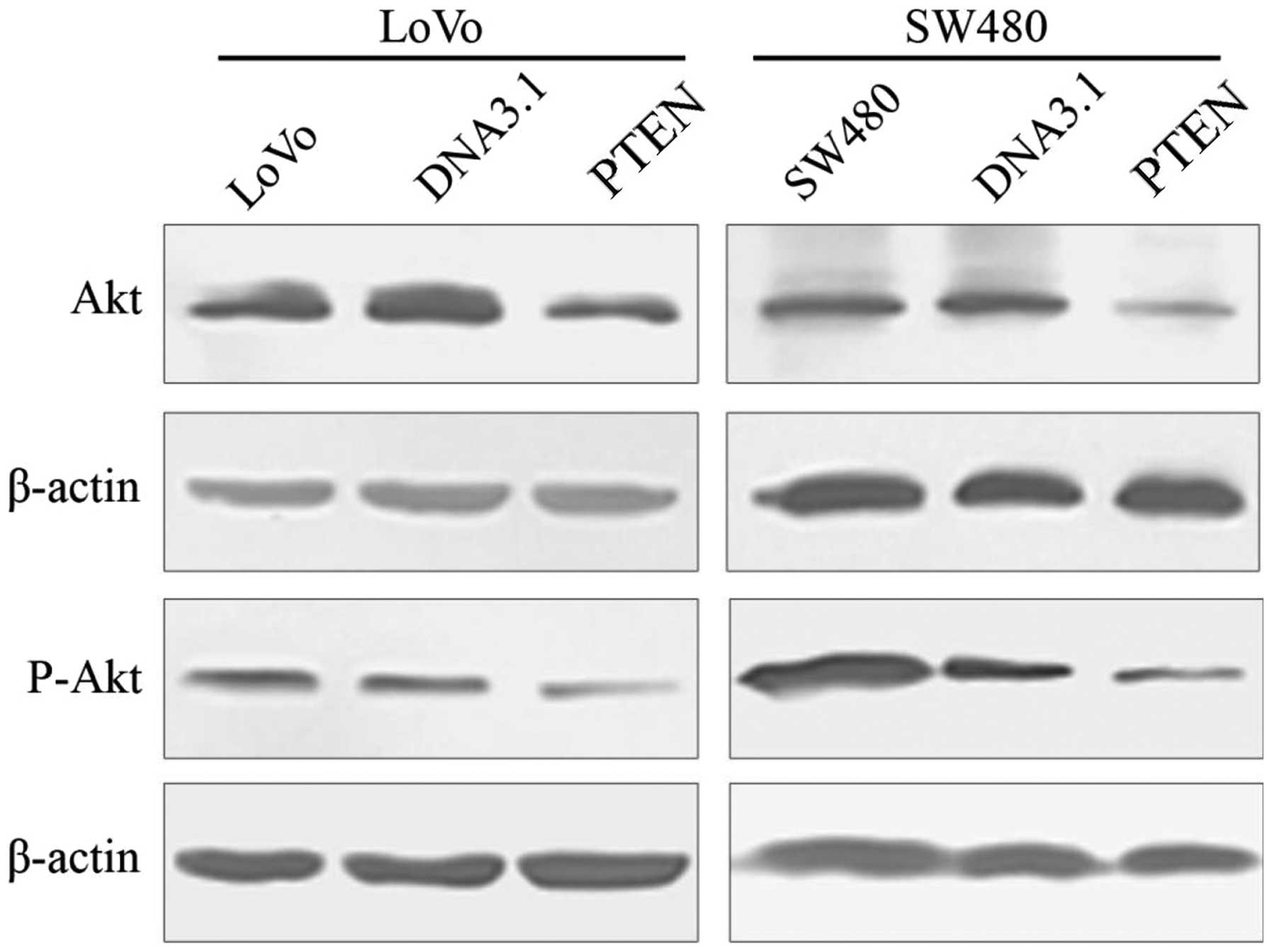
As shown in Fig. 5,
Akt and p-Akt protein expression levels were significantly reduced
in both the LoVo cells and the SW480 cells transfected with
pc-DNA3.1-PTEN compared to their respective control cells (LoVo:
Akt, F=420.337, P<0.001; p-Akt, F=262.269, P<0.001 and SW480:
Akt, F=2300.660, P<0.001; p-Akt, F=109.584, P<0.001). No
significant change was found in the LoVo or the SW480 cells
compared to these cells transfected with pc-DNA3.1 (P>0.05).
These data revealed that upregulation of PTEN protein reduced the
expression of Akt.
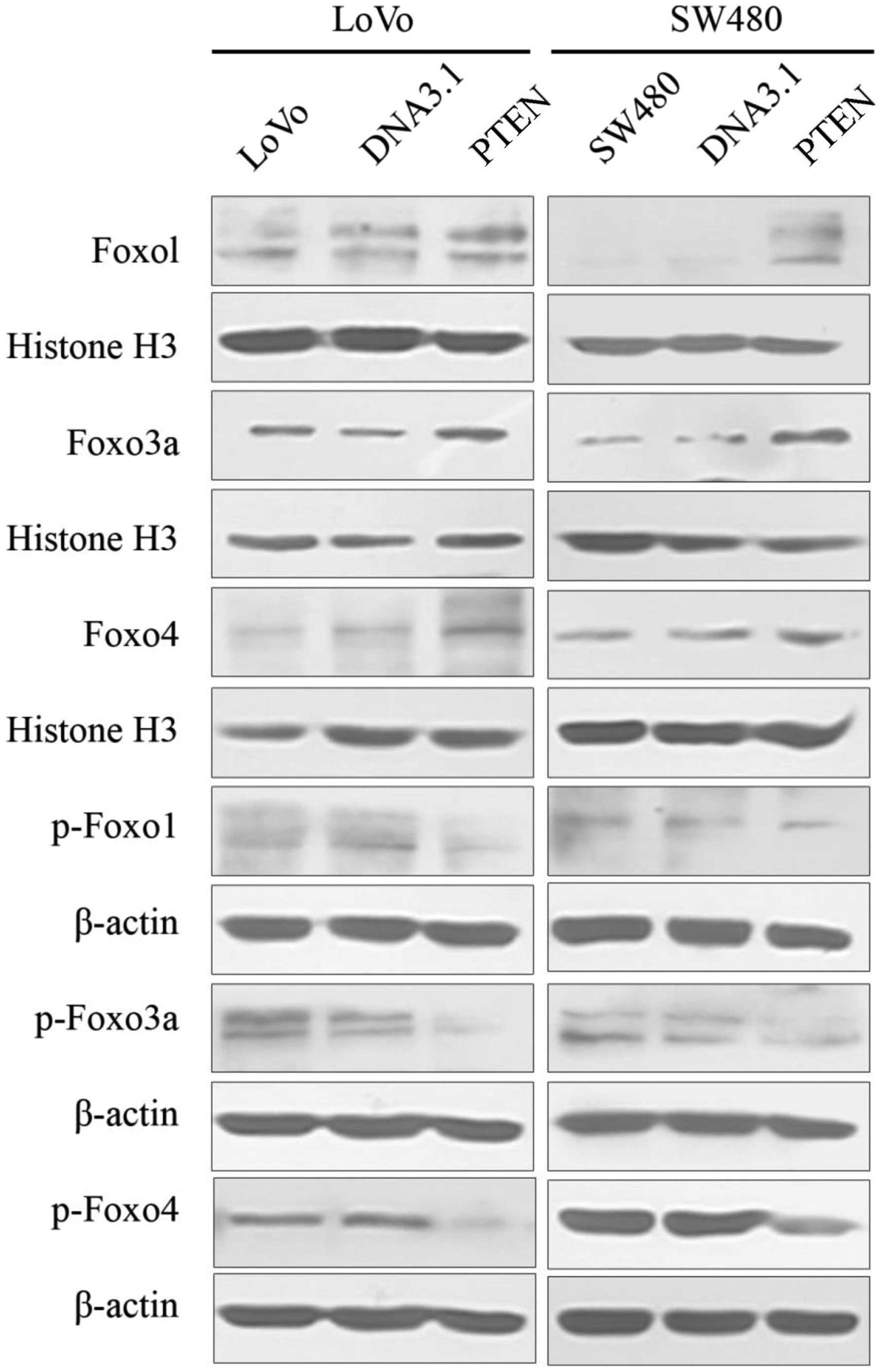
Nuclear proteins FoxO1 (FKHR), FoxO3a (FKHRL1), and
FoxO4 (AFX) were significantly increased in both the LoVo and SW480
cells transfected with pc-DNA3.1-PTEN compared to their respective
control cells (LoVo: FoxO1, F=37.414, P<0.001; FoxO3a,
F=117.618, P<0.001; FoxO4, F=392.631, P<0.001 and SW480:
FoxO1, F=200.868, P<0.001; FoxO3a, F=2837.989, P<0.001;
FoxO4, F=41.280, P<0.001) (Fig.
6). Moreover, cytosolic phosphoproteins p-FoxO1, p-FoxO3a, and
p-FoxO4 were significantly decreased in both the LoVo and SW480
cells transfected with pc-DNAA3.1-PTEN compared to their respective
control cells (LoVo: p-FoxO1, F=104.490, P<0.001; p-FoxO3a,
F=382.393, P<0.001; p-FoxO4, F=52.316, P<0.001 and SW480:
p-FoxO1, F=53.754, P<0.001; p-FoxO3a, F=39.003, P<0.001;
p-FoxO4, F=454.873, P<0.001). No significant changes were found
between cells alone and cells transfected with pc-DNA3.1.
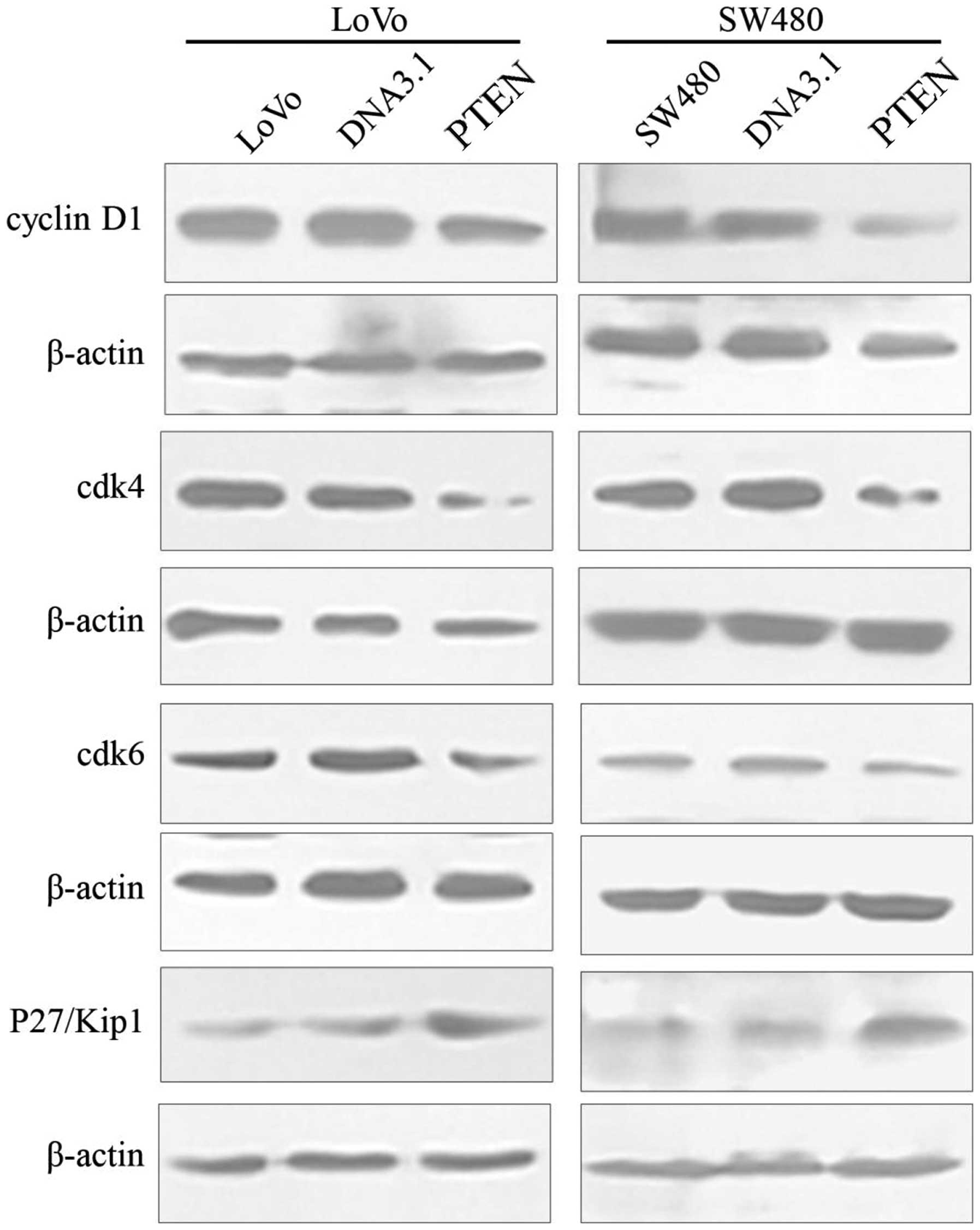
Cell cycle-related proteins, including cyclinD1,
cdk4 and cdk6 were significantly reduced in the both the LoVo and
SW480 cells transfected with pc-DNA3.1-PTEN compared to their
respective control cells (LoVo: cyclin D1, F=151.738, P<0.001;
cdk4, F=177.511, P<0.001; cdk6: F=1661.422, P<0.001 and
SW480: cyclin D1, F=848.596, P<0.001; cdk4, F=1228.556,
P<0.001; cdk6, F=99.949, P<0.001). Cyclin-dependent kinase
inhibitor p27Kip1 was significantly increased in both the LoVo and
SW480 cells transfected with pc-DNA3.1-PTEN compared to their
respective control cells (LoVo: F=854.323, P<0.001 and SW480:
F=140.832, P<0.001). No significant changes in cyclinD1, cdk4,
cdk6, or cyclin-dependent kinase inhibitor p27Kip1 were found
between the cells alone and the cells transfected with pc-DNA3.1
(P>0.05) (Fig. 7).
Discussion
PTEN expression and the progression and
metastasis of colorectal cancers (CRCs)
CRC is a common malignant cancer. The onset and
progression of CRCs are closely related to activation of oncogenes,
inactivation of tumor-suppressor genes, gene rearrangement, as well
as cell proliferation and apoptosis. Inactivation of
tumor-suppressor genes is a very common phenomenon in the
progression of CRC, and therefore these proteins are considered as
potential molecular targets for CRC treatment.
In 1974, Morson (11) proposed that CRC originated from
colorectal adenoma. Research has also suggested that CRC is formed
stepwise from normal colon mucosa becoming colorectal adenomas,
then adenomas with atypical hyperplasia, and finally carcinoma
(12–14). Progression from adenoma to carcinoma
generally takes 5–10 years or even longer, and different genes are
involved in the formation of carcinoma.
PTEN is a newly discovered tumor-suppressor gene
with phosphatase activity. PTEN protein acts as a phosphatase to
dephosphorylate phosphatidylinositol (3,4,5)-trisphosphate.
PTEN specifically catalyzes the dephosphorylation of the 3′
phosphate of the inositol ring in PIP3, resulting in the
biphosphate product PIP2, and inhibiting the phosphorylation of PI3
kinase, thereby blocking Akt and its downstream kinase activity and
inducing apoptosis (15).
In order to investigate the relationship between
PTEN expression and the progression of CRC, different tissues
including normal colon mucosa, colorectal adenomas, adenomas with
atypical hyperplasia, and carcinomas were collected to detect PTEN
expression using immunohistochemical staining. Our data revealed
that PTEN was mainly localized in the cytoplasm of the colorectal
epithelial cells and its expression levels in the normal colorectal
mucosa and hyperplastic polyps were higher than that in the
adenomas. PTEN expression in the CRC tissues was extremely low
(Fig. 1, Table I). This suggests that PTEN may play
an important role in the evolution of CRC. Of note, downregulation
of PTEN was found in adenomas, suggesting that PTEN is involved in
an early event during transformation and might be a useful marker
for the early diagnosis of CRC. Although a decreasing trend in PTEN
expression was observed through different Dukes’ stages, this
difference was not statistically significant due to the low number
of specimens for Dukes’ A and Dukes’ D stages. In conclusion, these
results strongly suggest that PTEN expression might be used as a
marker for early diagnosis of CRC and may provide a new target for
cancer treatment.
Effects of the upregulation of PTEN on
proliferation and apoptosis in CRC cells
Cell proliferation, differentiation and apoptosis
are essential to maintain cell homeostasis. The PI3K/Akt pathway is
closely associated with tumor development. The tumor-suppressor
gene PTEN is highly expressed in a variety of normal human tissues,
such as placenta, kidney, liver and brain. Mutations in the PTEN
gene are closely related to cancer-predisposing genetic diseases
including Cowden’s BRRS syndrome (16). PTEN mutations have been found in a
variety of tumors including prostate cancer, endometrial cancer,
glioma, lung cancer and liver cancer (17).
In the present study, in order to investigate
whether upregulation of PTEN affects the cell cycle, proliferation
and apoptosis, PTEN was overexpressed in LoVo and SW480 CRC cell
lines. Our data indicated that the proliferation of LoVo and SW480
cells transfected with pc-DNA3.1-PTEN was significantly decreased
when compared to the control cells. These data revealed that
upregulation of PTEN could inhibit the proliferation of tumor
cells. Moreover, significant increases were observed in the number
of cells in the G1 phase, while the number of cells in the S phase
was decreased in the LoVo and SW480 cells transfected with
pc-DNA3.1-PTEN compared to the control cells. This indicated that
upregulation of PTEN could inhibit cell proliferation though
blockage of G1/S phase transition.
When these cells were treated with 5-FU, increased
apoptosis was observed in the LoVo and SW480 cells transfected with
pc-DNA3.1-PTEN compared to the control cells. This indicated that
upregulation of PTEN could increase the sensitivity of colon cancer
cells to chemotherapeutic drugs such as 5-FU by inducing apoptosis.
The mechanism behind PTEN-induced apoptosis is not yet fully
understood, but may be related to the PI3K/Akt signaling pathway.
Since PTEN can regulate PIP3 levels, this may subsequently affect
the PIP3-PKB/Akt apoptotic pathway. Mutations in the PTEN gene in
tumor cells could decrease its phosphatase activity, leading to
cell proliferation and a reduction in apoptosis (18).
PTEN and the PI3K/Akt/FoxO signaling
pathway in CRC
The PI3K/Akt signaling pathway is closely related to
tumor onset and tumor development. PI (3–5) P3, the catalytic
product of PI3K, can activate downstream signal transduction
molecules, inducing tumor proliferation and inhibition of
apoptosis. PTEN suppresses tumor formation by restraining the
PI3K/AKT pathway (19). PTEN can
inhibit the phosphorylation of PIP3 kinase, blocking Akt and its
downstream kinase activity and inducing apoptosis. Mutation of the
PTEN gene can lead to abnormal activation of PIP3 and prevention of
cell death. In agreement with previous studies, expression levels
of Akt and P-Akt were significantly decreased in the Lovo and SW480
cells transfected with pc-DNA3.1-PTEN compared to the control
cells.
FoxO, a subgroup of the forkhead family, is a
transcription factor that plays an important role in regulating the
expression of genes involved in cell growth, proliferation and
apoptosis. The phosphorylation of FoxO is necessary for its
activity and is regulated by a variety of kinases (20). The protein kinase Akt can regulate
phosphorylation of FoxO at serine and threonine residues (T1, T2,
S1, S2 and S3), and inhibit FoxO transcriptional activity (21). FoxO is mainly localized in the
nucleus, but Akt-mediated phosphorylation of FoxO induces FoxO
transportation to the cytoplasm (21,22).
Inhibition of Akt/PKB retains FoxO in the nucleus, whereas
activation of PKB/Akt releases FoxO to the cytoplasm and leads to
inhibition of gene transcription (23–25).
Medema et al (26) reported that infection of mouse
embryonic fibroblasts (MEFs) with Foxo4 retrovirus could induce the
expression of P27kip (cyclin-dependent kinase inhibitor, CDKI) and
increase P27kip1 and cyclin E/cdk2 complex formation, causing G1
phase arrest. Kops et al (27) reported that infection of NIH 3T3
mouse fibroblasts and MEFs with FoxO4 and FoxO3 (FKHR-L1)
retroviruses induced expression of retinoblastoma-like p130 protein
Rb, which forms a complex with E2F-4 and reduces the level of free
E2F-4 in the nucleus to inhibit cell cycle progression. With the
exception of CDK, FoxO regulates expression of cyclins, including
cyclins A, B, D, E, G, and H and inhibits cell cycle progression.
FoxO factors can inhibit expression of cyclins D1 and D2, and
Foxo3a can elevate the expression of cyclin G2 (28,29).
In other words, FoxO proteins play important roles in the onset and
progression of tumors by regulating the cell cycle and apoptosis.
Mutations or decreases in FoxO can inhibit apoptosis and promote
cell overgrowth, leading to tumor expansion (9).
The inhibition of G1-S phase transition by PTEN is
dependent on Akt/PKB and can be restored by active Akt/PKB
(30). Overexpression of PTEN
increases the expression of P27kip and P27kip1/cyclin E/cdk2
complex formation and decreases cyclin D3 levels and CDK2 kinase
activity, eventually halting the cell cycle at the G1 phase
(18,31,32).
Moreover, PTEN can overcome the phosphorylation of glycogen
synthase kinase 3 (GSK3) by Akt/PKB. The phosphorylation of GSK3
promotes degradation of cyclinD1 and arrests the cell cycle and
inhibits cell growth (33). Wu
et al found that PTEN can induce three cyclin-dependent
kinase inhibitors, P21waf1, P27kip1 and P57 kip1, thereby
regulating the cell cycle (34).
Research has indicated that PI3-K/Akt can overcome NF-κB and
CD95/Fas-induced apoptosis, while PTEN can inhibit TNF-mediated
NF-κB activity (35,36). PTEN can also induce apoptosis by
activating caspase-3, caspase-8 and FADD-dependent pathways.
Overexpression of PTEN in SHG-44 glioma cells can induce apoptosis
(37). It has been reported that
PTEN can activate the P53 signaling pathway in glioma cells,
negatively regulating cell division and proliferation (38). In turn P53 can induce PTEN
expression and cell apoptosis (39,40).
In the present study, we first addressed whether
upregulation of PTEN activates the downstream transcription factor
FoxO. Our data indicated that nuclear FoxO1 (FKHR), FoxO3a (FKHRL1)
and FoxO4 (AFX) were significantly increased in both LoVo cells and
SW480 cells transfected with pc-DNA3.1-PTEN, while cytosolic
p-FoxO1 (FKHR, Ser256), p-FoxO3a (FKHRL1,
Ser253) and p-FoxO4 (AFX, Ser193) were
significantly decreased. PTEN overexpression induced FoxO
transportation from the nucleus to the cytoplasm and promoted the
transcription of downstream genes involved in cell proliferation
and apoptosis. Levels of cell cycle-related proteins cyclinD1, cdk4
and cdk6 were significantly decreased in the LoVo and SW480 cells
transfected with pc-DNA3.1-PTEN, while cyclin-dependent kinase
inhibitor p27/Kip1 expression was increased significantly. These
results revealed that upregulation of PTEN protein reduced Akt
activity and phosphorylated FoxO, which further inhibited cell
cycle progression. This further confirmed that upregulation of PTEN
can inhibit CRC cell proliferation and normal cell cycle
progression. Reagan-Shaw and Ahmad (41) showed that RNAi against PI3K in
breast cancer cells enhanced FoxO activation and eliminated the
inhibition of FoxO-mediated cell growth and apoptosis. Upregulation
of PTEN can induce the apoptosis of colon cancer cells treated with
5-FU, which may be related to FoxO activity.
In conclusion, this is the first report of a gradual
decrease in expression of PTEN from normal colon epithelial tissues
to colon hyperplastic polyps, colorectal adenomas and finally CRC.
Overexpression of PTEN in CRC cell lines inhibited cell
proliferation and cell cycle progression and increased 5-FU-induced
apoptosis. Upregulation of PTEN inhibited the activity of the Akt
pathway and regulated downstream genes involved in the cell cycle.
This suggests that inhibition of CRC cell proliferation and cell
cycle arrest by PTEN is closely related to PI3K/Akt/FoxO. This
study elucidated the molecular mechanism behind the malignant
proliferation of colon cancer cells and provides a new target for
the gene therapy and prognosis of CRC.
Acknowledgements
The present study was supported by the Science and
Information Technology Foundation of Guangzhou (grant nos. 2010J
E141 and 2011J4100051), the Guangdong Provincial Science and
Technology Department (grant no. 2011B0904005260) and the Natural
Science Foundation of Guangdong Province (grant no.
S2012040006707).
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2012. CA Cancer J Clin. 62:10–29. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
You WC, Jin F, Devesa S, et al: Rapid
increase in colorectal cancer rates in urban Shanghai, 1972–97, in
relation to dietary changes. J Cancer Epidemiol Prev. 7:143–146.
2002.
|
|
3
|
Cerottini JP, Caplin S, Pampallona S and
Givel JC: Prognostic factors in colorectal cancer. Oncol Rep.
6:409–414. 1999.PubMed/NCBI
|
|
4
|
Parsons DW, Wang TL, Samuels Y, et al:
Colorectal cancer: mutations in a signalling pathway. Nature.
436:7922005. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lee JO, Yang H, Georgescu MM, et al:
Crystal structure of the PTEN tumor suppressor: implications for
its phosphoinositide phosphatase activity and membrane association.
Cell. 99:323–334. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Di Cristofano A and Pandolfi PP: The
multiple roles of PTEN in tumor suppression. Cell. 100:387–390.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cully M, You H, Levine AJ and Mak TW:
Beyond PTEN mutations: the PI3K pathway as an integrator of
multiple inputs during tumorigenesis. Nat Rev Cancer. 6:184–192.
2006. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Accili D and Arden KC: FoxOs at the
crossroads of cellular metabolism, differentiation, and
transformation. Cell. 117:421–426. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Greer EL and Brunet A: FOXO transcription
factors at the interface between longevity and tumor suppression.
Oncogene. 24:7410–7425. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Brunet A, Bonni A, Zigmond MJ, et al: Akt
promotes cell survival by phosphorylating and inhibiting a Forkhead
transcription factor. Cell. 96:857–868. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Morson BC: The evolution of colorectal
carcinoma. Clin Radiol. 35:425–431. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sherman DW, Ye XY, McSherry C, Parkas V,
Calabrese M and Gatto M: Quality of life of patients with advanced
cancer and acquired immune deficiency syndrome and their family
caregivers. J Palliat Med. 9:948–963. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang SQ: Screening and prevention of
colorectal cancer in Haining County. Dis Colon Rectum. 28:300–304.
1985. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang SQ, Zhu SX and Wu JM: Screening and
prevention of colorectal cancer in Haining county. Chin Med J.
93:843–848. 1980.PubMed/NCBI
|
|
15
|
Stambolic V, Suzuki A, de la Pompa JL, et
al: Negative regulation of PKB/Akt-dependent cell survival by the
tumor suppressor PTEN. Cell. 95:29–39. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Eng C: PTEN: one gene, many syndromes. Hum
Mutat. 22:183–198. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rustia A, Wierzbicki V, Marrocco L,
Tossini A, Zamponi C and Lista F: Is exon 5 of the PTEN/MMAC1 gene
a prognostic marker in anaplastic glioma? Neurosurg Rev. 24:97–102.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cheney IW, Neuteboom ST, Vaillancourt MT,
Ramachandra M and Bookstein R: Adenovirus-mediated gene transfer of
MMAC1/PTEN to glioblastoma cells inhibits S phase entry by the
recruitment of p27Kip1 into cyclin E/CDK2 complexes.
Cancer Res. 59:2318–2323. 1999.PubMed/NCBI
|
|
19
|
Cantley LC and Neel BG: New insights into
tumor suppression: PTEN suppresses tumor formation by restraining
the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci USA.
96:4240–4245. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Burgering BM and Kops GJ: Cell cycle and
death control: long live Forkheads. Trends Biochem Sci. 27:352–360.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Burgering BM and Medema RH: Decisions on
life and death: FOXO Forkhead transcription factors are in command
when PKB/Akt is off duty. J Leukoc Biol. 73:689–701. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Brownawell AM, Kops GJ, Macara IG and
Burgering BM: Inhibition of nuclear import by protein kinase B
(Akt) regulates the subcellular distribution and activity of the
forkhead transcription factor AFX. Mol Cell Biol. 21:3534–3546.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shi F and LaPolt PS: Relationship between
FoxO1 protein levels and follicular development, atresia, and
luteinization in the rat ovary. J Endocrinol. 179:195–203. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Brunet A, Kanai F, Stehn J, et al: 14-3-3
transits to the nucleus and participates in dynamic
nucleocytoplasmic transport. J Cell Biol. 156:817–828. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ramaswamy S, Nakamura N, Sansal I,
Bergeron L and Sellers WR: A novel mechanism of gene regulation and
tumor suppression by the transcription factor FKHR. Cancer Cell.
2:81–91. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Medema RH, Kops GJ, Bos JL and Burgering
BM: AFX-like Forkhead transcription factors mediate cell-cycle
regulation by Ras and PKB through p27kip1. Nature.
404:782–787. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kops GJ, Medema RH, Glassford J, et al:
Control of cell cycle exit and entry by protein kinase B-regulated
forkhead transcription factors. Mol Cell Biol. 22:2025–2036. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Schmidt M, Fernandez de Mattos S, van der
Horst A, et al: Cell cycle inhibition by FoxO forkhead
transcription factors involves downregulation of cyclin D. Mol Cell
Biol. 22:7842–7852. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Martinez-Gac L, Marques M, Garcia Z,
Campanero MR and Carrera AC: Control of cyclin G2 mRNA expression
by forkhead transcription factors: novel mechanism for cell cycle
control by phosphoinositide 3-kinase and forkhead. Mol Cell Biol.
24:2181–2189. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ramaswamy S, Nakamura N, Vazquez F, et al:
Regulation of G1 progression by the PTEN tumor suppressor protein
is linked to inhibition of the phosphatidylinositol 3-kinase/Akt
pathway. Proc Natl Acad Sci USA. 96:2110–2115. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Di Cristofano A, De Acetis M, Koff A,
Cordon-Cardo C and Pandolfi PP: Pten and p27KIP1
cooperate in prostate cancer tumor suppression in the mouse. Nat
Genet. 27:222–224. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhu X, Kwon CH, Schlosshauer PW, Ellenson
LH and Baker SJ: PTEN induces G(1) cell cycle arrest and decreases
cyclin D3 levels in endometrial carcinoma cells. Cancer Res.
61:4569–4575. 2001.PubMed/NCBI
|
|
33
|
Weng LP, Brown JL and Eng C: PTEN
coordinates G(1) arrest by down-regulating cyclin D1 via its
protein phosphatase activity and up-regulating p27 via its lipid
phosphatase activity in a breast cancer model. Hum Mol Genet.
10:599–604. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wu RC, Li X and Schonthal AH:
Transcriptional activation of p21WAF1 by PTEN/MMAC1 tumor
suppressor. Mol Cell Biochem. 203:59–71. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Romashkova JA and Makarov SS: NF-kappaB is
a target of AKT in anti-apoptotic PDGF signalling. Nature.
401:86–90. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gustin JA, Maehama T, Dixon JE and Donner
DB: The PTEN tumor suppressor protein inhibits tumor necrosis
factor-induced nuclear factor kappa B activity. J Biol Chem.
276:27740–27744. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yuan XJ and Whang YE: PTEN sensitizes
prostate cancer cells to death receptor-mediated and drug-induced
apoptosis through a FADD-dependent pathway. Oncogene. 21:319–327.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Huang H, Cheville JC, Pan Y, Roche PC,
Schmidt LJ and Tindall DJ: PTEN induces chemosensitivity in
PTEN-mutated prostate cancer cells by suppression of Bcl-2
expression. J Biol Chem. 276:38830–38836. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Mayo LD, Dixon JE, Durden DL, Tonks NK and
Donner DB: PTEN protects p53 from Mdm2 and sensitizes cancer cells
to chemotherapy. J Biol Chem. 277:5484–5489. 2002. View Article : Google Scholar
|
|
40
|
Stambolic V, MacPherson D, Sas D, et al:
Regulation of PTEN transcription by p53. Mol Cell. 8:317–325. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Reagan-Shaw S and Ahmad N: RNA
interference-mediated depletion of phosphoinositide 3-kinase
activates forkhead box class O transcription factors and induces
cell cycle arrest and apoptosis in breast carcinoma cells. Cancer
Res. 66:1062–1069. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Carter JH, Douglass LE, Deddens JA, et al:
Pak-1 expression increases with progression of colorectal
carcinomas to metastasis. Clin Cancer Res. 10:3448–3456. 2004.
View Article : Google Scholar : PubMed/NCBI
|