Introduction
Lung cancer is a common malignant tumor. Patients
particularly with human non-small cell lung carcinomas (NSCLCs),
which constitute 80% of total lung cancer cases worldwide, present
with an extremely low survival rate due to the poor sensitivity of
NSCLCs to traditional chemotherapy and radiotherapy. Thus, new
antitumor chemicals and therapeutic targets must be urgently
developed. Statins, 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA)
reductase inhibitors, are chemotherapeutic agents for lowering
plasma cholesterol. Extensive studies, however, have demonstrated
the role of statins in cancer therapies, particularly the
inhibition of HMG-CoA reductase by statins based on the suppression
of tumor growth, stimulation of cell cycle arrest, and induction of
cancer cell apoptosis in vivo and in vitro. Moreover,
it was reported that patients who were treated with statins prior
to cancer diagnosis exhibited a reduced cancer-related mortality of
up to 15% (1).
Lovastatin, a first generation statin drug, has been
suggested as a promising potential therapeutic agent in cancer.
Bruemmer et al reported that atorvastatin inhibits vascular
smooth muscle cell DNA synthesis by blocking E2F and thereby
decreasing minichromosome maintenance (MCM) 6 and MCM7 expression
(2). Recent studies have also shown
that MCM2, MCM4 and MCM7 are potential therapeutic targets in
NSCLCs (3–5). Although the effects of lovastatin on
the anti-proliferation in cancer cells have been confirmed,
lovastatin’s effects on MCM2 have not yet been fully
elucidated.
DNA replication licensing in eukaryotic cells is a
strictly regulated process that ensures proper genome replication
and inheritance. MCM complexes, including MCM2, MCM3, MCM4, MCM5,
MCM6 and MCM7, bind to replication origins in the G1 phase of the
cell cycle and are the key regulatory components for DNA
replication licensing. Deregulation of these MCM proteins have been
linked to tumor formation, progression and malignant
transformation. Moreover, MCM2 has been considered a valuable
proliferation marker in many types of cancers, including cervical
progressive disease (6), colonic
adenoma and adenocarcinoma (7),
meningiomas (8), gingival
fibromatosis (9), non-melanoma
epithelial skin cancers (10), and
gastric cardiac cancer (11). Thus,
inhibiting MCM2 may present an attractive opportunity for the
development of effective anticancer drugs with few side effects.
Indeed, the silencing of MCM2 has resulted in anti-proliferation,
cell cycle arrest, and apoptosis in several types of cancers, such
as colon cancer (12), yet the
anti-proliferative mechanism of MCM2 under the conditions of
lovastatin treatment for NSCLCs remains unknown. In the present
study, we aimed to explore the possible molecular mechanism of
lovastatin related to MCM2 as a therapeutic target in human
non-small cell lung carcinomas.
Materials and methods
Antibodies and reagents
Lovastatin, purchased from Sigma Chemical Co. (St.
Louis, MO, USA) was dissolved into a refrigerated absolute alcohol
stock solution. The rabbit anti-MCM2 moloclonal antibody, the
anti-β-actin antibody, and secondary antibodies were purchased from
Abcam (Cambridge, UK). CDK4, cyclin D1, protein retinoblastoma
(Rb), Bax, Bcl-2, p21 and p53 antibodies were purchased from
Bioworld Technology Inc. (Visalia, CA, USA). SP60012 was purchased
from Beyotime Biotechnology Inc. (Nantong, China).
Cell lines and culture and drugs
NSCLC cell lines, A549 and GLC-82, provided by the
American Type Culture Collection (ATCC; Manassas, VA, USA), were
cultured in Dulbecco’s modified Eagle’s medium (DMEM) and
supplemented with 10% fetal bovine serum (Hyclone Laboratories,
Logan, UT, USA) containing 5% CO2 at 37°C. The two cell
lines, treated with different concentrations of lovastatin (0, 2.5,
5, 10 and 20 μM), were assessed using the colorimetric
method of MTT [3-(4,5-methylthiazol-2-yl)-2,5-diphenyltetrazolium
bromide] (5 mg/ml; Sigma Chemical Co.) assay. The cells were
incubated at a density of 1×106 cells/well in plates of
24-wells, and allowed to adhere overnight. Then lovastatin
treatment was carried out for 24, 48, 72 and 96 h, respectively.
MTT solution (400 μg/ml) was added and incubated for 1 h.
The formazan crystals were dissolved using dimethylsulfoxide
(DMSO). Quantification was provided by an absorbance reading at a
wavelength of 540 nm. Each test was performed at least 3 times.
RNA interference (RNAi)
A549 and GLC-82 cells were transfected with siRNA
against MCM2 or control siRNA using Lipofectamine 2000™
(Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s
instructions. We designed the MCM2 siRNA-targeting sequence,
5′-GACCUGAGGAAAG AAUCUATT, and a random siRNA sequence, 5′-UAGAUUCU
UUCCUCAGGUCTT, as the control-targeted sequence. For follow-up
tests, RNAi oligonucleotides were performed 48 h later using
Lipofectamine 2000. Cells were harvested and used for real-time PCR
and western blotting 48 h after transfection. A549 and GLC-82 cells
were labeled with propidium iodide (PI) for the cell cycle assay,
or stained with Annexin V for the cell apoptosis assay.
Western blot analysis
Total protein extracts were separated by
electrophoresis on 10% SDS-PAGE amide gels, and then transferred to
PVDF membranes. The PVDF membranes were blocked with 5% skim milk
in Tris-buffered saline containing 0.1% Tween-20 (TBST) and then
incubated with primary antibodies overnight at 4°C, washed with
TBST 3 times for 10 min, then exposed to peroxidase-conjugated
secondary antibodies for 2 h at room temperature and washed with
TBST 3 times for 10 min. Proteins were detected using enhanced
chemiluminescence (ECL; Pierce, Rockford, IL, USA).
RNA extraction and real-time PCR
Total RNA was extracted from lovastatin-treated and
untreated cells using TRIzol reagent (Invitrogen) according to the
manufacturer’s instructions. RNA levels were measured with the SYBR
Premix ExTaq Quantitative PCR kit (Takara, Ossu, Japan). Primers
for MCM2 and GAPDH genes were designed as follows: MCM2 forward,
AGAATCTATGGCGACAG and reverse, ACCTGC TCTGCCACTAACTG; GAPDH
forward, GGAAAGCCTG CCGGTGAC and reverse, ACCTGCTCTGCCACTAACTG.
MCM2 gene expression was analyzed using the Applied Biosystems 7500
Real Time PCR machine. The expression of the target gene relative
to GAPDH was determined using the formula 2−ΔΔCt. The
experiment was repeated 3 times for each group and the mean values
were calculated.
Apoptosis detection
The apoptosis of lovastatin-treated or untreated
cells was detected with the Annexin V fluorescein isothiocyanate
(FITC) apoptosis kit (Beyotime Biotechnology) according to the
manufacturer’s instructions. Differently treated cells were
trysinized and washed 3 times with cold PBS, and then 5 μM
Annexin V-FITC was added to the differently treated cells for 10
min at room temperature and analyzed on a FACSort
(Becton-Dickinson, San Jose, CA, USA).
Flow cytometric analysis
Our cell cycle analysis was performed on a flow
cytometer and analyzed using the ModFit LT2.0 software (Coulter).
A549 and GLC-82 cells were synchronized overnight in serum-free
medium which was replaced by complete medium. The two cell lines
were treated with lovastatin for 24, 48, 72 and 96 h, respectively
prior to being harvested. The treated and control cells
(5×105) were washed with ice-cold PBS 3 times for 10 min
and fixed in cold 70% ethanol solution, then washed with PBS,
treated with RNase (1 μg/ml), and stained with PI (50
μg/ml) (both from Sigma Chemical Co.) for 30 min at 37°C.
DNA distributions were analyzed on a FACSort with Cell Quest
software (version 313).
Statistical analysis
Statistical analysis was performed using SPSS
software, version 12.0 (SPSS, Chicago, IL, USA). Each experiment
was repeated at least 3 times, and the results are expressed as
means ± SD. We considered P<0.05 to indicate a statistically
significant result.
Results
Lovastatin induces anti-proliferation,
G1/S phase arrest, and apoptosis in NSCLC cells
To confirm the anti-proliferative effects of
lovastatin, we cultured two NSCLC cell lines, A549 and GLC-82, and
treated them with lovastatin at various concentrations (0, 2.5, 5,
10 and 20 μmol/l) for 1 to 4 days, respectively (Fig. 1A). The MTT assay showed that
lovastatin treatment produced time- and dose-dependent growth
inhibition in the two cell lines, which was apparent 48 h after
treatment and this time period was used for further
experiments.
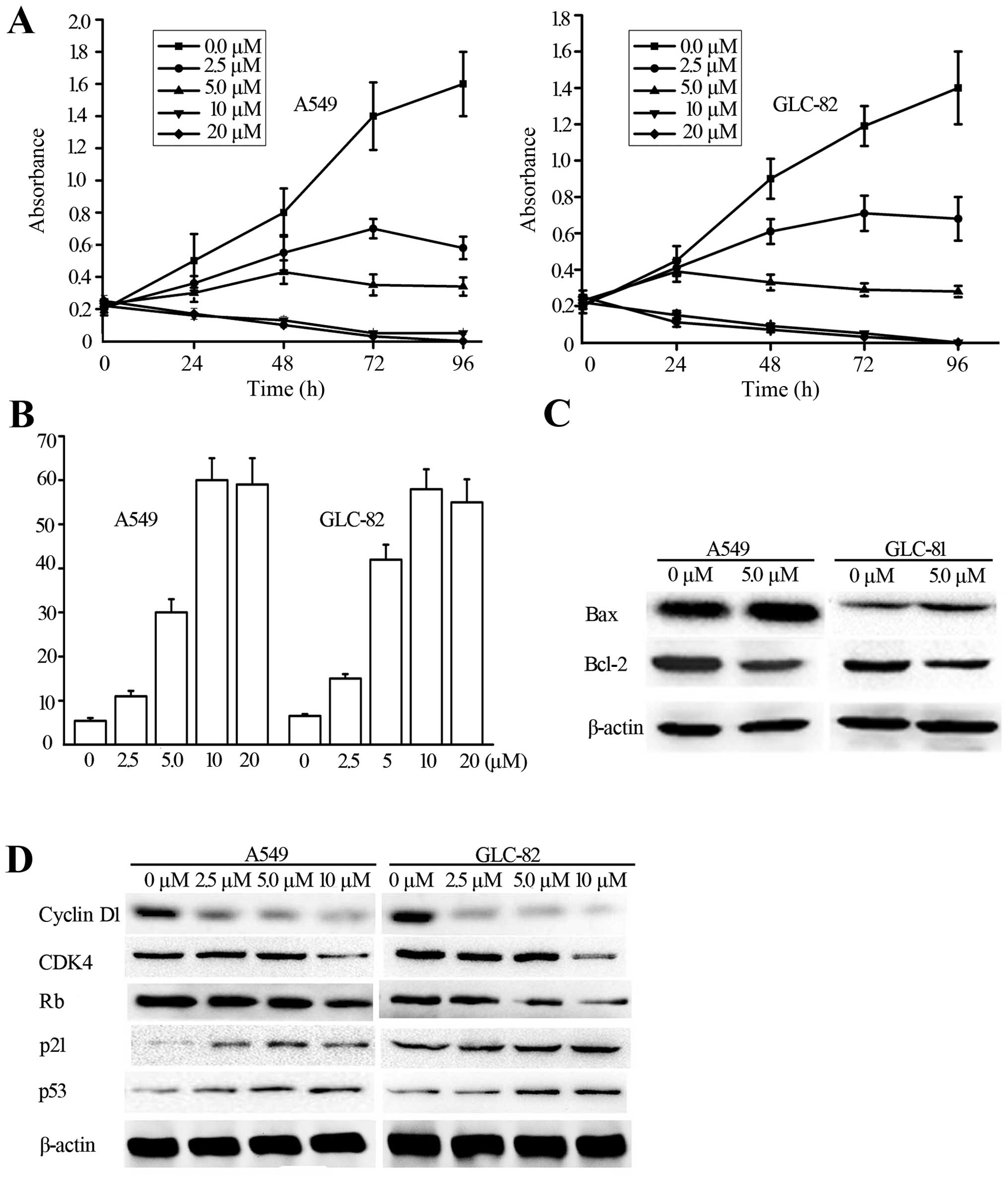 | Figure 1Effects of lovastatin on cell growth,
cell cycle progression and apoptosis in NSCLC cells. (A) Human
NSCLC A549 and GLC-82 cells were treated with different
concentrations (0, 2.5, 5, 10 and 20 μM) of lovastatin for
1, 2, 3 and 4 days, and cell viability was measured by MTT assay.
Data represent means ± SD of triplicate experiments;
*P<0.05, significantly different compared with the
alcohol-treated control. (B) Cell apoptosis was analyzed in A549
and GLC-82 cells following treatment with lovastatin at serial
concentrations (0, 2.5, 5, 10 and 20 μM), as measured by
Annexin V staining. (C) Protein levels of Bcl-2 and Bax were
determined by western blotting in the A549 and GLC-82 cells, and
β-actin was used as the sample loading control. (D) A549 and GLC-82
cells were treated with lovastatin at a series of concentrations of
0, 2.5, 5 and 10 μM for 48 h. Protein levels of CDK4, cyclin
D1, Rb, p53 and p21 were determined by western blot analysis, and
β-actin was used as the sample loading control. All experiments
were performed in triplicates. NSCLCs, human non-small cell lung
carcinomas. |
To investigate the exact mechanism involved in the
anti-proliferative effects of lovastatin, cell cycle progression in
the NSCLC cells was determined. We examined the effect of
lovastatin on cell cycle distribution with PI staining and flow
cytometry. As shown in Table I, for
the A549 cells, the cell population in the G1 phase was 50.1, 56.8,
64.7 and 73.4% at concentrations of lovastatin 0, 2.5, 5.0 and 10
μM, respectively, and, for GLC-82 cells, 59.5, 64.2, 67.2
and 73.5% at concentrations of 0, 2.5, 5.0 and 10 μM,
respectively. Lovastatin treatment markedly increased the G1 phase
population, causing G1/S arrest.
 | Table ILovastatin-induced cell cycle arrest
at the G1 phase. |
Table I
Lovastatin-induced cell cycle arrest
at the G1 phase.
| Cells | Treatment | Cells in each phase
of the cell cycle (%)
|
|---|
| G0/G1 | S | G2/M |
|---|
| A562 | 0 | 50.1±1.5 | 32.8±0.5 | 14.1±0.2 |
| 2.5 μM
lovastatin |
56.8±1.3 | 28.5±1.6 | 14.7±0.5 |
| 5.0 μM
lovastatin |
64.7±0.8 | 23.9±2.3 | 11.4±0.5 |
| 10.0 μM
lovastatin |
73.4±1.1 | 17.7±1.1 | 8.9±0.6 |
| GLC-82 | 0 | 59.5±2.1 | 29.5±2.1 | 11.0±0.4 |
| 2.5 μM
lovastatin |
64.2±1.4 | 24.5±1.1 | 11.3±0.5 |
| 5.0 μM
lovastatin |
67.2±1.6 | 22.9±1.8 | 9.9±0.2 |
| 10.0 μM
lovastatin |
73.5±1.2 | 17.7±0.9 | 8.8±0.3 |
We also aimed to ascertain whether lovastatin
treatment is involved in apoptosis. Annexin V was determined in the
A549 cells after lovastatin treatment, and the percentages of
apoptotic cells were increased after 48 h of lovastatin treatment
(Fig. 1B). To validate the results
further, western blot analysis of several apoptosis-related
markers, such as Bcl-2 and Bax was carried out. As shown in
Fig. 1C, Bax was found to increase
quickly and Bcl-2 was downregulated. The results showed that
apoptosis could be responsible for lovastatin-mediated growth
inhibition.
Lovastatin treatment affects G1/S
transition-related regulators
The G1/S transition of the cell cycle is regulated
by the CDK/cyclin complex, CKI, and Rb. To further clarify the
mechanism of G1/S arrest following lovastatin treatment, we
examined the expression of G1/S transition-related regulators.
First, we used a western blot analysis to test the cell
cycle-related genes CDK4, cyclin D1, Rb, p53, and p21 following
lovastatin treatment. As summarized in Fig. 1D, among these genes, cyclin D, CDK4,
and Rb were greatly inhibited, whereas p21 and p53 were profoundly
upregulated.
Lovastatin treatment inhibits MCM2
expression
MCM2 plays an important role in the origination of
eukaryotic genome replication. However, the exact mechanisms of MCM
members related to lovastatin treatment remain unknown. We
investigated MCM2 mRNA following lovastatin treatment for 48 h by
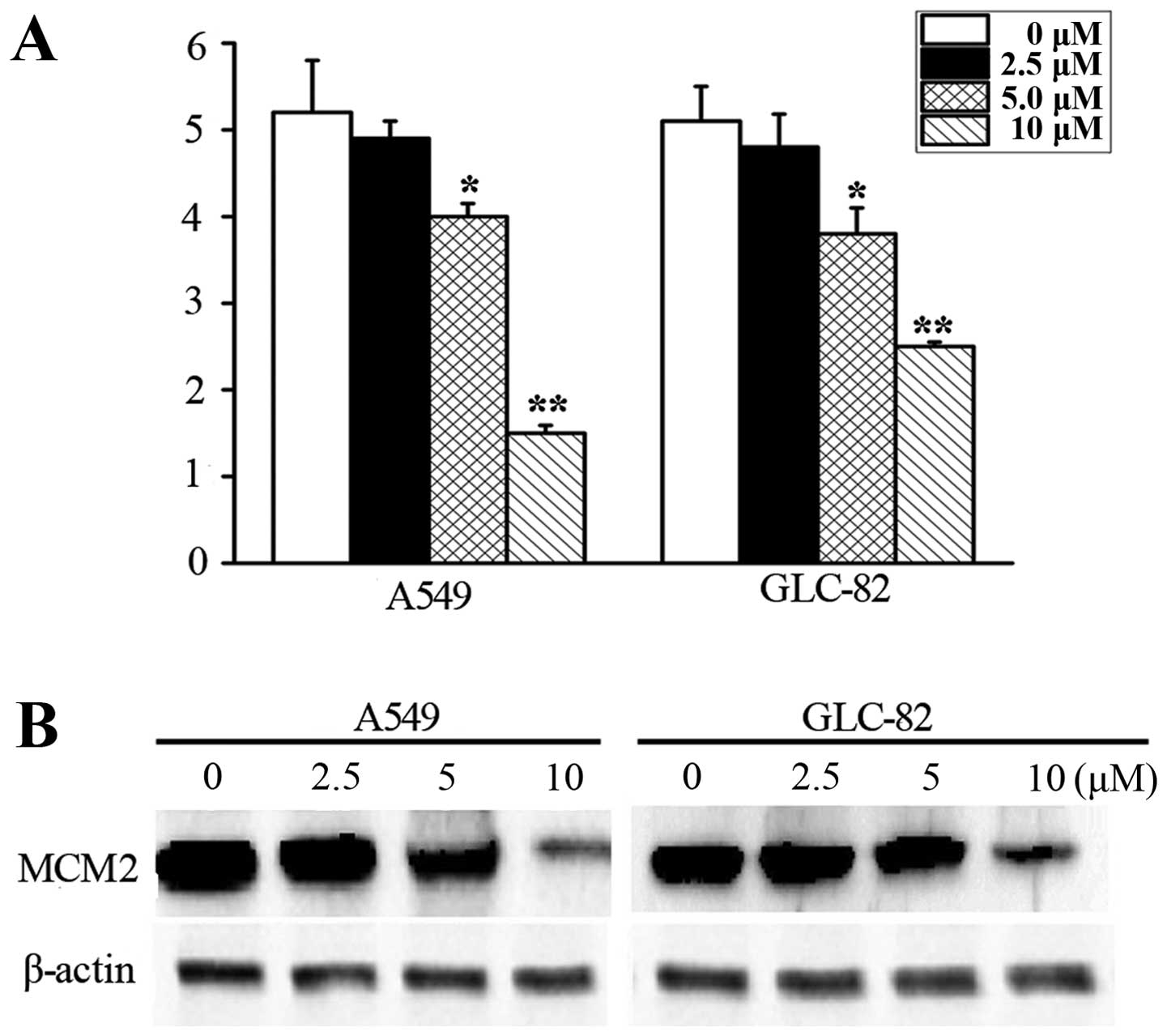
RT-PCR in the A549 and GLC-82 cells. As shown in Fig. 2A, we observed that lovastatin
treatment induced a significant decrease in MCM2. We then confirmed
the inhibition of MCM2 following lovastatin treatment by measuring
the protein levels. Our western blot analysis data showed that MCM2
expression was substantially downregulated after lovastatin
treatment in the A549 and GLC-82 cells (Fig. 2B).
MCM2 knockdown decreases proliferation
and induces G1/S transition arrest in NSCLC cells
Lovastatin treatment induces anti-proliferation and
cell cycle arrest, as well as the downregulation of MCM2
expression. Thus, we aimed to ascertain whether siMCM2 could induce
similar effects. First, we transfected siMCM2 oligonucleotide
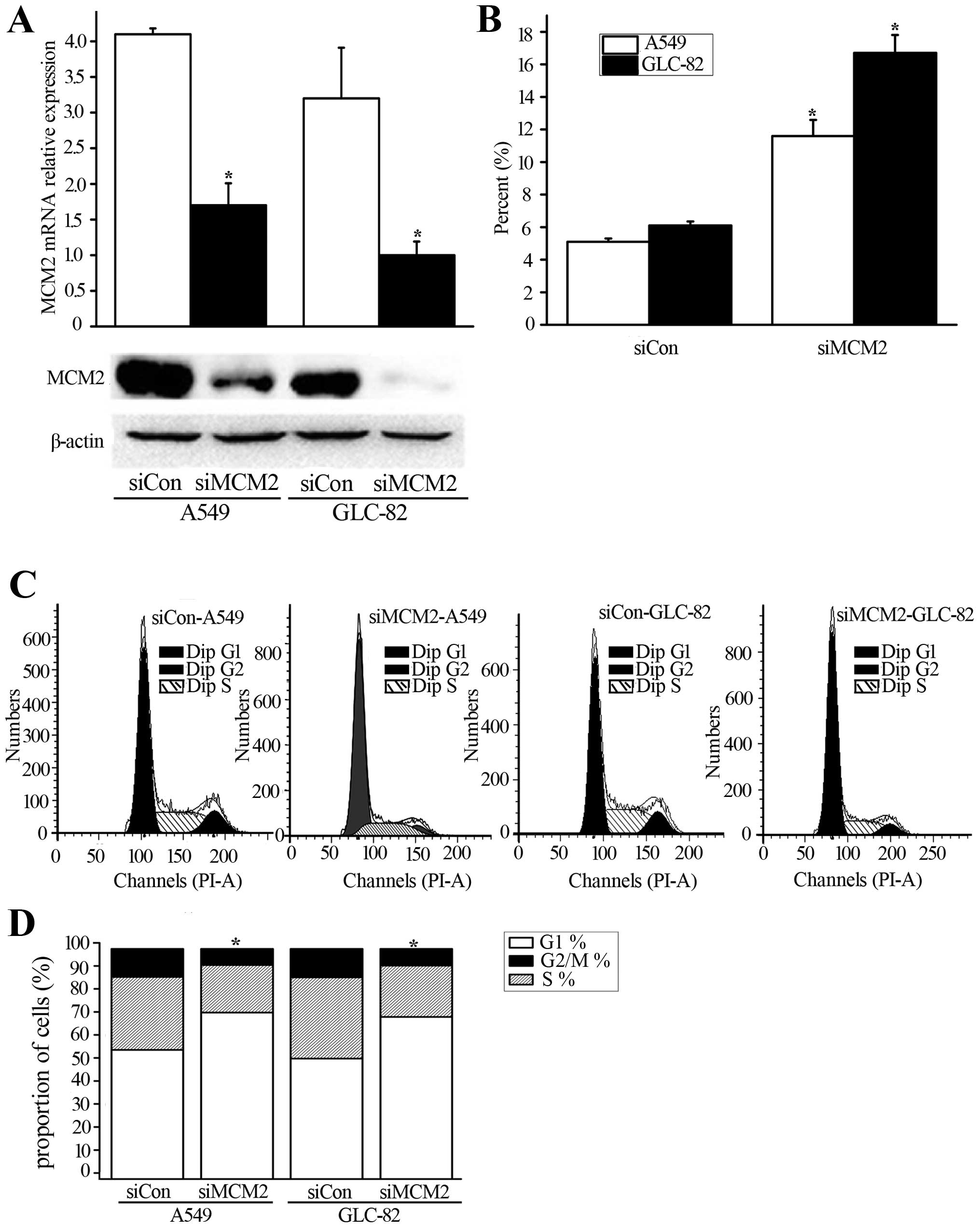
duplexes into A549 and GLC-82 cells. As a result, we determined the
efficacy of anti-proliferation and cell cycle arrest following
siMCM2. As shown in Fig. 3A, MCM2
expression was suppressed after MCM2 knockdown at the mRNA and
protein levels in the A549 and GLC-82 cells. Next, we evaluated the
apoptosis of cancer cells after MCM2 knockdown. siMCM2 was found to
increase the apoptosis of cancer cells compared with the control
(Fig. 3B). Moreover, our assessment
of PI staining and flow cytometry showed that G1/S arrest occurred
following MCM2 knockdown at 48 h (Fig.
3C and D). Thus, MCM2 may be associated with G1/S arrest
following lovastatin treatment.
Changes in G1/S transition-related
regulators and apoptosis factors after silencing MCM2 in NSCLC
cells
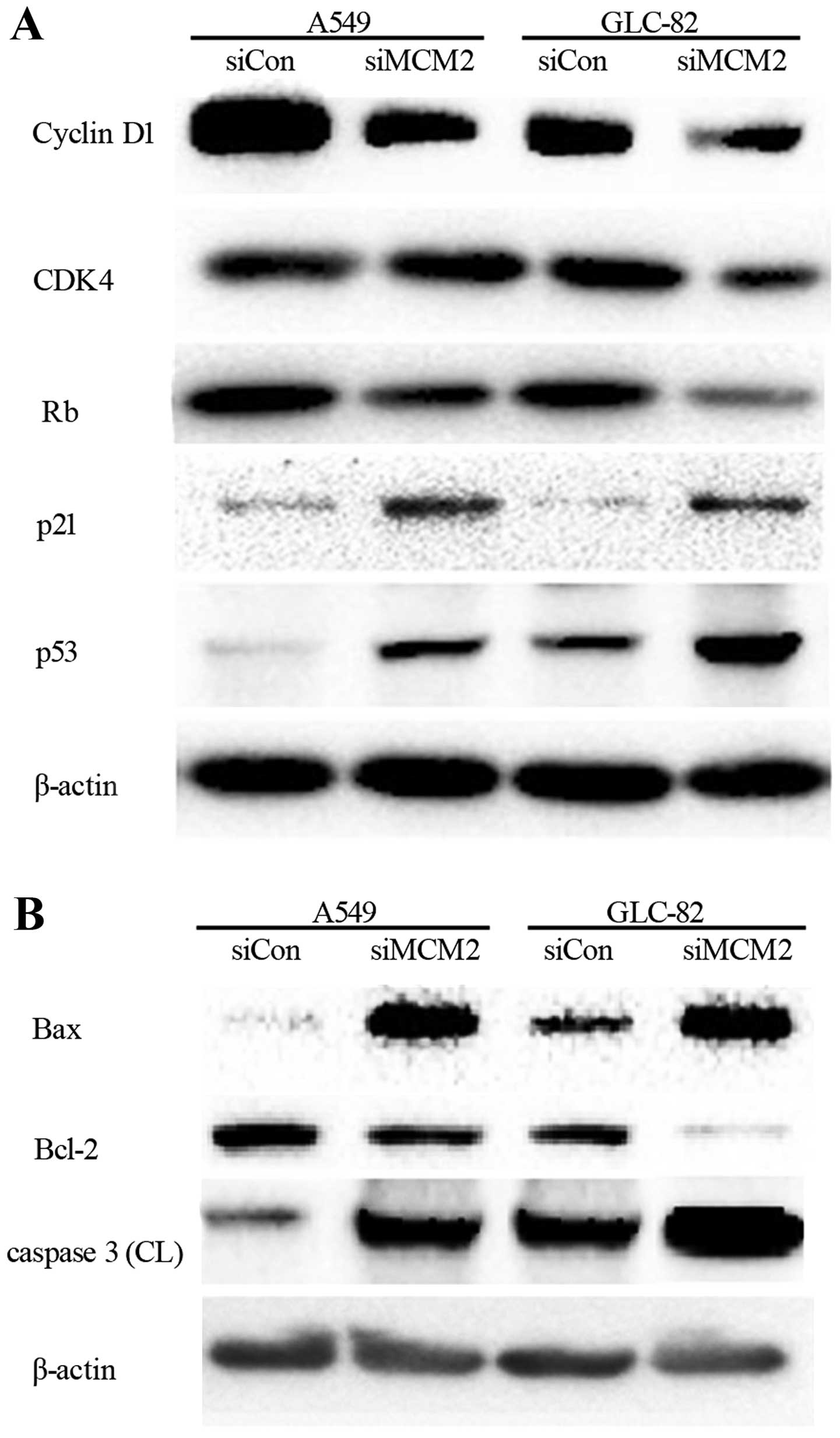
Rb inactivation is a key event in G1 to S
transition, mediated by CDK4/cyclin D1, where the expression of Rb,
CDK4, and cyclin D1 were detected. We found that Rb was markedly
inhibited by MCM2 knockdown in a concentration-dependent manner
after 48 h (Fig. 4A). In addition,
cyclin D1 and CDK4 were significantly downregulated in both cell
lines (Fig. 4A). p21 and p53
proteins, meanwhile, were significantly upregulated in the
MCM2-knockdown cell lines when compared to levels in the cells with
siCon. In conclusion, MCM2 knockdown affected Rb expression, cyclin
D1 and CDK4. In contrast, p21 and p53 increased the rates of G1/S
arrest.
To investigate MCM2 knockdown-induced
apoptosis-related proteins, the activity of caspase-3 was examined
after lovastatin treatment for 48 h. As shown in Fig. 4B, caspase-3 was significantly
increased. We also examined the expression of Bax and Bcl-2
(Fig. 4B). The results showed that
Bax was upregulated and Bcl-2 was downregulated.
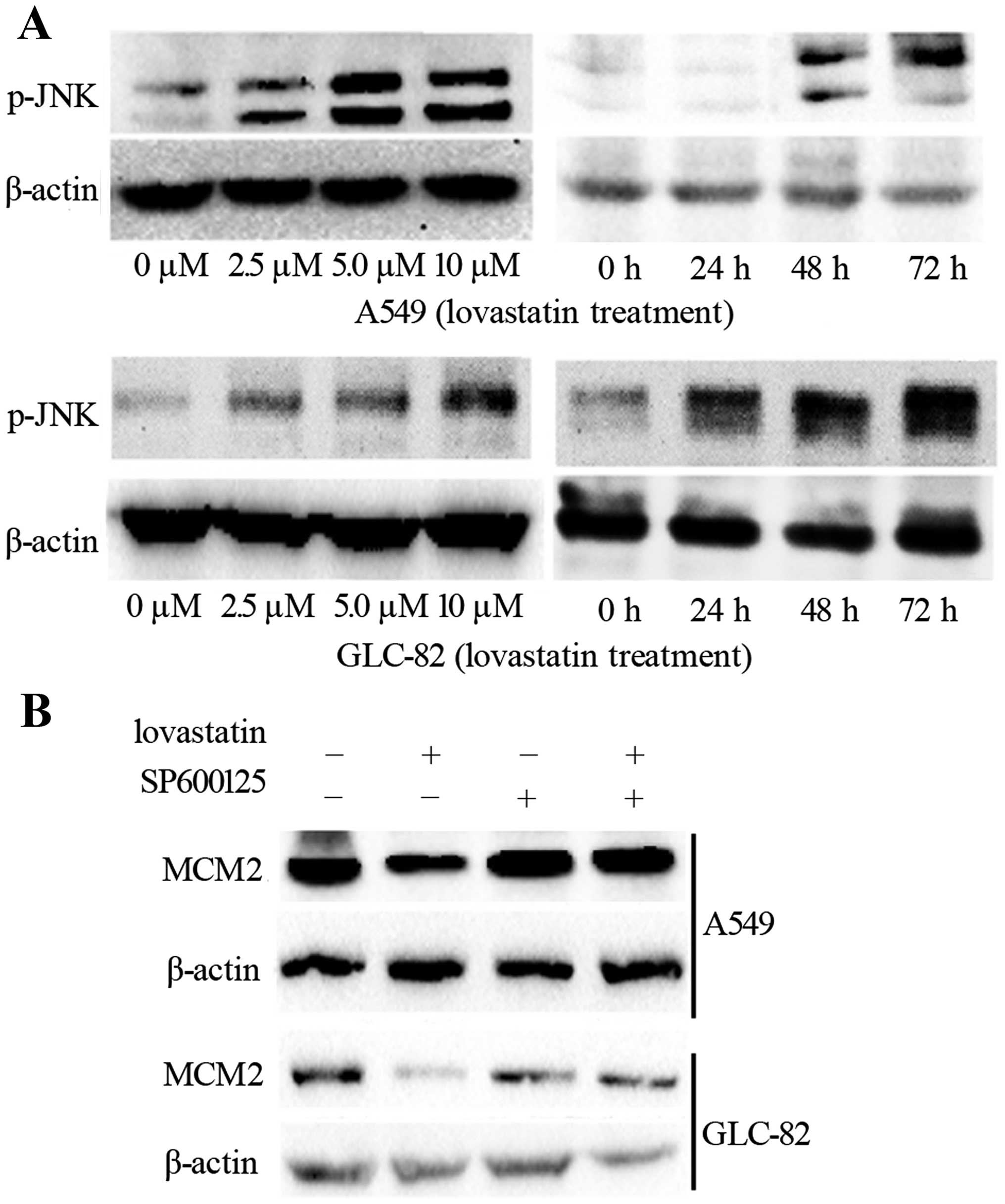
JNK pathway activation is involved in the
downregulation of MCM2 by lovastatin treatment
It has been confirmed in previous studies that JNK
pathway activation is involved in statin treatment in many types of
cancers. To further clarify the mechanism involved in the
proliferation of the inhibitory effects of MCM2, we investigated
whether the JNK pathway is involved in the lovastatin-induced
downregulation of MCM2 in NSCLC cells. We investigated the
expression of p-JNK under the effects of lovastatin treatment. From
the observations (Fig. 5A), p-JNK
was inhibited in a dose- and time-dependent manner. We then used a
specific inhibitor of JNK, SP600125, to treat the lung cancer
cells. As expected, the protein MCM2 was downregulated by
lovastatin treatment alone, while MCM2 was not changed when treated
with the inhibitor SP600125 alone (Fig.
5B). When we combined these two factors, downregulation of MCM2
by lovastatin was restored (Fig.
5B). These results indicated that the JNK pathway is associated
with lovastatin treatment in NSCLC cells.
Discussion
Many studies have confirmed that statins, including
lovastatin, inhibit cell growth and induce cell cycle arrest and
apoptosis in many cancer types, such as human hepatocellular
carcinoma (13,14), Barrett’s esophageal adenocarcinoma
(15), melanoma (15,16),
ARH77 multiple myeloma (17), colon
cancer (18), murine melanoma
(19), prostate (20) and breast cancer (21), and NSCLCs in vitro and in
vivo (3,22–25).
Recent data have demonstrated that atorvastatin combined with
carboplatin or lovastatin combined with ionizing radiation may
offer an effective strategy against NSCLCs (22). Pelaia et al showed that
simvastatin induced pro-apoptotic and anti-proliferative activity
in NSCLC cells by inhibition of the Ras/Raf/MEK/ERK signaling
cascade (25). Furthermore, Falcone
et al used lung cancer tissues to explore the inhibitory
effects of simvastatin and rosuvastatin on MMP-2, MMP-9 and NF-κB
expression (23). Although statins
have been shown to inhibit NSCLC cells growth in vitro and
in vivo via different pathways, the particular mechanism of
anti-proliferation involving lovastatin remains to be elucidated
comprehensively.
We, first, confirmed the effects of lovastatin on
the anti-proliferation of NSCLCs. The results demonstrated that
lovastatin indeed triggered cell proliferation inhibition, cell
cycle arrest and apoptosis (Fig.
1). Previous studies reported that CDK4 and cyclin D activation
of the G1 phase and Rb protein depend on the full expression of
MCMs and fully forming pre-RC (26). Cyclin D1 and CDK4 particularly are
linked to replication licensing. After lovastatin treatment, Rb,
cyclin D1 and CDK4 were inhibited in NSCLC cells. Therefore, we
inferred that lovastatin treatment deactivated the cyclin D1/CDK4
complex, which repressed Rb protein expression p53 can lead to the
stabilization of this tumor suppressor, and it triggers the arrest
of G1/S or G2/M through the induction of cyclin-dependent kinase
inhibitor (CDKI) p21cip14, which can bind to cyclin D/CDK4 to
repress their activity, leading to cell cycle arrest. In the
present study, p21 and p53 were upregulated in siMCM2 cells, which
contributed to G1/S arrest, but not in the siCon-transfected NSCLC
cells (Fig. 4A). The induction of
these kinase inhibitors and inhibition of cell cycle-related
kinases by lovastatin could thus contribute to the observed cell
cycle arrest.
MCMs, a highly conserved helicase and key regulatory
component of eukaryotic DNA replication, are overexpressed in many
types of human malignancies and have been important targets for
cancer chemotherapy. Thus inhibiting eukaryotic replicative
helicase activity of MCM2, would be of significant research
utility. Some small molecular inhibitors of specific replication
factors, such as ciprofloxacin and Trichostatin A, have been
reported as potential therapeutic targets of MCMs (12,27).
In the present study, we explored whether lovastatin can be a
therapeutic target of MCM2. We surprisingly found that MCM2
expression decreased at the mRNA and protein levels (Fig. 2A and B) after lovastatin treatment
in two NSCLC cell lines. We suggest that lovastatin
treatment-mediated cell cycle arrest and cell apoptosis may be
bound up with the inhibition of MCM2, which in turn regulates the
sets of genes responsible for DNA replication.
The relationship between MCM2 expression and
apoptosis is controversial. One study found that MCM2
overexpression induced apoptosis in HL60 cells (28). Another showed that there is no
relationship between MCM2 expression and apoptosis in malignant
fibrous histiocytomas (29). Liu
et al, however, demonstrated that the silencing of MCM2
expression by siRNA induced apoptosis in HCT116 cells (12). Caspases are essential for the
apoptosis process, and apoptosis signals can activate caspase-9,
which in turn activates effector caspases, such as caspase-3. These
caspases can mediate cellular destruction. The conflicting results
in regards to apoptosis and MCM2 expression may be due to the cell
type examined (12). Our data
showed that MCM2 knockdown activated caspase-3, upregulated Bax
expression and downregulated Bcl-2 expression, which led to NSCLC
cell apoptosis.
JNK, one subgroup of MAPKs, is activated in response
to a variety of extracellular signals. It has been demonstrated
that JNKs may act as tumor suppressors (30). Previous studies revealed that statin
treatment could increase p-JNK in many cancers, including ovarian
(31) and human breast cancer
(32). Ogunwobi et al
confirmed that blocking the p-JNK pathway induces apoptosis
(15). Our data demonstrated that
p-JNK was increased after lovastatin treatment alone for 48 h, but
that the pretreatment of NSCLC cells with SP600125 (a JNK
inhibitor) significantly reduced the lovastatin-induced expression
of MCM2, confirming the role of p-JNK in the induction of MCM2 in
NSCLC cells.
In conclusion, the present study describes a novel
anti-proliferation mechanism of MCM2 under the conditions of
lovastatin treatment for NSCLCs. We found that lovastatin treatment
significantly altered expression of cell cycle-related regulators,
and MCM2 knockdown resulted in i) G1/S cell cycle arrest and ii)
activation of apoptosis. In addition, we found that lovastatin
activated the JNK pathway involved in the downregulation of MCM2.
We suggest that MCM2 may be a potential target of lovastatin
treatment in NSCLCs.
Acknowledgments
This study was financially supported by the Natural
Science Foundation of Heilongjiang Province in China (no.
D201111).
References
|
1
|
Nielsen SF, Nordestgaard BG and Bojesen
SE: Statin use and reduced cancer-related mortality. N Engl J Med.
367:1792–1802. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bruemmer D, Yin F, Liu J, Kiyono T, Fleck
E, Van Herle A, Graf K and Law RE: Atorvastatin inhibits expression
of mini-chromosome maintenance proteins in vascular smooth muscle
cells. Eur J Pharmacol. 462:15–23. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Toyokawa G, Masuda K, Daigo Y, et al:
Minichromosome maintenance protein 7 is a potential therapeutic
target in human cancer and a novel prognostic marker of non-small
cell lung cancer. Mol Cancer. 10:652011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kikuchi J, Kinoshita I, Shimizu Y, et al:
Minichromosome maintenance (MCM) protein 4 as a marker for
proliferation and its clinical and clinicopathological significance
in non-small cell lung cancer. Lung Cancer. 72:229–237. 2011.
View Article : Google Scholar
|
|
5
|
Yang J, Ramnath N, Moysich KB, Asch HL,
Swede H, Alrawi SJ, Huberman J, Geradts J, Brooks JS and Tan D:
Prognostic significance of MCM2, Ki-67 and gelsolin in non-small
cell lung cancer. BMC Cancer. 6:2032006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yang QC, Zhu Y, Liou HB, Zhang XJ, Shen Y
and Ji XH: A cocktail of MCM2 and TOP2A, p16INK4a and Ki-67 as
biomarkers for the improved diagnosis of cervical intraepithelial
lesion. Pol J Pathol. 64:21–27. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang Y, Li Y, Zhang WY, Xia QJ, Li HG,
Wang R, Yang L, Sun XF and Zhou ZG: mRNA expression of
minichromosome maintenance 2 in colonic adenoma and adenocarcinoma.
Eur J Cancer Prev. 18:40–45. 2009. View Article : Google Scholar
|
|
8
|
Saydam O, Senol O, Schaaij-Visser TBM,
Pham TV, Piersma SR, Stemmer-Rachamimov AO, Wurdinger T, Peerdeman
SM and Jimenez CR: Comparative protein profiling reveals
minichromosome maintenance (MCM) proteins as novel potential tumor
markers for meningiomas. J Proteome Res. 9:485–494. 2010.
View Article : Google Scholar :
|
|
9
|
Martelli-Júnior H, Santos CO, Bonan PR,
Moura PF, Bitu CC, León JE and Coletta RD: Minichromosome
maintenance 2 and 5 expressions are increased in the epithelium of
hereditary gingival fibromatosis associated with dental
abnormalities. Clinics (Sao Paulo). 66:753–757. 2011. View Article : Google Scholar
|
|
10
|
Abdou AG, Elwahed MG, Serag El-Dien MM and
Eldien DS: Immunohistochemical expression of MCM2 in nonmelanoma
epithelial skin cancers. Am J Dermatopathol. 36:959–964. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pang C, Yuan H, Ren S, Chen Y, An H and
Zhan Y: TMEM16A/B associated CaCC: structural and functional
insights. Protein Pept Lett. 21:94–99. 2014. View Article : Google Scholar
|
|
12
|
Liu Y, He G, Wang Y, Guan X, Pang X and
Zhang B: MCM-2 is a therapeutic target of trichostatin A in colon
cancer cells. Toxicol Lett. 221:23–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Relja B, Meder F, Wilhelm K, Henrich D,
Marzi I and Lehnert M: Simvastatin inhibits cell growth and induces
apoptosis and G0/G1 cell cycle arrest in hepatic cancer cells. Int
J Mol Med. 26:735–741. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sutter AP, Maaser K, Höpfner M, Huether A,
Schuppan D and Scherübl H: Cell cycle arrest and apoptosis
induction in hepatocellular carcinoma cells by HMG-CoA reductase
inhibitors. Synergistic antiproliferative action with ligands of
the peripheral benzodiazepine receptor. J Hepatol. 43:808–816.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ogunwobi OO and Beales ILP: Statins
inhibit proliferation and induce apoptosis in Barrett’s esophageal
adenocarcinoma cells. Am J Gastroenterol. 103:825–837. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kidera Y, Tsubaki M, Yamazoe Y, et al:
Reduction of lung metastasis, cell invasion, and adhesion in mouse
melanoma by statin-induced blockade of the Rho/Rho-associated
coiled-coil-containing protein kinase pathway. J Exp Clin Cancer
Res. 29:1272010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tu YS, Kang XL, Zhou JG, Lv XF, Tang YB
and Guan YY: Involvement of Chk1-Cdc25A-cyclin A/CDK2 pathway in
simvastatin induced S-phase cell cycle arrest and apoptosis in
multiple myeloma cells. Eur J Pharmacol. 670:356–364. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xiao H, Zhang Q, Lin Y, Reddy BS and Yang
CS: Combination of atorvastatin and celecoxib synergistically
induces cell cycle arrest and apoptosis in colon cancer cells. Int
J Cancer. 122:2115–2124. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Favero GMF, F Otuki M, Oliveira KA,
Bohatch MS Jr, Borelli P, Barros FE, Maria DA, Fernandes D and
Bydlowski SP: Simvastatin impairs murine melanoma growth. Lipids
Health Dis. 9:1422010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hoque A, Chen H and Xu XC: Statin induces
apoptosis and cell growth arrest in prostate cancer cells. Cancer
Epidemiol Biomarkers Prev. 17:88–94. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Campbell MJ, Esserman LJ, Zhou Y, et al:
Breast cancer growth prevention by statins. Cancer Res.
66:8707–8714. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sanli T, Liu C, Rashid A, Hopmans SN,
Tsiani E, Schultz C, Farrell T, Singh G, Wright J and Tsakiridis T:
Lovastatin sensitizes lung cancer cells to ionizing radiation:
modulation of molecular pathways of radioresistance and tumor
suppression. J Thorac Oncol. 6:439–450. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Falcone D, Gallelli L, Di Virgilio A,
Tucci L, Scaramuzzino M, Terracciano R, Pelaia G and Savino R:
Effects of simvastatin and rosuvastatin on RAS protein, matrix
metalloproteinases and NF-κB in lung cancer and in normal pulmonary
tissues. Cell Prolif. 46:172–182. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hanai J, Doro N, Sasaki AT, Kobayashi S,
Cantley LC, Seth P and Sukhatme VP: Inhibition of lung cancer
growth: ATP citrate lyase knockdown and statin treatment leads to
dual blockade of mitogen-activated protein kinase (MAPK) and
phosphati-dylinositol-3-kinase (PI3K)/AKT pathways. J Cell Physiol.
227:1709–1720. 2012. View Article : Google Scholar :
|
|
25
|
Pelaia G, Gallelli L, Renda T, et al:
Effects of statins and farnesyl transferase inhibitors on ERK
phosphorylation, apoptosis and cell viability in non-small lung
cancer cells. Cell Prolif. 45:557–565. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu P, Slater DM, Lenburg M, Nevis K, Cook
JG and Vaziri C: Replication licensing promotes cyclin D1
expression and G1 progression in untransformed human cells. Cell
Cycle. 8:125–136. 2009. View Article : Google Scholar
|
|
27
|
Simon N, Bochman ML, Seguin S, Brodsky JL,
Seibel WL and Schwacha A: Ciprofloxacin is an inhibitor of the
Mcm2–7 replicative helicase. Biosci Rep. 33:783–795. 2013.
View Article : Google Scholar
|
|
28
|
Suzuki S, Kurata M, Abe S, Miyazawa R,
Murayama T, Hidaka M, Yamamoto K and Kitagawa M: Overexpression of
MCM2 in myelodysplastic syndromes: association with bone marrow
cell apoptosis and peripheral cytopenia. Exp Mol Pathol.
92:160–166. 2012. View Article : Google Scholar
|
|
29
|
Osaki M, Osaki M, Yamashita H, Shomori K,
Yoshida H and Ito H: Expression of minichromosome maintenance-2 in
human malignant fibrous histiocytomas: correlations with Ki-67 and
p53 expression, and apoptosis. Int J Mol Med. 10:161–168.
2002.PubMed/NCBI
|
|
30
|
Bogoyevitch MA, Boehm I, Oakley A,
Ketterman AJ and Barr RK: Targeting the JNK MAPK cascade for
inhibition: basic science and therapeutic potential. Biochim
Biophys Acta. 1697:89–101. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liu H, Liang SL, Kumar S, Weyman CM, Liu W
and Zhou A: Statins induce apoptosis in ovarian cancer cells
through activation of JNK and enhancement of Bim expression. Cancer
Chemother Pharmacol. 63:997–1005. 2009. View Article : Google Scholar
|
|
32
|
Koyuturk M, Ersoz M and Altiok N:
Simvastatin induces apoptosis in human breast cancer cells: p53 and
estrogen receptor independent pathway requiring signalling through
JNK. Cancer Lett. 250:220–228. 2007. View Article : Google Scholar
|