Introduction
Ovarian cancer is one of the leading causes of
cancer-related deaths among women worldwide (1). Poor prognosis is often associated with
the advanced stages of ovarian cancer wherein angiogenesis and
distant metastases are observed (2,3).
Angiogenesis, a key step in tumor growth, can be induced by
pro-angiogenic factors such as the fibroblast growth factor,
angiopoietin, platelet-derived growth factor and vascular
endothelial growth factor (VEGF) (4). VEGF has been shown to support both
physiological vasculogenesis and cancer vascular network and is
upregulated in many cancer cell types (5). The VEGF family members induce the
proliferation, migration and differentiation of endothelial cells,
the major component of angiogenesis and lymphogenesis, by binding
to VEGF receptor tyrosine kinases on endothelial cells (6,7). The
expression of VEGF and the microvessel density are regulated by
NF-κB (8–10). NF-κB is an important and ubiquitous
transcription factor that often dictates cellular transformation,
proliferation, apoptosis, invasion and angiogenesis. In the
canonical pathway of NF-κB, the activation of an IKK complex
phosphorylates IκB proteins that bind and inhibit NF-κB/Rel
proteins. Phosphorylation of IκB subsequently leads to the
activation of NF-κB/Rel complexes and their translocation to the
nucleus (11). Since the IκB family
has been linked to the development of cancer (12), a super-engineered repressor of IκBα,
i.e. IκBαM (S32A,S36A) was generated (13). IκBαM has been shown to inhibit the
activity of IκBα through blocking the phosphorylation of endogenous
IκBα, prevent the translocation of p65 into the nucleus (14–16)
and reduce angiogenesis and metastases in ovarian cancer cell lines
and other human cancer cell lines (8,17,18).
Rhizoma paridis, a stem of Paris
polyphylla Smith var. chinensis (Franch.) Hara or Paris
polyphylla Smith var. yunnanensis (Franch.) Hand-Mazz., is
known for its many clinical applications in traditional Chinese
medicine. Its active components have been used to treat traumatic
bleeding, inflammation and microbial infection (19) and most recently, cancer (20). Of the five main active components of
Rhizoma paridis (21–23),
Paris saponin I (PSI) (polyphyllin D) and Paris saponin II (PSII)
(formosanin C), the steroidal saponins, have displayed potent
albeit selective cytotoxic effects on tumor cells (24). In our previous study, we
demonstrated that PSII suppressed the growth of human ovarian
cancer cells via multiple mechanisms including regulation of ERK1/2
activity, promotion cell cycle arrest and activation of the
mitochondrial apoptotic pathway (25). We also observed that PSII treatment
reduced the expression of NF-κB-downstream targets such as VEGF,
Bcl-2 and Bcl-xL. These observations prompted us, in the present
study, to examine the possibility that PSII modulates NF-κB
activity and VEGF-mediated angiogenesis. We also attempted to
elucidate the molecular mechanisms underlying such effects in the
present study. Our studies revealed that PSII rendered its
inhibitory effects by suppressing NF-κB signaling in ovarian cancer
cells. We also showed that the combination treatment of PSII
treatment and the transfection of a super-engineered repressor
IκBαM into SKOV3 cells markedly reduced angiogenesis and tumor
growth in xenograft mouse models.
Materials and methods
Reagents
Purified PSII, isolated from Rhizoma paridis
(25), was provided by the
Department of Pharmacology at Sichuan University (Chengdu, Sichuan,
China). VEGF was obtained from R&D Systems (Minneapolis, MN,
USA). VEGF ELISA kit was purchased from R&D Systems. Growth
factor-reduced Matrigel was from BD Biosciences (San Jose, CA,
USA). Antibodies against VEGFR2, VEGF, Bcl-2 and Bcl-xL were
obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA).
β-actin antibody was purchased from Sigma Chemical Co. (St. Louis,
MO, USA). CD31 antibodies were from Epitomics, Inc. (Burlingame,
CA, USA).
Cell lines and cell culture
Primary human umbilical vascular endothelial cells
(HUVECs) were from Sichuan university, China. The human high-grade
serous ovarian cancer SKOV3 cell line was purchased from the
American Type Culture Collection (ATCC; Rockville, MD, USA).
SKOV3/vector and SKOV3/IκBαM cells were obtained from the
University of Texas M.D. Anderson Cancer Center. HUVECs were
maintained in endothelial cell culture medium (ECM) (Lonza,
Walkersville, MD, USA) as per the manufacturer’s protocol. The
SKOV3 cell line was cultured in RPMI-1640 medium (Gibco-BRL, Life
Technologies Gaithersburg, MD, USA) supplemented with 10% fetal
bovine serum (FBS) (HyClone, Logan, UT, USA) at 37°C under a
humidified 95:5% (v/v) mixture of air and CO2.
Cell viability assay
Cells (5×103 cells/well) were directly
incubated with indicated concentrations of PSII for indicated
durations. Carrier dimethyl sulfoxide (DMSO) (<0.1%) was used as
a negative control. The cell viability was examined by the
3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT;
Sigma) assay (26). The cytotoxic
effects of PSII were expressed as the 50% inhibitory concentration
(IC50).
Terminal deoxynucleotidyl
transferase-mediated dUTP nick end labeling (TUNEL) assay
The terminal deoxynucleotidyl transferase-mediated
dUTP nick end labeling assay was performed as previously described
(25). Briefly, cells were treated
with PSII after seeding on coverslides for 24 h. All cell layers
were fixed by a 4% paraformaldehyde solution, washed in
phosphate-buffered saline (PBS), and then permeated by a
permeabilization solution (0.1% Triton X-100 solution) for 2 min at
4°C. The apoptosis index was determined as the percentage of
TUNEL-positive cells/1,000 4,6-diamidino-2-phenylindole
(DAPI)-stained nuclei. Random fields were recorded using
fluorescence microscopy (confocal scanning laser microscopy, Leica
TCS4D; Germany).
Endothelial cell Transwell migration
assay
The chemotactic motility was carried out using a
Transwell migration assay with 8.0-μm pore polycarbonate
filter inserts as previously described (27). Briefly, the well was coated with
0.1% gelatin. HUVECs (2×104 cells/well) in ECM medium
supplemented with 0.5% FBS were placed in the top chamber. In the
bottom chamber, VEGF (50 ng/ml) in ECM medium was used as a
positive control. Conditioned media from SKOV3/vector or
SKOV3/IκBαM cells cultured in the presence or absence of PSII were
used as a chemoattractant. After 18 h of incubation, the migrated
cells were fixed with 4% paraformaldehyde and stained with 1%
crystal violet. Images were captured using an Olympus inverted
microscope (Olympus; magnification, ×200). The migrated cells were
counted. Three independent experiments were carried out.
Endothelial cell tube-like network
formation assay
A tube formation assay was carried out as previously
described (28). Briefly, each well
of pre-chilled 24-well plates was coated with 100 μl reduced
growth factor Matrigel (2.8 mg/ml; BD Biosciences). Matrigel was
polymerized at 37°C for 45 min. SKOV3 cells were treated with or
without PSII and then incubated with fresh media without PSII for
24 h. The conditioned media were collected and used to study the
in vitro tube formation assay. Endothelial cells
(1.5×104 cells/ml) in ECM medium supplemented with 0.5%
FBS were placed onto the layer of Matrigel. VEGF (50 ng/ml) was
used as a positive control for tube formation. After 20 h of
incubation at 37°C, tubulogenesis was fixed and photographed using
a Zeiss microscope equipped with a digital camera and Matrox
Intellicam imaging software. Tubular structures were quantified
using AngioSys software (TCS Cellworks) as per the manufacturer’s
instructions.
Western blot analysis
The whole-cell lysates were obtained using
radioimmunoprecipitation assay (RIPA; Sigma) buffer (20 mmol/l
Tris, 2.5 mmol/l EDTA, 1% Triton X-100, 1% deoxycholate, 0.1% SDS,
40 mmol/l NaF, 10 mmol/l Na4P2O7
and 1 mmol/l phenylmethylsulfonyl fluoride). Protein concentrations
were determined using the Bradford assay (Bio-Rad, Hercules, CA,
USA) and equalized before loading. Twenty five micrograms of total
proteins were resolved using 12% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Primary
antibodies against Bcl-2 (1:1,000), Bcl-xL (1:1,000), VEGF
(1:2,000), IκBα (1:1,000) p-IκBα (1:1,000, Ser32), phosphorylated
P65 (1:2,000, Ser536), P65 (1:2,000), IκκB (1:1,000) (all from Cell
Signaling, Danvers, MA, USA), and HRP-conjugated secondary
antibodies were used for the studies. Relative optical density of
the protein signals of interest were qualified by ImageJ software
(NIH).
Electrophoretic mobility shift assay
(EMSA)
EMSA study was carried out as previously described
(13). Briefly, SKOV3 cells were
pretreated with various concentrations of PSII for 24 h. Nuclear
extracts were prepared. EMSA was carried out using Ds-Cold-NF-κB
probes: 5′-AGT TGA GGG GAC TT CCC AGG C-3′ and 5-TGG GGA ACC TGT
GCT GAG TCA CTG GAG-3′. The positive control was the nuclear
extracts from the SKOV3 cells treated with 50 ng/ml TNF for 30 min.
The negative control was the extracts from the SKOV3 cells without
any treatment.
Immunoprecipitation (IP) and in vitro
kinase assay
The IP and in vitro kinase assays were
carried out as previously described (29). Briefly, reaction mixtures (25
μl) contained 40 mM β-glycerophosphate, 7.5 mM
MgCl2, 5% glycerol, 7.5 mM EGTA, [γ-32P] ATP
(0.2 mM, 1 μCi), 1 mM orthovanadate, 50 mM NaF and 0.1%
(v/v) β-mercaptoethanol. The cytoplasmic extract (2 mg)
immunoprecipitated with the appropriate antibody was used for the
phosphorylation reaction and was washed with lysis buffer
containing 50 mM Tris-HCl (pH 7.5), 120 mM NaCl, 5 mM EDTA, 50 mM
NaF, 0.2 mM Na3VO4, 1 mM DTT, 0.5% NP-40 and
protease inhibitors (Protease Inhibitor Cocktail Tablets;
Boehringer, Mannheim; 1 tablet/50 ml) or with 1 μg of
purified recombinant GST-IκBα (Cell Signaling) at 37°C for 1 h.
Reactions were stopped by adding 1 volume of Laemmli sample buffer
containing 5% β-mercaptoethanol and resolved on a 4–20% SDS/PAGE
gel. Gels were autoradiographed and bands were counted using a
Molecular Dynamics PhosphorImager software.
Histology and immunohistochemistry
Paraffin tumor sections (5 μm) were derived
from solid tumor sections that were resected, fixed with 10%
formaldehyde and embedded in paraffin. Sections were treated with
0.3% hydrogen peroxide at room temperature to block endogenous
peroxidase activities ensued by a 5% bovine serum albumin
incubation. Tumor sections were exposed to antibodies against CD31
(1:100) and VEGFR2 (1:100). Images were captured by a Leica DM
4000B photomicroscope (Solms, Germany; magnification, ×200 and
×400). The microvessel density was based on CD31
immunohistochemical signals calculated by Image-Pro Plus 6.0
program (Media Cybernetics) (n=5). Hematoxylin and eosin (H&E)
staining was carried out using standard techniques.
Xenograft mouse model of human ovarian
tumor
The human ovarian mouse tumor model has been
previously described (26).
Briefly, 4- to 6-week-old female Balb/c nude mice (Chengdu
Experimental Animal Center, Chengdu, China) were randomly divided
into 6 groups (n=5). SKOV3/vector or SKOV3/IκBαM cells
(5×106 cells/100 μl) were injected subcutaneously
into the mice. One week after the implantation, mice were treated
with PSII (15 and 25 mg/kg) by daily intraperitoneal injections.
The administrations were carried out on 4 consecutive days/week for
4 weeks (between day 8 and 35). Control groups received the control
solution containing the same amount of DMSO (v <0.1%) without
PSII. The body weights and tumor volumes were recorded twice
weekly. The volume (V) of the solid tumors was measured by a
caliber and calculated according to the formula: V = length ×
width2 × 0.52. The implanted tumors and vessels were
monitored by a Philips HD11 ultrasound scanner (Philips Medical
Systems, Best, The Netherlands) equipped with a 11 MHz linear array
transducer. The volume of solid tumors (expressed in millimeters)
was documented in three dimensions (length, width and height). The
minimum diameter of the lesion that can be detected by ultrasound
is 0.01 cm. At the termination of the present study, all mice were
euthanized using carbon dioxide asphyxiation. All experiments were
conducted based on the National Institutes of Health Guidelines for
the Care and use of Experimental Animals. All protocols were
approved by the Animal Investigation Committee of the Institute for
Nutritional Sciences (Shanghai, China).
Statistical analysis
Statistical significance of differences between
groups was performed using one-way ANOVA followed by the Student’s
t-test. Data are presented as mean ± standard error (SE).
IC50 values were calculated by SPSS software version
13.0 (SPSS, Inc., China). A value of p≤0. 05 was considered to
indicate a statistically significant result.
Results
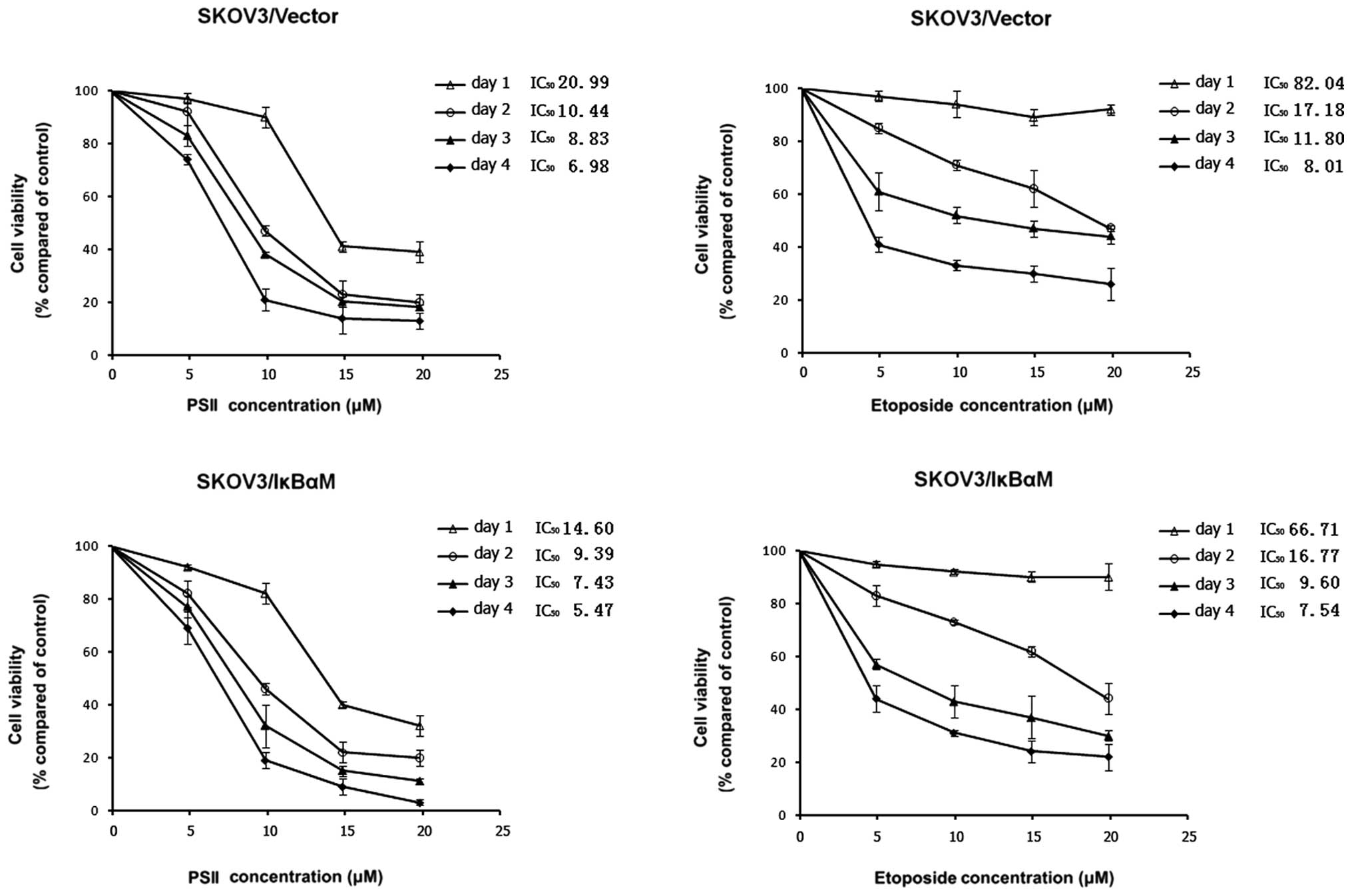
PSII treatment inhibits the growth of
human tumor cell lines
In our previous study, we demonstrated that PSII had
no effects on the survival of non-tumorigenic human vascular smooth
muscle, human bronchial or ovarian surface epithelial (OSE) cells.
However, PSII decreased the cell viability and inhibited the growth
of human tumor cell lines including a high-grade serous ovarian
cancer cell line SKOV3 in a concentration-dependent manner
(28). Here, we further
demonstrated that PSII exhibited more potent antitumorigenic
activity than VP16-etoposide. Kinetic studies showed that PSII
treatment inhibited SKOV3 cell growth in a concentration-dependent
manner. Most important, at the same concentration and at the same
time point, PSII killed more cells than VP16, a positive control
(Fig. 1). PSII treatment resulted
in lower IC50 values (20.99, 10.44, 8.83 and 6.98
μM, day 1–4, respectively) than those of VP16 (82.04, 17.18,
11.80 and 8.01 μM, day 1–4, respectively).
A previous study showed that NF-κB plays a crucial
role in SKOV3 tumor cell growth (13). The transfection of SKOV3 cells with
IκBαM reduced the DNA binding and gene transcription activity of
NF-κB. using this model system, we wanted to further characterize
the antitumorigenic property of PSII under a condition wherein the
NF-κB signaling pathway is compromised. Kinetic studies (Fig. 1) demonstrated that the combination
treatment (PSII and the transfection of IκBαM into SKOV3 cells)
rendered marked inhibitory effects on tumor cell growth. The
antitumorigenic effect was more effective compared to the PSII only
treatment. Notably, this combination (PSII and IκBαM transfection)
was more potent than a combination treatment of IκBαM transfection
and VP16, an antimicrotubule agent (our positive control). For
example, the maximum inhibitory ratio achieved with 10 μM
PSII treatment and IκBαM transfection following a 4-day treatment
was 82% compared with the 65% inhibition produced by 10 μM
VP16 treatment and IκBαM transfection. In this model, PSII
treatments also yielded lower IC50 values compared to
those of the VP16 treatments. Since our interest focused on the
effects of PSII on NF-κB activity, VEGF-mediated angiogenesis, and
the molecular mechanisms underlying such effects but not cell
death, the above studies allowed us to identify the sub-cytotoxic
concentrations of PSII to be used for most of the subsequent
studies (2.5 and 5 μM).
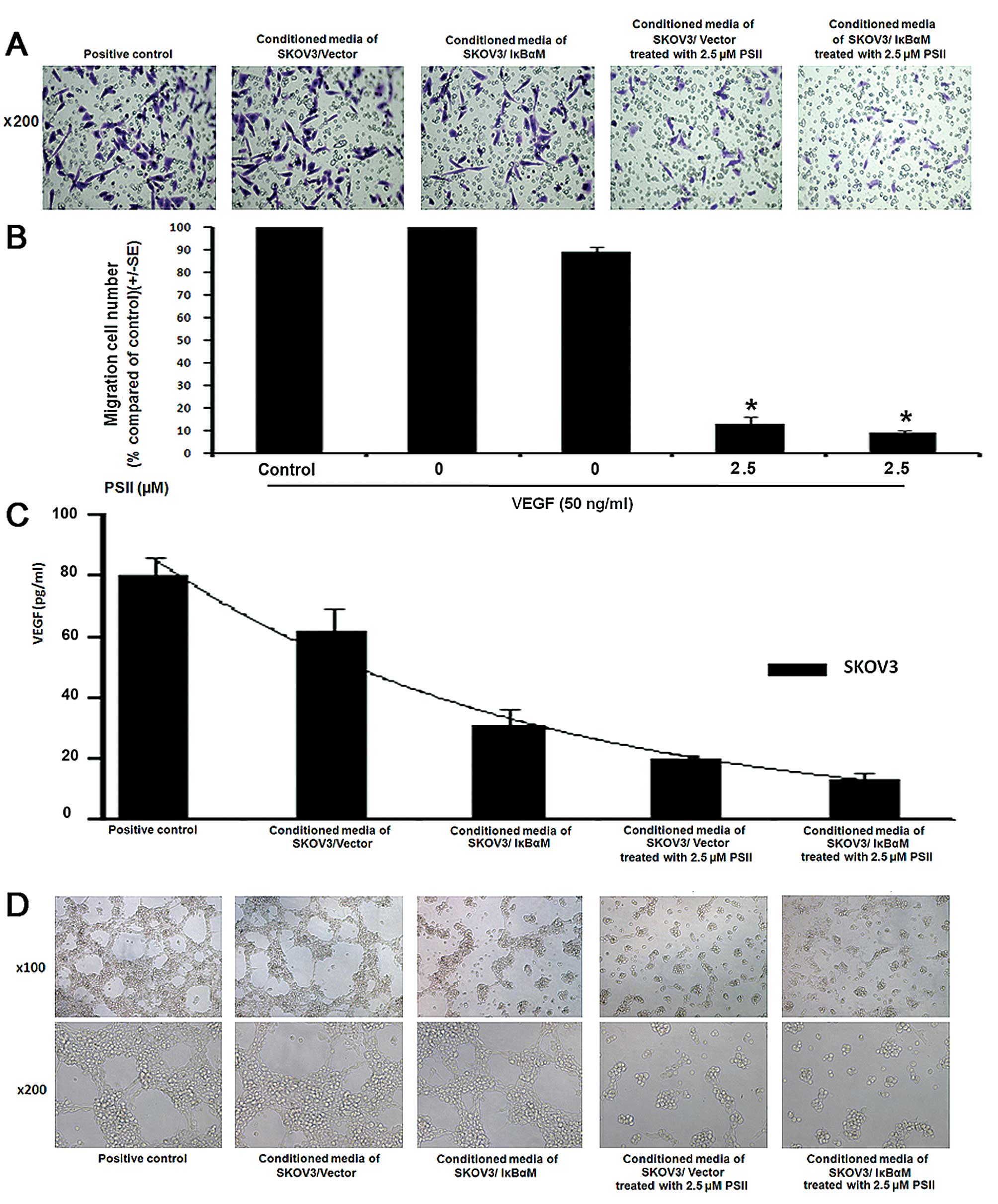
PSII inhibits SKOV3 cell-induced HUVEC
motility and tube-like network formation
Cell motility and tubulogenesis are crucial steps in
angiogenesis (30). Therefore, we
wanted to determine whether PSII treatment affects the ability of
tumor cells to induce endothelial cell migration and tube
formation. First, using the Boyden chamber assay, we showed that
conditioned media from the PSII-treated SKOV3 cells failed to
induce HUVEC migration as compared to the control-treated group
(carrier DMSO, <0.1%) and the VEGF-induced migration group
(Fig. 2A and B). We further
demonstrated that PSII treatment also modulated SKOV3 cell-induced
tubulogenesis. As shown in Fig. 2D,
conditioned media from the PSII-treated SKOV3 cells also failed to
induce tube formation in a concentration-dependent manner compared
to the control treatment (VEGF only, 50 ng/ml; and DMSO carrier,
<0.1%). The disruption of tube formation was observed in HUVECs
following treatment with cytotoxic levels of PSII (2.5 μM)
(28). We also measured the VEGF
level in the conditioned medium obtained from the PSII-treated
SKOV3 cells. A marked reduction in VEGF levels was observed in the
conditioned media of the PSII-treated SKOV3 cells compared to the
control (Fig. 2C). We further
examined the possibility that the reduced VEGF level may be
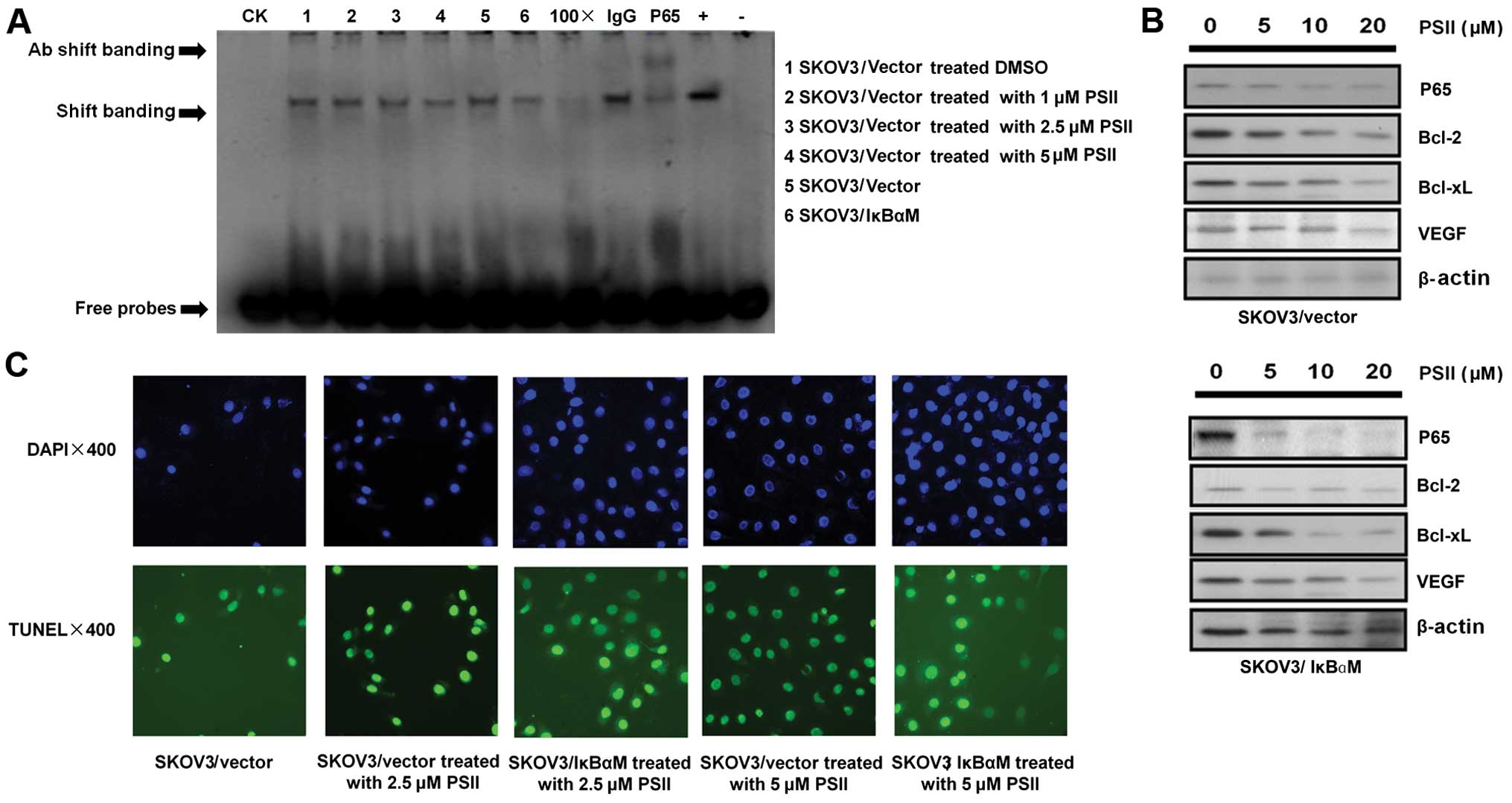
attributed to cell death. As shown in Fig. 3C, TUNEL staining studies showed a
minimal increase in the percentage of TUNEL-positive SKOV3 cells
following treatment with 2.5 μM PSII compared to the
control. Together, the results suggest that PSII treatment
modulates VEGF levels in the tumor microenvironment leading to
reduced capillary formation.
PSII inhibits the activity of NF-κB in
human ovarian cancer cells
Since a previous study (13) demonstrated that NF-κB plays an
important role in SKOV3 tumor cell growth and PSII treatment
affects the levels of SKOV3-secreted VEGF, a target of the NF-κB
signaling pathway (31), we wanted
to examine whether PSII modulates NF-κB activity in SKOV3 cells. In
Fig. 3A, EMSA data indicated that
the binding of NF-κB to its promoter DNA consensus sequence was
reduced in the PSII- treated cells at a concentration as low as 2.5
μM. Notably, PSII treatment (5 μM) and the
transfection of IκBαM into SKOV3 cells yielded a comparable
reduction in DNA binding. Such observable effects of PSII on NF-κB
transcriptional activity led us to examine potential changes in the
expression of downstream NF-κB targets. In Fig. 3B, we showed that PSII treatment
modulated the expression of well-known NF-κB targets, e.g. Bcl-2,
Bcl-xL and VEGF (31). PSII
treatment led to the downregulation of these targets in a
concentration-dependent manner. This finding supports the previous
experiment showing the reduced levels of secreted VEGF in the
conditioned media of PSII-treated cells (Fig. 2C). Together, the results indicated
that PSII treatment compromises NF-κB activation and reduces the
expression of several NF-κB-downstream targets known to play
pivotal roles in tumor growth biology.
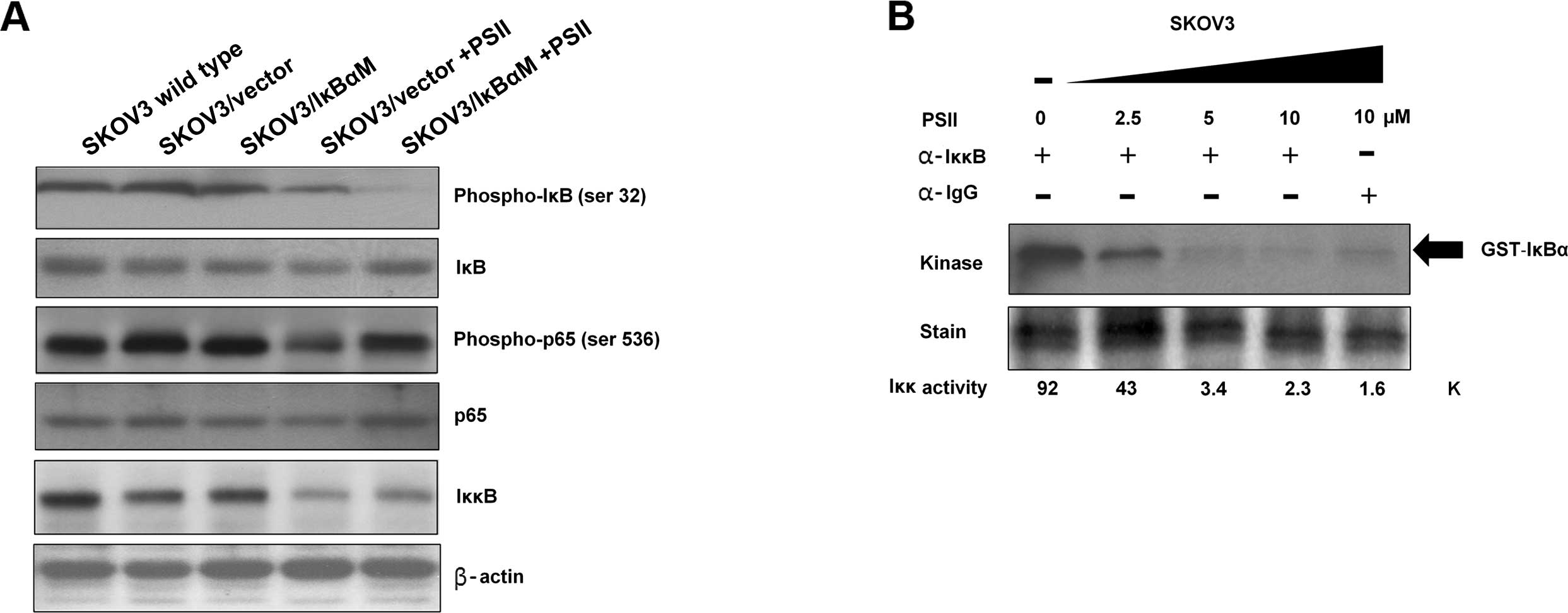
Effect of PSII on components of the NF-κB
canonical pathway
Since the binding of NF-κB to its promoter DNA
consensus sequence and the expression of several NF-κB-downstream
targets were altered by the treatment of PSII, we wanted to
determine whether IκBα or P65 levels were altered in PSII-treated
cells. Protein signals of IκBα, phosphorylated-IκBα, P65,
phosphorylated-P65 and IKKβ were assessed. Protein signals from the
extracts of SKOV3, SKOV3/vector and SKOV3/IκBαM cells were used as
controls. The results from Fig. 4A
showed that while there was no change in the total IκBα and P65
levels in all conditions, unexpectedly, PSII treatment (2.5
μM) appeared to reduce IKKβ expression. Consistent with this
observation, PSII treatment also reduced phosphorylation of IκBα
and P65 on Ser32 and Ser536, respectively.
Next, we examined whether PSII treatment also
affects IKKβ kinase activity on its substrate IκBα.
Immunoprecipitated IKKβ from the extracts of the PSII-SKOV3 treated
cells and carrier DMSO-treated cells were used in an in
vitro kinase assays. Fig. 4B
showed that control cells had stronger IKKβ kinase activity as
compared to that of the PSII-treated SKOV3 cells. Indeed, PSII
treatment suppressed IKKβ activity in a concentration-dependent
manner. Together, these results indicate that PSII targets IKKβ
leading to a reduction in NF-κB signaling and the expression of
NF-κB-downstream targets.
PSII and mutant IκBα treatment inhibit
tumor growth and angiogenesis in a xenograft mouse model of human
ovarian cancer
Our previous study showed that PSII suppresses tumor
growth in a xenograft mouse model of ovarian cancer (25). To characterize the anti-angiogenic
property of PSII in vivo and the relationship with NF-κB
signaling pathway, we continued to use this xenograft model
employing SKOV3/vector and SKOV3/IκBαM cells. Consistent with our
previous finding (28), PSII
treatments suppressed tumor growth rates and reduced the tumor
weights and tumor sizes as compared to the control treatment
(Fig. 5A and C). In our previous
study, we demonstrated that color Doppler ultrasound can be used as
a non-invasive method to assess angiogenesis (28). using this approach (Fig. 5B), here, we confirmed that, on day
35, before the termination of the in vivo tumor growth
study, PSII rendered profound inhibitory effects on neovascularity
refected by the stark reduction in spectral and color Doppler
signals in the PSII-treated groups compared to that of the control
mice. The maximum diameter of blood vessels and microvessel density
were significantly reduced (p<0.05) (Fig. 5C). The aforementioned reductions
were also correlated with the suppression of tumor growth evident
by a >50% reduction in tumor wet weights as compared to that of
the controls (p<0.05) (Fig.
5C).
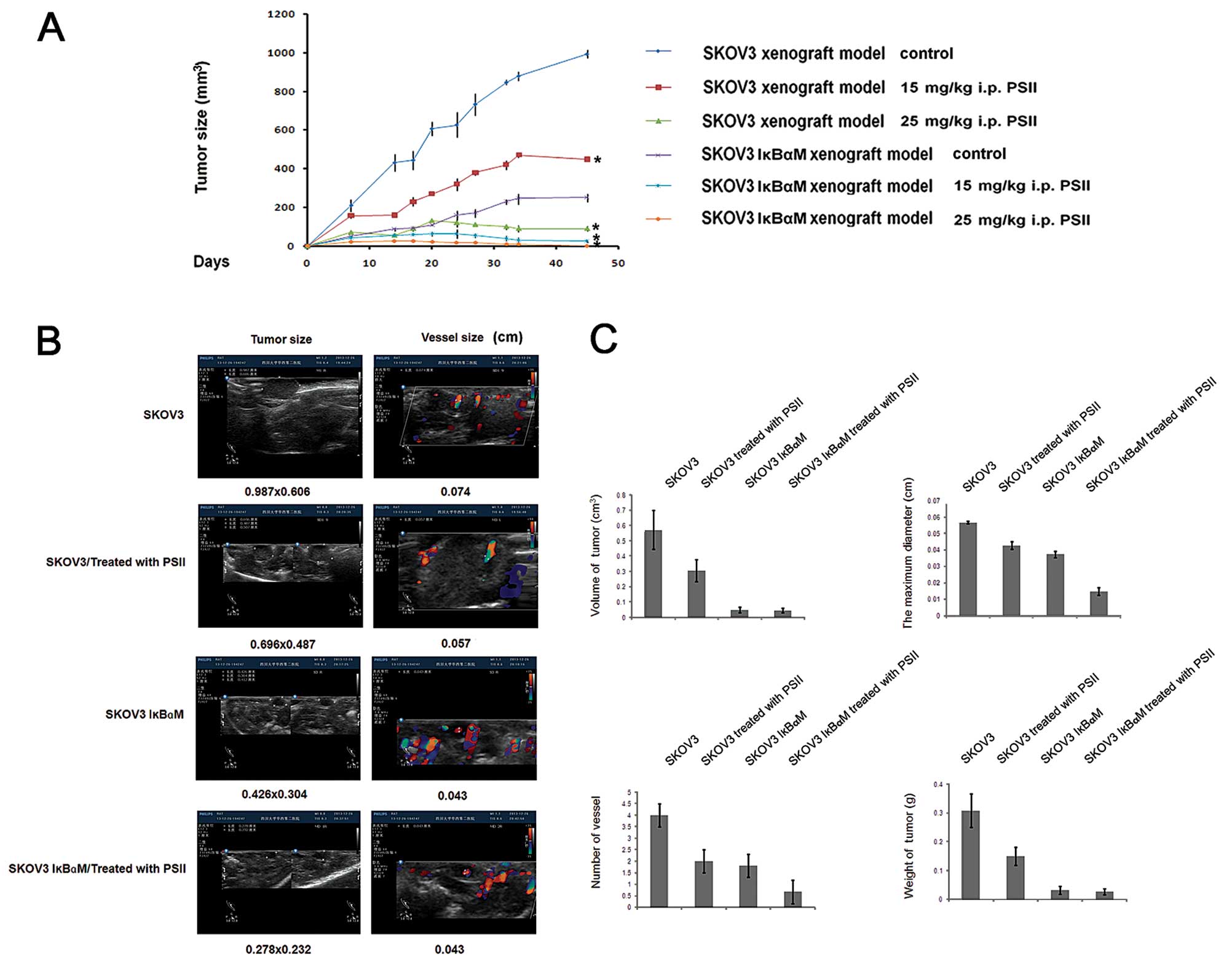 | Figure 5PSII suppresses tumor growth and
angiogenesis in a xenograft mouse model of ovarian cancer. (A) Mice
were implanted with 5×106 SKOV3/vector or SKOV3/IκBαM
cells on day 0 and were randomly divided into various treatment and
control groups (n=5). Eight days after the implantation, the
tumor-bearing mice were treated according to the protocols.
Briefly, tumor-bearing mice were treated with PSII or received the
vehicle (DMSO, <0.1%) in saline solution by intraperitoneal
administration for 4 weeks, 4 consecutive days/week with two
different doses of PSII, 15 or 25 mg/kg. Columns, mean; bars, SD,
*p<0.05, compared to the control. (B) Implanted
tumors, delined by the yellow lines, and vessels in the
PSII-treated and control mice were monitored by a Philips HD11
ultrasound scanner. The flows toward and away from the ultrasound
transducer were noted by the red and blue color, respectively. (C)
Measurement of the tumor wet weight, tumor volume, the maximum
diameter of blood vessels and the number of vessels are presented.
Columns, mean (n=3); bars, SE. PSII, Paris saponin II. |
Notably, in combination with the transfection of the
super-engineered repressor of IκBα, i.e. IκBαM (S32A and S36A)
known to inhibit NF-κB activity, the antitumorigenic effects of
PSII were enhanced significantly. While the effects of PSII
treatment alone or in combination with IκBαM rendered similar
inhibitory effects on tumor wet weights and tumor volumes, the
combination treatment markedly suppressed the growth rate and
reduced the number of vessels and the maximum diameter of blood
vessels (by >50%) compared to that of the PSII only treatment
(Fig. 5C). Clearly, the combination
treatment exerts marked and extremely effective anti-angiogenic
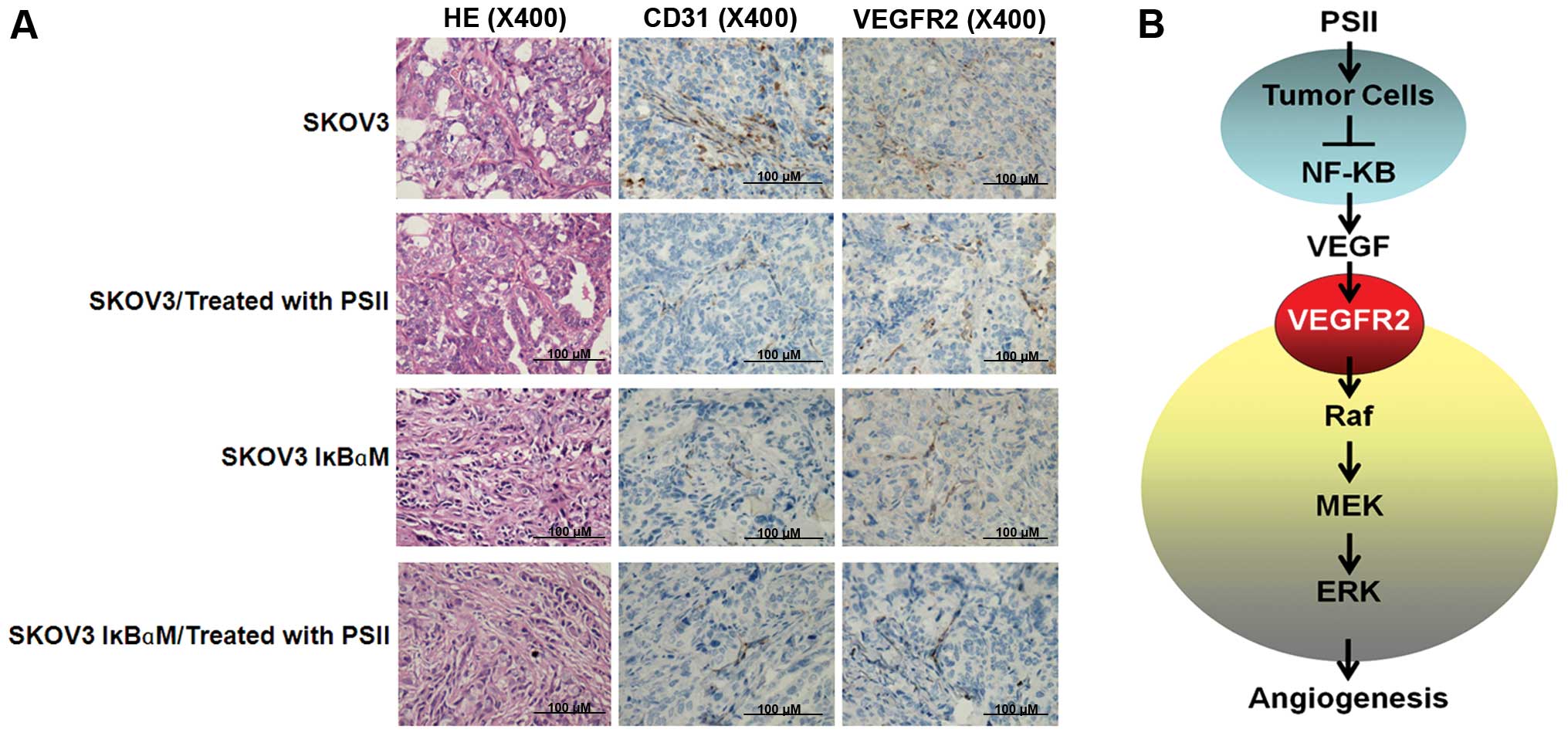
effects. To confirm this observation, we used immunohistochemistry
to assess signals of angiogenesis markers, i.e. VEGFR2 and CD31, on
tumor sections of the grafts from the treated or control mice. Our
results confirmed that PSII treatment clearly reduced the
expression of VEGFR2, the key player of VEGF signaling and
microvessel density refecting by CD31 signals (Fig. 6A). These data also suggest the
possibility of using PSII in combination with a drug that inhibits
NF-κB signaling. Together, the data in the present study allowed us
to elucidate a novel antitumorigenic and anti-angiogenic feature of
PSII (Fig. 6B). For the first time,
we provide evidence showing that PSII potently inhibits
angiogenesis and the growth of human ovarian cancer by suppressing
NF-κB signaling in cancer cells leading to the suppression of
pro-angiogenic factors of the tumor microenvironment, e.g.
VEGF.
Discussion
Saponin II (formosanin C), a steroidal saponin, is
one of the main active components of Rhizoma paridis
(21–23,32).
In our previous study, we characterized the antitumorigenic and
anti-angiogenic effects of PSII in a mouse model of ovarian cancer.
We showed that PSII not only induced tumor cell death but also
compromised endothelial cell activity leading to inhibitory effects
on angiogenesis in different in vivo, ex vivo and in
vitro model systems of angiogenesis (25). In the present study, we demonstrated
that PSII also modulated angiogenesis indirectly by targeting VEGF
expression in tumor cells. Specifically, PSII reduced IKKβ
expression and reduced its kinase activity. While the reduction of
IKKβ kinase activity can be attributed to the degradation of IKKβ
upon PSII treatment, most importantly, this degradation did affect
downstream expression of anti-apoptotic molecules and
pro-angiogenic VEGF in tumor cells. Our results were also
consistent with previous studies showing that NF-κB regulated VEGF
expression and the microvessel density (8–10).
The binding of VEGF to its receptors causes receptor
dimerization and auto-phosphorylation leading to the activation of
several downstream kinases and the expression of anti-apoptotic
proteins such as Bcl-2. The expression of Bcl-2 in tumor-associated
endothelial cells could be induced by VEGF secreted from tumor
cells and endothelial cells in the tumor microenvironment (33). Studies have shown that the
upregulation of expression of Bcl-2 in microvascular endothelial
cells could also promote intratumoral angiogenesis and tumor growth
(34,35). Therefore, targeting the VEGF
signaling pathway in either tumor cells or endothelial cells may
compromise angiogenesis. As shown in the present study, PSII
reduced VEGF expression and tumor cell-secreted VEGF levels at a
subcytotoxic level. Nevertheless, at such a concentration, PSII
still reduced NF-κB activities in tumor cells resulting in low VEGF
levels in the tumor microenvironment. The result demonstrated the
therapeutic potential of PSII in anti-angiogenic therapy. PSII, at
low-doses, prevented tumor-induced angiogenesis without damaging
the healthy endothelial cells. At such low doses, patients may
avoid side-effects often observed in anti-angiogenic therapy
(36).
Our previous study demonstrated that PSII exhibited
antitumorigenic ability in human ovarian cancer cells by inducing
apoptosis (25). Here, we confirmed
that such induction was the result of the PSII treatment. PSII
treatment altered the expression of anti-apoptotic proteins Bcl-2
and Bcl-xL in the SKOV3 cell line in a concentration-dependent
manner. Notably, such downregulation was even more prominent when
the NF-κB activation in the SKOV3 cells transfected with IκBαM was
compromised. Our results were consistent with previous studies
showing that suppression of the NF-κB activation could be
attributable to increased levels of apoptosis (8).
In conclusion, in the present study, for the first
time, we identified molecular targets of a steroidal saponin family
member, PSII. PSII modulated IKKβ expression and kinase activity
leading to a reduction in NF-κB transactivation. As a result, the
treatment altered the expression of several downstream targets of
NF-κB, i.e. VEGF, Bcl-2 and Bcl-xL. Most importantly, we continued
to demonstrate the therapeutic potential of a combination treatment
using PSII. PSII can be used in combination with other drugs/agents
that modulate NF-κB transactivation in cancer cells.
Acknowledgments
The present study was supported by the Fund of the
National Nature Science Foundations grant no. 81001159/81202387 and
the Sichuan Province Science and Technology Plan Project no.
2013JY0013/2014JY0213 and the Scientific Research Foundation of
Sichuan university for outstanding young scholars no. 2013SCu04A22.
The authors would like to thank Dr Meifang Xiao and Dr Hong Zou for
providing us with the PSI and PSII compounds, and also like to
thank Diane Hackett and Maude E. Veech for their technical
support.
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
McGuire WP, Hoskins WJ, Brady MF, et al:
Cyclophosphamide and cisplatin compared with paclitaxel and
cisplatin in patients with stage III and stage IV ovarian cancer. N
Engl J Med. 334:1–6. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Eckstein N: Platinum resistance in breast
and ovarian cancer cell lines. J Exp Clin Cancer Res. 30:912011.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Carmeliet P: Angiogenesis in life, disease
and medicine. Nature. 438:932–936. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kerbel RS: Tumor angiogenesis. N Engl J
Med. 358:2039–2049. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xu W, Liu LZ, Loizidou M, Ahmed M and
Charles IG: The role of nitric oxide in cancer. Cell Res.
12:311–320. 2002. View Article : Google Scholar
|
|
7
|
Ferrara N and Kerbel RS: Angiogenesis as a
therapeutic target. Nature. 438:967–974. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Huang S, Robinson JB, Deguzman A, Bucana
CD and Fidler IJ: Blockade of nuclear factor-κB signaling inhibits
angiogenesis and tumorigenicity of human ovarian cancer cells by
suppressing expression of vascular endothelial growth factor and
interleukin 8. Cancer Res. 60:5334–5339. 2000.PubMed/NCBI
|
|
9
|
Aggarwal BB: Nuclear factor-kappaB: the
enemy within. Cancer Cell. 6:203–208. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yu HG, Yu LL, Yang Y, et al: Increased
expression of RelA/nuclear factor-κB protein correlates with
colorectal tumorigenesis. Oncology. 65:37–45. 2003. View Article : Google Scholar
|
|
11
|
Biswas DK, Shi Q, Baily S, et al: nf-κb
activation in human breast cancer specimens and its role in cell
proliferation and apoptosis. Proc Natl Acad Sci USA.
101:10137–10142. 2004. View Article : Google Scholar
|
|
12
|
Gilmore TD, Koedood M, Piffat KA and White
DW: Rel/NF-kappaB/IkappaB proteins and cancer. Oncogene.
13:1367–1378. 1996.PubMed/NCBI
|
|
13
|
Yang G, Xiao X, Rosen DG, et al: The
biphasic role of nf-κb in progression and chemoresistance of
ovarian cancer. Clin Cancer Res. 17:2181–2194. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Scherer DC, Brockman JA, Chen Z, Maniatis
T and Ballard DW: Signal-induced degradation of IκBα requires
site-specific ubiquitination. Proc Natl Acad Sci USA.
92:11259–11263. 1995. View Article : Google Scholar
|
|
15
|
Brown K, Gerstberger S, Carlson L,
Franzoso G and Siebenlist U: Control of I kappa B-alpha proteolysis
by site-specific, signal-induced phosphorylation. Science.
267:1485–1488. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Brockman JA, Scherer DC, McKinsey TA, et
al: Coupling of a signal response domain in iκbα to multiple
pathways for nf-κb activation. Mol Cell Biol. 15:2809–2818.
1995.PubMed/NCBI
|
|
17
|
Huang S, Pettaway CAUH, Bucana CD and
Fidler IJ: Blockade of nf-κb activity in human prostate cancer
cells is associated with suppression of angiogenesis, invasion, and
metastasis. Oncogene. 20:4188–4197. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fujioka S, Sclabas GM, Schmidt C, et al:
Function of nuclear factor κb in pancreatic cancer metastasis. Clin
Cancer Res. 9:346–354. 2003.PubMed/NCBI
|
|
19
|
Ho JW, Leung YK and Chan CP: Herbal
medicine in the treatment of cancer. Curr Med Chem Anticancer
Agents. 2:209–214. 2002. View Article : Google Scholar
|
|
20
|
Man S, Gao W, Zhang Y, et al: Antitumor
and antimetastatic activities of Rhizoma Paridis saponins.
Steroids. 74:1051–1056. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cosgrove D: Angiogenesis imaging -
ultrasound. Br J Radiol. 76(Spec No 1): S43–S49. 2003. View Article : Google Scholar
|
|
22
|
Howard-Claudio C: MRI methods for the
detection of angiogenesis. Supplement to Appl Radiol. 34:2005.
|
|
23
|
Man S, Gao W, Zhang Y, et al:
Characterization of steroidal saponins in saponin extract from
Paris polyphylla by liquid chromatography tandem multi-stage mass
spectrometry. Anal Bioanal Chem. 395:495–505. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xiao X, Bai P, Bui Nguyen TM, et al: The
antitumoral effect of Paris Saponin I associated with the induction
of apoptosis through the mitochondrial pathway. Mol Cancer Ther.
8:1179–1188. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xiao X, Zou J, Bui-Nguyen TM, et al: Paris
saponin II of Rhizoma Paridis - a novel inducer of apoptosis in
human ovarian cancer cells. Biosci Trends. 6:201–211. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Schmitt J and Matei D: Targeting
angiogenesis in ovarian cancer. Cancer Treat Rev. 38:272–283. 2012.
View Article : Google Scholar
|
|
27
|
Nicosia RF and Ottinetti A: Growth of
microvessels in serum-free matrix culture of rat aorta. A
quantitative assay of angiogenesis in vitro. Lab Invest.
63:115–122. 1990.PubMed/NCBI
|
|
28
|
Xiao X, Yang M, Xiao J, et al: Paris
Saponin II suppresses the growth of human ovarian cancer xenografts
via modulating VEGF-mediated angiogenesis and tumor cell migration.
Cancer Chemother Pharmacol. 73:807–818. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Agbottah E, Yeh WI, Berro R, et al: Two
specific drugs, BMS-345541 and purvalanol A induce apoptosis of
HTLV-1 infected cells through inhibition of the NF-kappaB and cell
cycle pathways. AIDS Res Ther. 5:122008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Patan S: Vasculogenesis and angiogenesis.
Cancer Treat Res. 117:3–32. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Fujioka S, Sclabas GM, Schmidt C, et al:
Inhibition of constitutive nf-κb activity by iκbα M suppresses
tumorigenesis. Oncogene. 22:1365–1370. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wen F, Yin H, Chen C, et al: Chemical
characteristics of saponins from Paris fargesii var. brevipetala
and cytotoxic activity of its main ingredient, paris saponin H.
Fitoterapia. 83:627–635. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Nor JE, Christensen J, Mooney DJ and
Polverini PJ: Vascular endothelial growth factor (VEGF)-mediated
angiogenesis is associated with enhanced endothelial cell survival
and induction of Bcl-2 expression. Am J Pathol. 154:375–384. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Nör JE, Christensen J, Liu J, et al:
Up-Regulation of Bcl-2 in microvascular endothelial cells enhances
intratumoral angiogenesis and accelerates tumor growth. Cancer Res.
61:2183–2188. 2001.PubMed/NCBI
|
|
35
|
Karl E, Warner K, Zeitlin B, et al: Bcl-2
acts in a proangiogenic signaling pathway through nuclear factor-κB
and CXC chemokines. Cancer Res. 65:5063–5069. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lacouture ME, Lenihan DJ and Quaggin SE:
Antiangiogenic Therapy: Tolerability and Management of Side
Effects. 2009, http://www.angioorg/pdf/Angio_Poster_Final_6-30.pdfurisimplewww.angioorg/pdf/Angio_Poster_Final_6-30.pdf.
|