Introduction
Colon cancer is a leading cause of cancer and
cancer-related mortality in the Western world, and its incidence is
on the increase (1). Stress has
been associated with the increased incidence and development of
cancer (2). Adrenaline (AD) and
noradrenaline (NA), two of the most important mediators released in
response to stress, exert their effects through interaction with α-
and β-adrenergic receptors (ARs). ARs are targets for many
therapeutically important drugs, such as the ones used for
cardiovascular diseases, asthma, prostatic hypertrophy, nasal
congestion obesity and pain (3).
Expression of β-AR has been identified in colon cancer cells and
previous studies have shown that their activation has been
implicated in carcinogenesis and tumor progression (4–6).
Interest regarding the efficacy of β-AR blockers as possible
additions to cancer-treatment paradigms has increased. However,
which of these drugs are useful remains to be determined.
Previous findings suggest that drug efficacy may be
influenced by the signaling effectors engaged by a unique receptor
(7). For instance, some β-AR
blockers that are inverse agonists for G-protein-mediated functions
were found to be agonists, neutral blockers, or inverse agonists
for β-arrestin-mediated signaling, resulting in markedly different
effects in vivo (7). A
better description of the efficacy profiles for β-AR blockers may
be useful to explain the reason for individual members of a drug
class having different therapeutic indications.
Over the last years, the biological effects of
stress pathways on cancer progression have been focused on the
effects of stress hormones on tumor cell proliferation, apoptosis,
invasion, metastasis, angiogenesis, stroma-cell microenvironment
and cellular immune responses (8).
Several in vitro and in vivo studies have shown that
AD and NA can induce cell proliferation in different types of
cancer such as non-small cell lung carcinoma (9,10),
colon cancer (5), oral squamous
carcinoma (11), breast cancer
(12) and prostate cancer (13).
In the present study, we aimed to clarify the role
of AD, NA and ISO, and several β-blockers on colon-cancer cell
proliferation using a human colon adenocarcinoma cell line.
Materials and methods
Reagents
RPMI-1640 medium was purchased from Invitrogen
(Invitrogen Life Technologies, Paisley, UK). The following reagents
were purchased from Sigma (St. Louis, MO, USA): AD
(Adrenaline-L-adrenaline(+)-bitartrate salt), NA
(Noradrenaline-L-(−)-noradrenaline(+)-bitartrate salt monohydrate),
ISO (Iso prenaline-(−)-isoprenaline(+)-bitartrate salt), PRO
(Propranolol-DL-propranolol hydrochloride),
ICI-(±)-1-[2,3-(dihydro-7-methyl-1H-inden-4-yl)oxy]-3-[(1-methylethyl)
amino]-2-butanolhydrochloride) (ICI 118,551), ATE
(Atenolol-(±)-4-[2-hydroxy-3[(1methylethyl)amino]propoxy]benzeneace
tamide), penicillin, streptomycin, (FBS) fetal bovine serum,
trypsin-EDTA solution and MTT
[3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide]. CAR
{Carvedilol-1-(9H-carbazol-4-yloxy)-3-[(2-(2-methoxyphenoxy)ethyl)amino]-2-propanol}
was purchased from Enzo Life Sciences, Inc., (Farmingdale, NY,
USA). The cell proliferation ELISA BrdU kit (colorimetric) was
purchased from Roche Diagnosis GmbH (Mannheim, Germany) and Cell
Titer 96® Aqueous ONE Solution Reagent cell
proliferation assay (MTS
[3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium]
were purchased from Promega (Madison, WI, USA).
Cell culture
HT-29 human colon adenocarcinoma cells were kindly
provided by Professor Bruno Sarmento (Institute of Biomedical
Engineering-INEB) and Professor Fernando Magro (Faculty of Medicine
of the University of Porto).
HT-29 cells were cultured in RPMI-1640 medium
supplemented with 10% of FBS, 100 U/ml penicillin and 100 μg
streptomycin. The cells were grown at 37°C in a humidified 5%
CO2 atmosphere. Culture medium was changed every 2–3
days. When the cells reached 90–100% confluency, the medium was
removed, and the cell monolayer was washed once with PBS. The cell
monolayer was treated with 1 ml of 0.25% (w/v) trypsin-EDTA and
incubated for 2 min to ensure complete cell detachment. For
sub-culturing, the cells were sub-cultured in plastic culture
dishes (21 cm2, 60-mm diameter, Corning Costar, Corning,
NY, USA). For the experiments, HT-29 cells were seeded in 96-well
(0.37 cm2, 6.9 mm diameter, TPP) or 24-well plastic cell
culture clusters (2 cm2, 16-mm diameter, TPP) depending
on experimental conditions. Experiments were performed 4–5 days
after the initial seeding (90–100% confluency).
Viability experimental studies
Trypan blue exclusion assay
Prior to any experiment, cell viability was
determined by Trypan Blue exclusion assay. The experiments were
only performed, when viability was >90%. In brief, cells were
trypsinized and stained with 0.4% trypan blue and viable cells were
counted with a hemocytometer.
MTS assay
HT-29 cells were seeded at 1×105 cells/ml
in 96-well plates for 24 h and incubated with each treatment for 12
or 24 h, depending on experimental conditions. Cell viability assay
was assessed using Cell Titer 96 Aqueous ONE Solution Reagent cell
proliferation assay (MTS) according to the manufacturer’s
instructions. Briefly, the culture medium was removed and the cells
were pre-incubated with the compounds under study in culture medium
at 37°C for 12 or 24 h. This medium was removed and the cells were
incubated for 3 h with 100 μl of FBS-free culture medium and
20 μl of MTS. Optical density was measured at 492 nm.
Results were expressed as percentage of the control.
MTT assay
In order to determine the half maximal inhibitory
concentration (IC50), the concentration that reduces the
effect by 50%, and the half maximal effective concentration
(EC50) values, the concentration that yields half
maximal response, an MTT assay was performed. This method is based
on mitochondrial dehydrogenase activity. Mitochondrial
dehydrogenases of viable cells cleave the tetrazolium ring,
yielding purple formazan crystals insoluble compounds in aqueous
solutions. The amount of these compounds was determined
spectrophotometrically. The cells were seeded in 96-well plates at
a density of 1×104 cells/well for 24 h, and then
incubated for 24 h with increasing concentrations of the various
compounds under study (0.1, 1, 5, 10, 20, 50 and 100 μM).
The cells were then washed twice with PBS and incubated with MTT (5
mg/ml) for 2 h at 37°C. Blue formazan crystals were solubilized
with DMSO and the colored solution was subsequently read at 550 nm.
The samples were assayed in triplicate and at least in three
independent experiments, and the mean value for each experiment was
calculated. Results were presented as mean (± SEM) and are
expressed as percentage of the control [adapted from Stanojcovic
et al (14)].
Proliferation experimental studies
Cell proliferation was assessed as DNA synthesis. To
evaluate DNA synthesis, the incorporation of
[3H]-thymidine or 5′-bromodeoxyuridine (BrdU) into DNA
was determined, as detailed in the subsequent sections.
Incorporation of
[3H]-thymidine
HT-29 cells were seeded for attachment at
5×104 cells/well in 24-well (1.65 cm2, 14.5
mm diameter; Orange Scientific, Belgium) plastic cell-culture
clusters in a final volume of 0.5 ml culture medium containing 10%
FBS. After 24 h in culture, the cells were treated with several
concentrations of the adrenergic agonists dissolved in culture
medium (controls were produced in the presence of culture media).
After 24 h, the cells were incubated with 0.2 ml of
methyl-[3H]-thymidine (0.5 μCi/well) for 4 h. The
medium was removed and the cells were fixed by incubation in 0.3 ml
of 10% TCA for 1 h at 4°C. The cells were then washed twice with
0.3 ml of 10% TCA to remove unbound radioactivity. The plates were
air-dried and the cells lysed with 0.28 ml/well of 1 M NaOH. A
0.25-ml aliquot of the lysate was neutralized with 0.050 ml of HCl
prior to the addition of scintillation fluid. Radioactivity of the
samples was quantified by a liquid scintillation counter. The
counts (disintegrations/min) of each treatment were averaged and
expressed as percentage of the controls [adapted from Miranda et
al (15)].
Incorporation of BrdU
The incorporation of the BrdU assay is a method
based on the incorporation of BrdU, a thymidine analogue, instead
of thymidine into the DNA of proliferating cells. After its
incorporation into DNA, BrdU is detected by immunoassay.
HT-29 cells were grown at 1×105 cells/ml
in 96-well plates for 24 h, and proliferation was measured using
the Cell Proliferation ELISA BrdU kit (Roche Diagnostics GmbH),
according to the manufacturer’s instructions. Briefly, the cells
were labeled with BrdU at a final concentration of 10
μM/well), for 12 h at 37°C. The cells were then denatured
with FixDenat solution, and incubated for 120 min with 1:100
diluted mouse anti-BrdU conjugated to peroxidase. Following removal
the antibody conjugate and washing twice with washing solution (PBS
1X), the substrate solution was added for 25 min and, after this
period, the reaction was stopped with 1 M
H2SO4 solution. Absorbance was measured
within 5 min at 450 nm with a reference wavelength at 690 nm using
an ELISA plate reader. The blank corresponded to 100 μl of
culture medium without BrdU, and the control was produced in the
presence of culture media without any treatment. Results were
presented as mean (± SEM) and are expressed as percentage of the
control.
Another protocol for the BrdU assay was tested for
treatments at 24 h. Briefly, 5×103 cells/well were
seeded in 96-well plates. The medium was supplemented with
antibiotics plus 1% FBS for cell attachment. The cells were
subsequetly starved in serum-free medium for another 12 h to
synchronize the cell cycle. HT-29 cells were incubated with AD, ISO
or NA (0, 1 and 10 μM) for 24 h to study the
growth-promoting effect of these adrenergic agonists. To examine
the effects of various β-blockers, the cells were pretreated with
or without, PRO (50 μM), CAR (5 μM), ATE (50
μM) and ICI (5 μM) for 45 min prior to, and also
simultaneously with, AD or ISO treatment. Cell proliferation was
indicated by the amount of DNA synthesis measured with the BrdU
incorporation assay kit, according to the manufacturer’s
instructions. Briefly, the cells were labeled with 10
μl/well BrdU and incubated at 37°C for 4 h. Following
removal of the labeling medium, the cells were fixed and probed
with the anti-BrdU monoclonal antibody at 25°C for 2 h and its
substrate tetramethyl-benzidine (TMB) at 25°C for 30 min. After
removal of the unconjugated antibody, the cells were rinsed three
times with the washing solution and treated with 200 μl/well
substrate solution. After color development, 1 M
H2SO4 was added (25 μl/well) to stop
the substrate reaction, and the absorbance of each sample was
measured in an enzyme-linked immunosorbent assay (ELISA) microplate
reader at 450 nm (with a reference wavelength at 690 nm for blank
to disccount the non-specific binding to the anti-BrdU antibody).
The value from the non-specific binding was subtracted from all the
other values. The results were presented as mean (± SEM) and
expressed as percentage of the control.
Statistical analysis
The results were presented as arithmetic mean ± SEM.
Differences in cell proliferation, viability or cell growth between
treated and corresponding untreated cells (controls) were tested
using Student’s t-tests. For the calculation of EC50 and
IC50 values, the parameters of the Hill equation were
fitted to the experimental data by using a non-linear regression
analysis, using a computer-assisted method (15), with ‘n’ representing the number of
replicates of at least three different experiments. Comparisons
between ≥3 groups were performed with one-way analysis of variance
(ANOVA) followed by the Tamhane or Bonferroni test. Differences
were considered statistically significant when P<0.05.
Given the variability of the results on different
days, each experimental finding was adjusted to the respective
control.
Results
General
The initial aim of this study was to assess the
viability of HT-29 cells following chronic exposure to several AR
agonists/antagonists under evaluation. For this purpose, the MTS
(for treatment at 12 and 24 h with the AR agonists) and MTT (for
the determination of IC50 and EC50 values)
assays were used. After these initial experiments, we studied the
effects of the same drugs mentioned above on cellular proliferation
measured by DNA synthesis.
Effect of chronic treatment with the
adrenergic agonists on cell viability
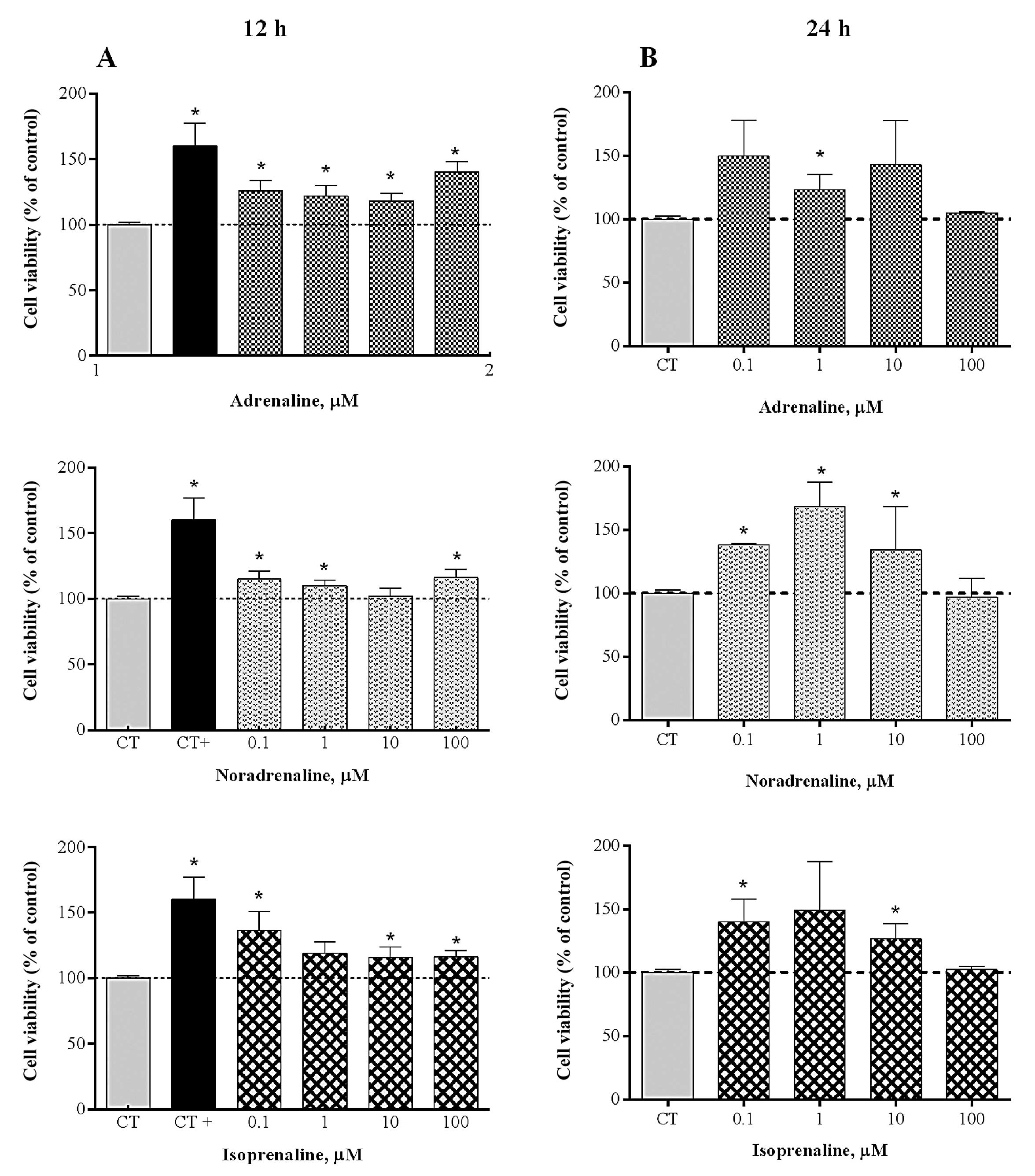
The MTS assays showed that none of the tested
adrenergic agonists used, at 12 or 24 h, affected the viability of
HT-29 cells (Fig. 1). By contrast,
for the two treatments carried out, we observed that for the
majority of the concentrations the agonists enhanced cell
viability.
Determination of EC50 values
for the adrenergic agonists in HT-29 cells
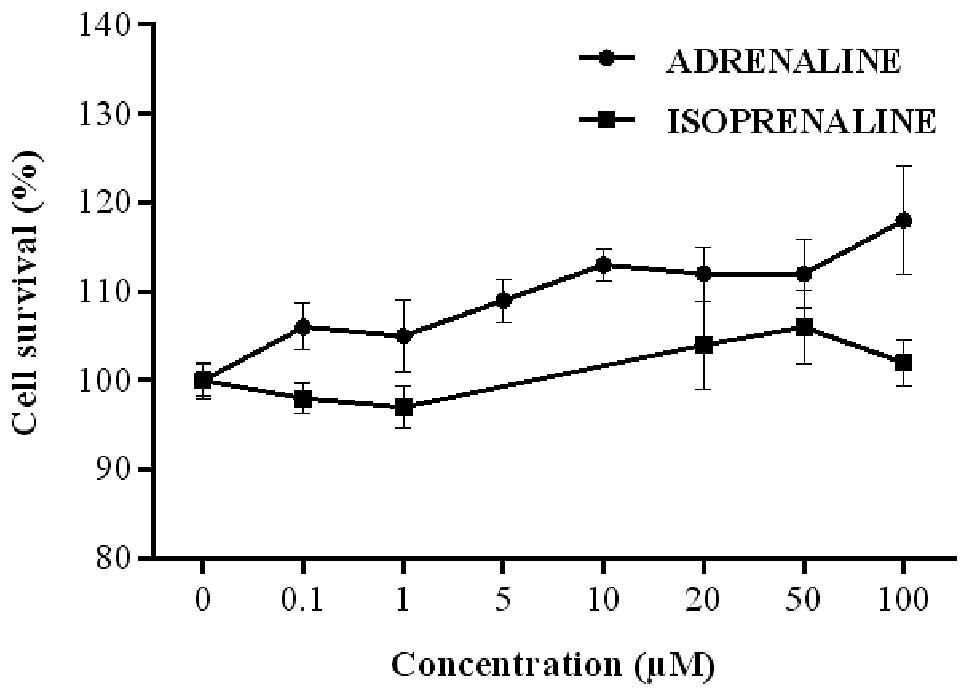
Cell exposure to AD and ISO generated
concentration-response curves for the two agonists (Fig. 2). Thus, the respective
EC50 values at 9.98 (0.51–197.2) and 29.27 (0.72–1194.0)
μM for AD and ISO, respectively, were calculated (Table I).
 | Table IEC50 values for adrenaline
and isoprenaline on HT-29 proliferation. |
Table I
EC50 values for adrenaline
and isoprenaline on HT-29 proliferation.
| Cell type | Agonist |
EC50/μM | 95% CI | n |
|---|
| HT-29 | Adrenaline | 9.98 | 0.51–197.2 | 10–11 |
| Isoprenaline | 29.27 | 0.72–1,194.0 | 8–12 |
Determination of IC50 values
for the β-blockers in HT-29 cells
PRO potently inhibited the viability of HT-29 cells
at concentrations >50 μM, with 65.4 μM being the
IC50 for this drug (Fig.
3). HT-29 viability was inhibited in a concentration-dependent
manner by CAR following exposure for 24 h. Among the β-blockers
tested, CAR was identified as the most potent blocker for this
effect, with an IC50 of 8.0 μM. ATE, when used at
the highest concentration (100 μM), significantly decreased
HT-29 viability, with 52.9 μM being its IC50
value. CAR and ICI showed similar IC50 values, 8.0 and
8.9 μM, respectively (Table
II).
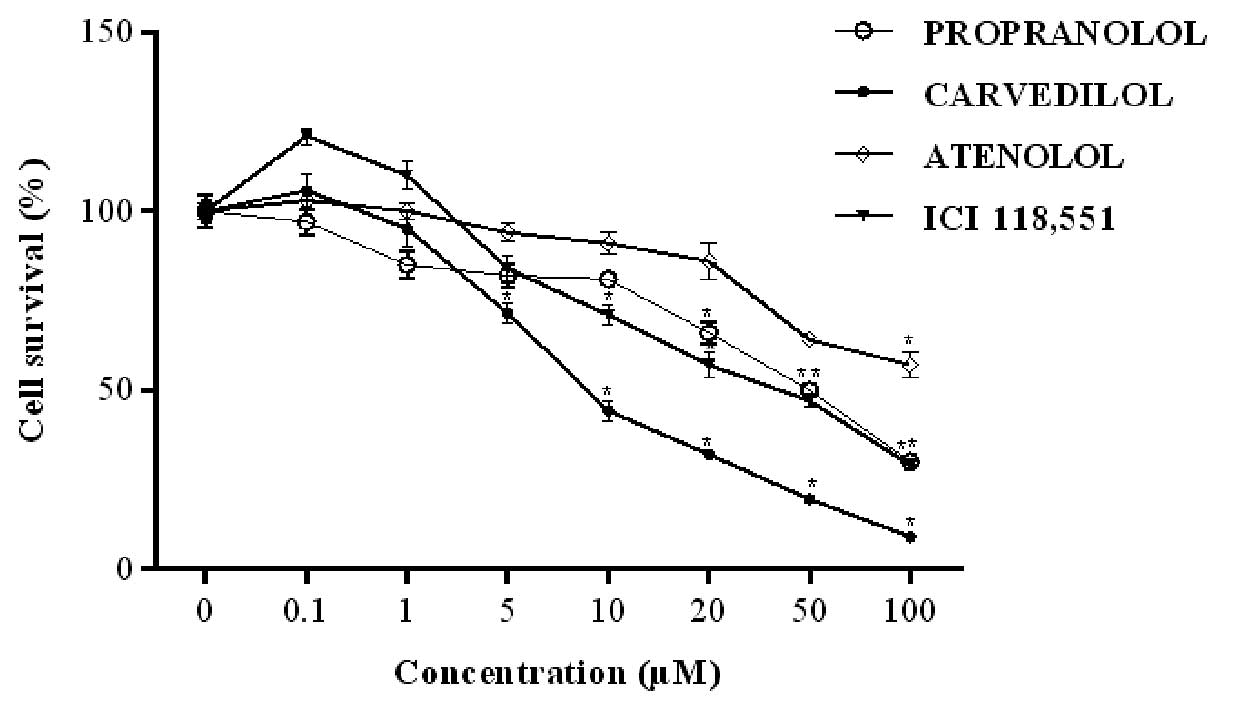 | Figure 3Concentration-response curves for
HT-29 cell survival. The cells were treated with increasing
concentrations (0, 0.1, 1, 5, 10, 20, 50 and 100 μM) of each
β-blocker, propranolol (PRO), carvedilol (CAR), atenolol (ATE) and
ICI 118,551 (ICI) for 24 h and viability was assessed by MTT assay.
Results are presented as mean ± SEM and normalized to 100% of the
control groups (without drugs). *P<0.05 compared to
the control. |
| Table IIIC50 of β-blockers on
HT-29 proliferation. |
Table II
IC50 of β-blockers on
HT-29 proliferation.
| Cell type | β-blockers |
IC50/μM | 95% CI | n |
|---|
| HT-29 | Propranolol | 65.4 | 33.7–126.9 | 10–12 |
| Carvedilol | 8.0 | 6.0–10.6 | 12 |
| Atenolol | 52.9 | 21.7–128.7 | 6–12 |
| ICI 118,551 | 8.9 | 6.5–12.0 | 12 |
Effect of chronic treatment with the
adrenergic agonists on HT-29 cell proliferation
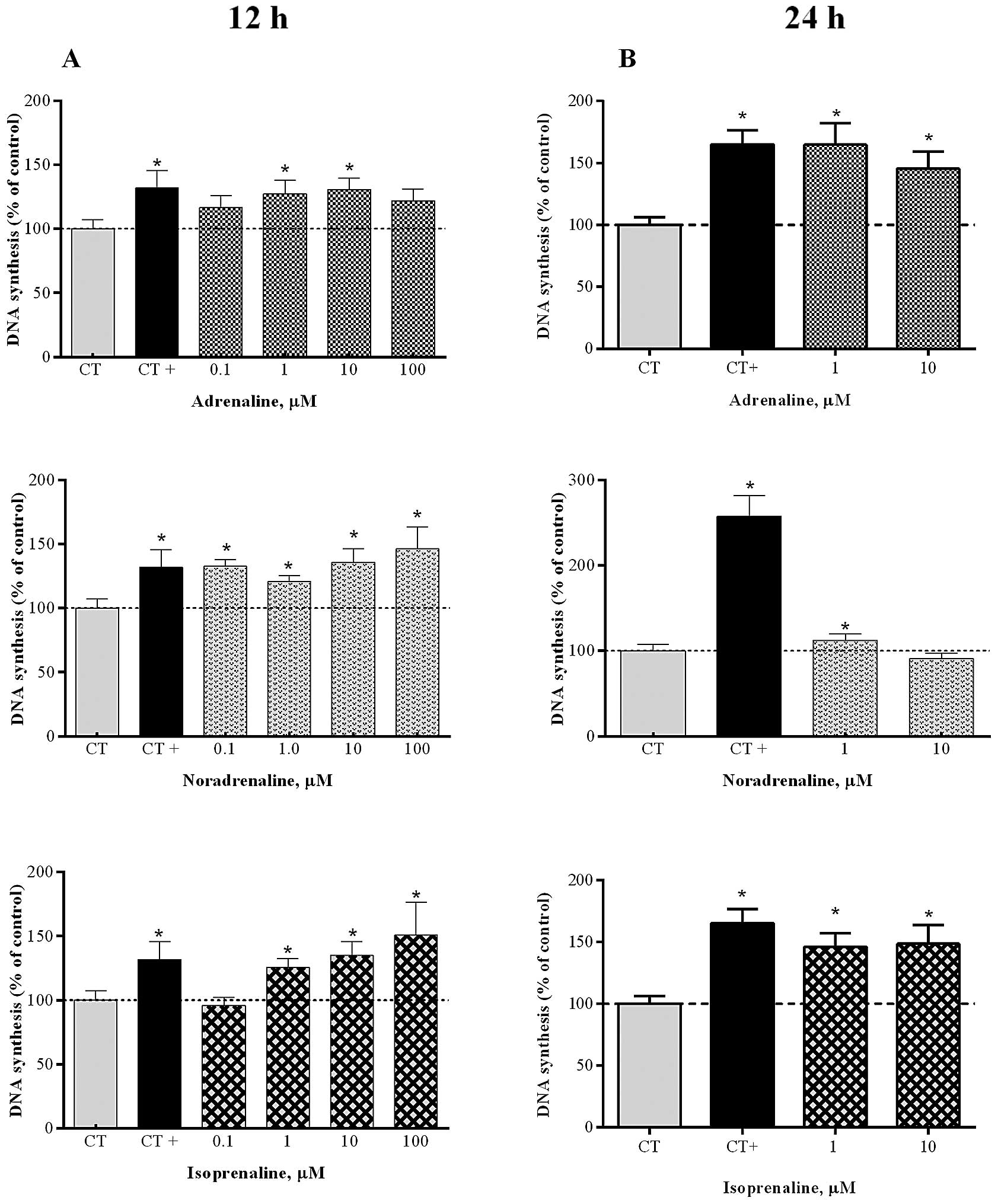
The exposure of HT-29 cells to the adrenergic
agonists AD, NA and ISO (at 0.1–100 μM) for 12 h markedly
increased proliferation of these cells (Fig. 4A). After this period, AD had its
maximum effect on proliferation at 10 μM (131.0±8.7%, n=10),
NA at 100 μM (146.3±17.1%, n=8) and ISO at 100 μM
(150.9±25.5%, n=7), relative to the controls. By contrast, chronic
treatment for 24 h with AD led to a significant increase of cell
proliferation by 164.7% (n=18) and 145.5% (n=18), when used at 1
and 10 μM, respectively (Fig.
4B), whereas ISO enhanced HT-29 cell proliferation by 146.1%
(n=10) and 148.6% (n=15), respectively, at 1 and 10 μM, when
compared to the controls.
Effect of β-blockers on HT-29 cell
proliferation
To elucidate the role of β-AR following cell
proliferation induced by AR activation, the agonists AD and ISO (a
non-selective vs. a β-selective agonist) were employed following
and simultaneously with the β-blockers PRO (50 μM), CAR (5
μM), ATE (50 μM) or ICI (5 μM) for 24 h.
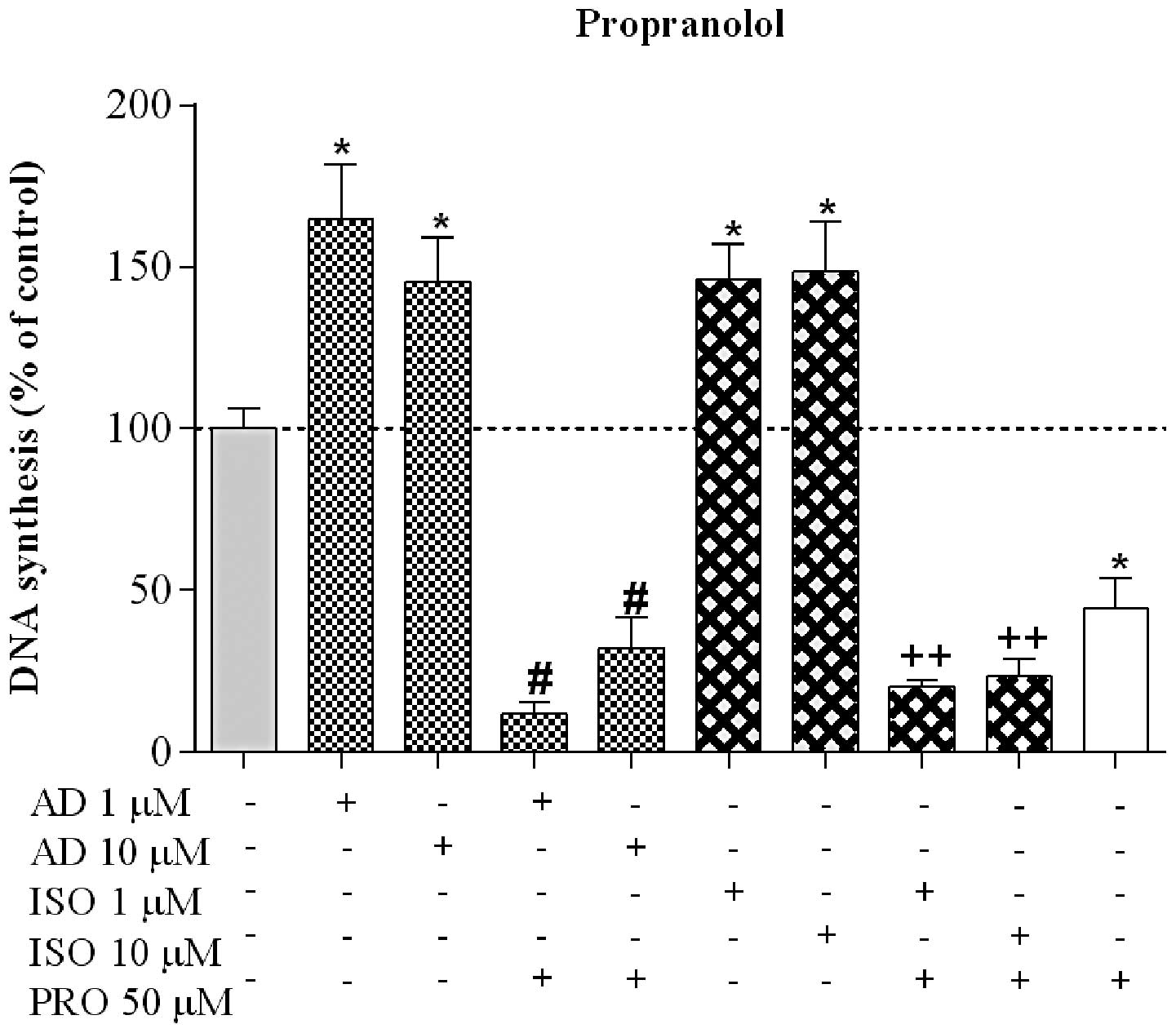
AD-induced cell proliferation was markedly reduced by PRO to
11.8±3.4% (n=6) and 32.0±9.7% (n=5), when AD was used at 1 and 10
μM, respectively. PRO markedly decreased cell proliferation
stimulated by ISO to 20.1±2.1% (n=5) and 23.3±5.2% (n=5), when the
agonist was used at 1 and 10 μM, respectively (Fig. 5). Furthermore, PRO induced a
significant proliferation decrease to 44.2±9.6% (n=6), when
compared with the control group (Fig.
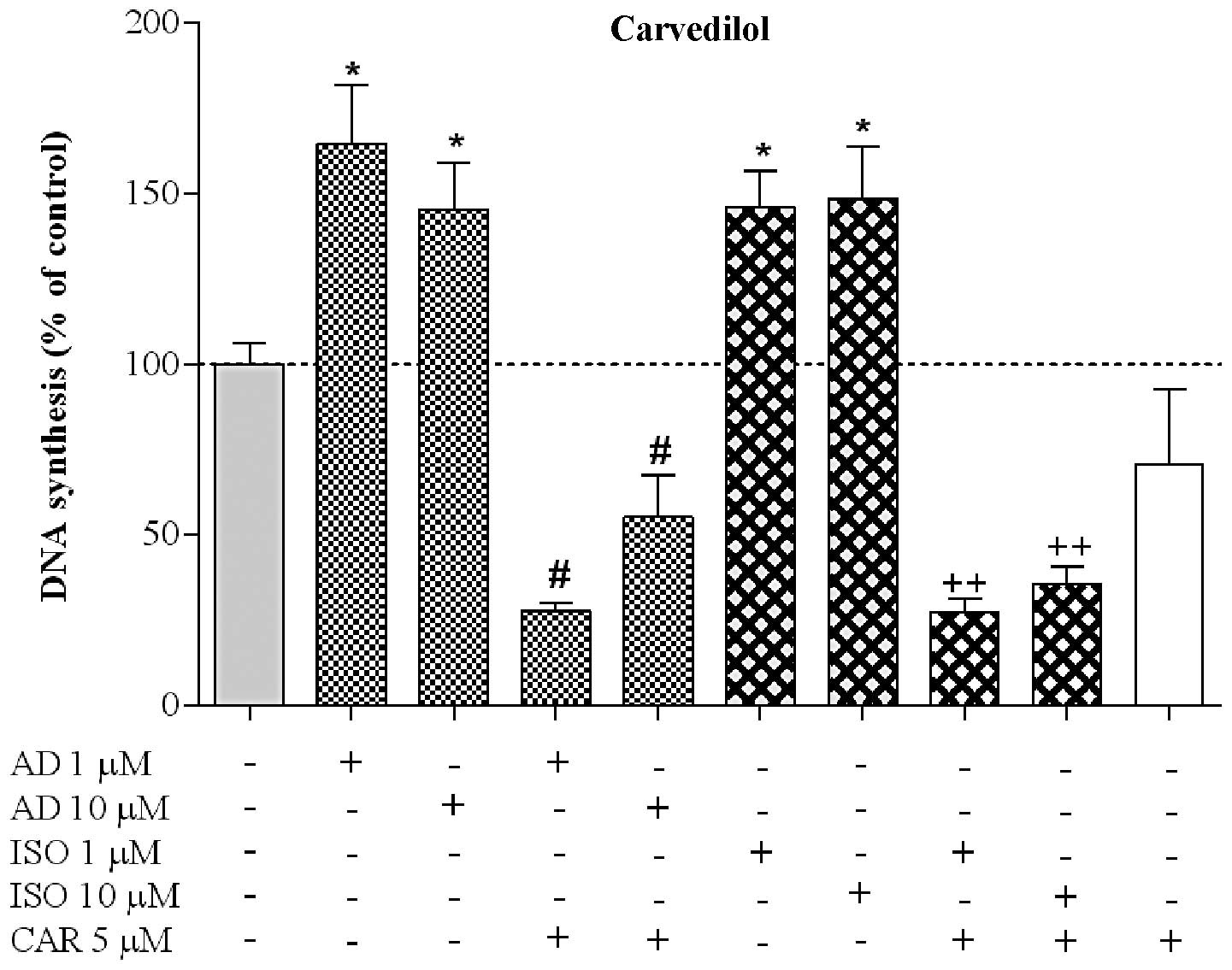
5). The response profile of CAR, a potent non-selective β- and
α1-AR antagonist, in reversing the proliferative effects
of AD and ISO, was similar to the results obtained with PRO. CAR
was able to markedly inhibit the proliferative effect induced by
the two agonists (Fig. 6). CAR
decreased the proliferation induced by AD to 28% (n=6) and 56%
(n=6), when AD was applied at 1 and 10 μM, respectively, and
to 27% (n=6) and 36% (n=6) when ISO was used at 1 and 10 μM,
respectively. In contrast to PRO, CAR did not significantly affect
the proliferation of HT-29 cells. To elucidate the role of
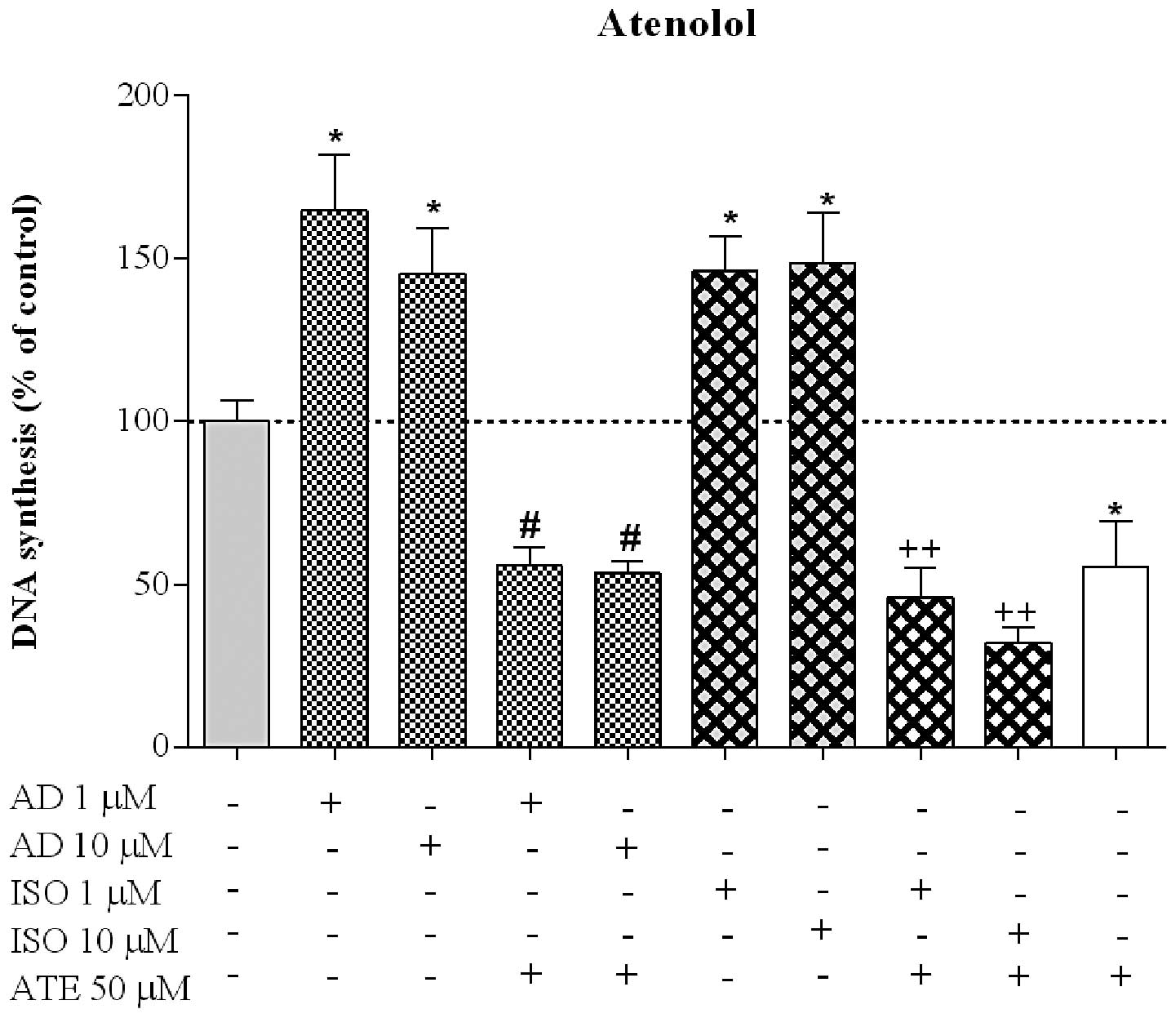
β1-AR in HT-29 proliferation, we used ATE, a
β1- selective antagonist. Fig. 7 shows that ATE significantly blocked
AD- and ISO-induced cell proliferation, confirming the involvement
of the β1 subtype in the proliferative effect, through
promotion of proliferation. ATE significantly decreased cell
proliferation induced by AD at 1 and 10 μM to 55.7±5.6%
(n=6) and 53.4±3.6% (n=6), respectively, and to 45.8±9.2% (n=6) and
32.1±4.6% (n=6) for ISO, respectively, at 1 and 10 μM.
Furthermore, when applied alone, ATE decreased proliferation to
55.4±13.9% (n=6), compared to the control. To investigate the
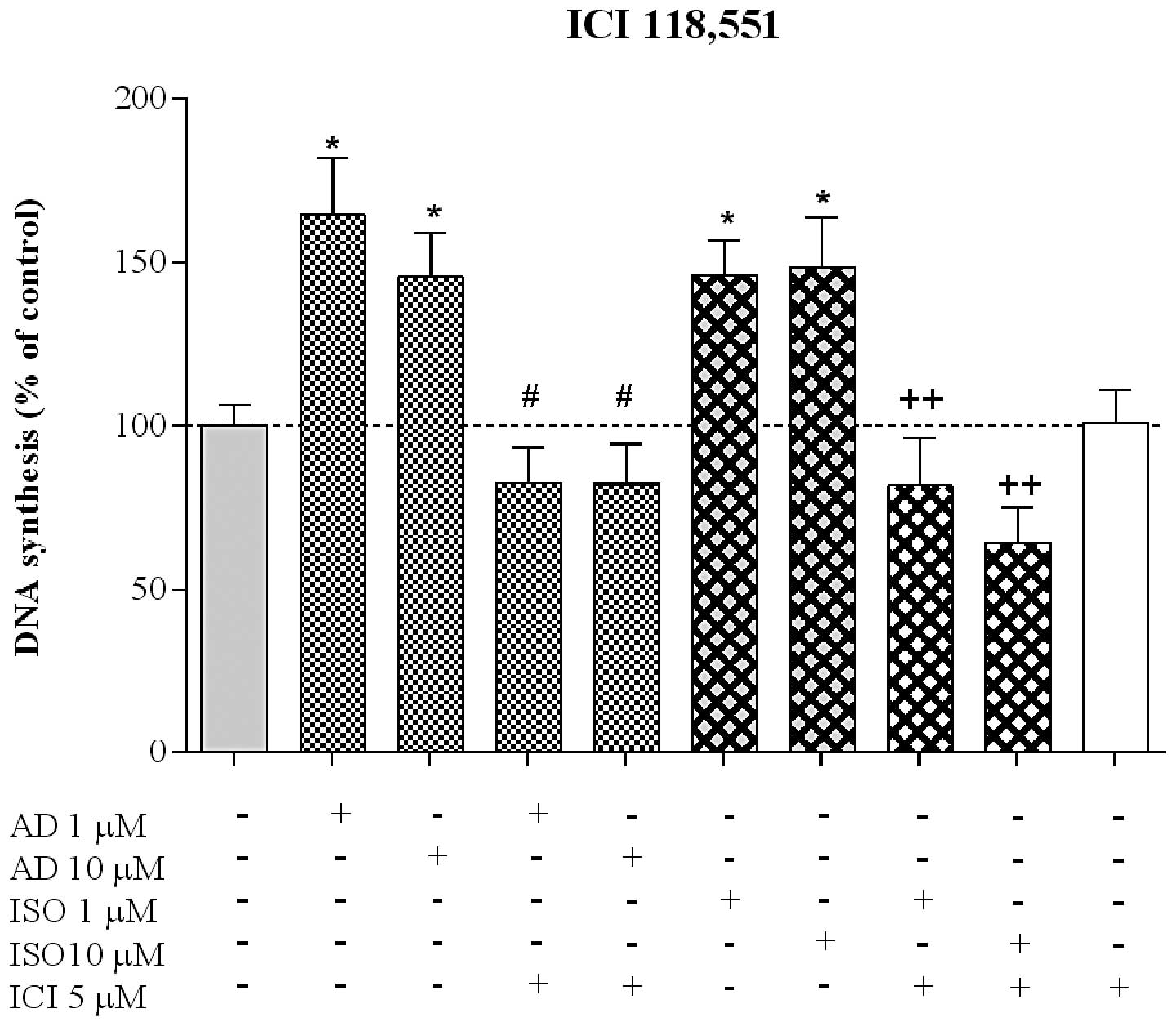
involvement of β2-AR in HT-29 proliferation, the cells
were incubated with ICI, a β2-selective antagonist,
either alone or with the AR agonists. As is evident in Fig. 8, ICI did not significantly affect
the proliferation induced by AD, whereas it reduced the effect
induced by ISO 10 μM to 64.1±11.1% (n=7), reinforcing the
involvement of the β2-AR subtype in this process. ICI
had no effect on HT-29 cell proliferation.
Discussion
Stress is considered to play a central role in the
incidence and development of cancer (16). However, the molecular and cellular
mechanisms by which stress increases the risk of certain types of
cancer and their prognosis remain understudied. It has been
postulated that endogenous CA mediate the association between
stress response and poor cancer outcomes (10,17).
Epidemiologic studies have associated the use of β-blockers in
clinical settings to reduce the rates of progression for several
solid tumors (18). Findings
suggest that β-AR blockers may be inexpensive and safe therapeutic
agents for cancer. The above studies mostly do not distinguish
between β1- or β2-activity blockers, and the
signaling pathways involved in these responses remain poorly
understood.
In the present study, we addressed the effect of the
stress hormones, AD and NA, and ISO, a synthetic β non-selective
agonist, and several β-blockers, on colon cancer cell
proliferation, a critical component of the carcinogenesis cascade.
In tumoral cells, β-AR is the key receptor in mediating the effects
of CA. The expression of β-AR has been identified in normal colon
tissue and in colon cancer cells, including in HT-29 cells, β2-AR
being the predominant receptor subtype in these cells (6). In fact, several studies have
recognized the activation of β-AR as a central mediator of stress
effects on cancer growth. The activation of these receptors is
involved in different tumorigenic processes including proliferation
(5), migration (4), apoptosis (19), angiogenesis (20) and differentiation in a various types
of cancer (21). Therefore, β-AR
blockade with pharmacological agents may be used to alleviate the
effects of stress following cancer growth and progression.
Concordant with this, findings of previous studies have shown that
β-AR blockade may suppress cancer-cell invasion and inhibit
adrenergic-driven metastasis (22).
In our study, the results obtained with the
adrenergic agonists, confirmed that stress hormones affect
colon-cancer cell proliferation, and suggest a prominent role for
β-AR in this process. AD and ISO, as previously shown by other
authors (5,6,23),
significantly enhanced HT-29 cell proliferation, most likely
through β-AR. Τhese agonists, both with a high affinity for β-AR
were strongly expressed in these cells, inducing a similar
proliferative response in the present study. Based on the above
observations, we explored the AR subtypes involved in AD and ISO
effects, using β-blockers with distinct profiles for AR. Taken
together, our results clearly indicate the involvement of the β-AR
subtypes, β1 and β2, in promoting
colon-cancer cell proliferation. PRO, the non-selective β-AR
antagonist, was the most potent β-blocker in reversing AD effects
following cell proliferation, by its ability to bind to the two
subtypes, as identified in previous studies (23). The proliferation increase stimulated
by AD was abolished by PRO, less by CAR, even less by ATE and was
not affected by ICI. According to β2 involvement, the
β1-blocker (ATE) was a weak antagonist for AD action,
while the selective β2-blocker (ICI) had no effect. By
contrast, with the exception of ICI, which had a moderate effect,
all the other β-blockers markedly inhibited the proliferation
induced by ISO. Thus, unlike other reports (5,6,23), the
β1 blockade by ATE was more effective in reverting AD-
and ISO-induced cell proliferation as compared to the β2
antagonism by ICI.
As previously mentioned, β-blockers are not solely
antagonists for the G-protein pathways, but they may independently
modulate more than one pathway, and behave as partial agonists,
inverse agonists or pure antagonists in each pathway, increasing
the complexity of their actions (24). Thus, biased agonism may be important
for the therapeutic use of β-AR blocker in cancer, since distinct
signaling through these pathways is considered to have specific
functional consequences (24). The
β-AR blockers examined were already recognized as being inverse
agonists (7). The finding that PRO
and ATE, when used alone, were able to decrease HT-29 cell
proliferation, suggests that they acted as inverse agonists, a
function already described in other experimental models for the two
drugs acting via β1- and β2-AR (7). The activation of β-ARs increases cAMP
intracellular concentrations and promotes cell proliferation, two
processes known to be reversed by treatment with either
β1-or β2-AR antagonists (25). PRO and ATE acting as inverse
agonists by binding to the β-ARs leads to a decrease of cAMP
accumulation (7), an outcome that
may explain the proliferation decrease induced by these drugs in
our study. However, β2 selective agonist (ICI) or CAR
did not exert an antiproliferative effect when used alone, although
both have been described as being able to decrease cAMP levels
(26). Thus, as suggested
previously (27), β-blockers seem
to have complex profiles for cAMP modulation and Erk1/2 activation
at β1- and β2-AR. Moreover, CAR is able to
activate different signaling pathways depending on the cell type
(26), although its effect on HT-29
proliferation was as yet unknown. CAR behaved similarly to PRO when
used simultaneously with the two agonists. However, in contrast to
data obtained with other cell types for β1 and
β2, CAR had no effect as inverse agonist (26).
Ongoing investigation have focused on the downstream
signaling pathways involved in β-AR-mediated tumor growth. Among
the mammalian MAPK pathways, ERK is the one that is most studied,
and the deregulation of this pathway occurs in approximately
one-third of all human cancers (28). It has been previously concluded that
β-AR-mediated ERK1/2 activation is a potential mechanism underlying
stress-induced cancer cell growth in vivo, suggesting, for
instance, that β-AR blockade may be an effective approach for
patients with stress-related colon cancer (23). However, in 2006, Shenoy et al
(30) showed in tumor models that
ISO, by binding to β2-AR leads to the activation of a
G-protein-independent ERK pathway, although this was dependent on
β-arrestin. The pathway whereby growth factors and mitogens
activate ERK signaling is of particular relevance to cancer
(28). The β-blockers used in our
study, with the exception of ICI, have already been described as
being capable of activating ERK pathway. However, as referred
before, when used alone, ATE and PRO decreased cell proliferation
and CAR had no effect.
CAR, PRO and ATE are widely used clinically. Several
studies have shown that these and other β-blockers reveal new
clinical applications in medicine, which is very attractive for
commercial purposes. However, gaining understanding of β signaling
pathways involved in cancer may allow the identification and
selection of the appropriate inhibitors to prevent and treat
cancer. Corroborating the putative use of β-blockers as therapeutic
agents in cancer, at least three phase II clinical studies
assessing the safety and efficacy of β-blockers in breast,
colorectal and ovarian cancers have been conducted (29).
Results of this study demonstrate the elucidation of
the most effective β-AR blockers in reverting the CA-induced
proliferative effects in colon cancer cells. Consequently, these
blockers may be used as promising strategies in cancer treatment
(in combination with other treatment paradigms).
Acknowledgments
This study was supported by the Rectory of the
University of Porto and Santander Totta (PP-IJUP2011-320), LPCC,
Research Department-Portuguese League Against Cancer (Núcleo
Regional do Norte).
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global Cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hamer M, Chida Y and Molloy GJ:
Psychological distress and cancer mortality. J Psychosom Res.
66:255–258. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Guimaraes S and Moura D: Vascular
adrenoceptors: an update. Pharmacol Rev. 53:319–356.
2001.PubMed/NCBI
|
|
4
|
Masur K, Niggemann B, Zanker KS and
Entschladen F: Norepinephrine-induced migration of SW 480 colon
carcinoma cells is inhibited by beta-blockers. Cancer Res.
61:2866–2869. 2001.PubMed/NCBI
|
|
5
|
Wong HP, Ho JW, Koo MW, et al: Effects of
adrenaline in human colon adenocarcinoma HT-29 cells. Life Sci.
88:1108–1112. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wu WK, Wong HP, Luo SW, et al:
4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone from cigarette smoke
stimulates colon cancer growth via beta-adrenoceptors. Cancer Res.
65:5272–5277. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Galandrin S and Bouvier M: Distinct
signaling profiles of beta1 and beta2 adrenergic receptor ligands
toward adenylyl cyclase and mitogen-activated protein kinase
reveals the pluridimensionality of efficacy. Mol Pharmacol.
70:1575–1584. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lutgendorf SK, Sood AK and Antoni MH: Host
factors and cancer progression: biobehavioral signaling pathways
and interventions. J Clin Oncol. 28:4094–4099. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Al-Wadei HA, Al-Wadei MH and Schuller HM:
Cooperative regulation of non-small cell lung carcinoma by
nicotinic and beta-adrenergic receptors: a novel target for
intervention. PLoS One. 7:e299152012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Thaker PH, Han LY, Kamat AA, et al:
Chronic stress promotes tumor growth and angiogenesis in a mouse
model of ovarian carcinoma. Nat Med. 12:939–944. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bernabe DG, Tamae AC, Biasoli ER and
Oliveira SH: Stress hormones increase cell proliferation and
regulate interleukin-6 secretion in human oral squamous cell
carcinoma cells. Brain Behav Immun. 25:574–583. 2011. View Article : Google Scholar
|
|
12
|
Perez Pinero C, Bruzzone A, Sarappa MG,
Castillo LF and Lüthy IA: Involvement of α2- and β2-adrenoceptors
on breast cancer cell proliferation and tumour growth regulation.
Br J Pharmacol. 166:721–736. 2012. View Article : Google Scholar
|
|
13
|
Zhang P, He X, Tan J, Zhou X and Zou L:
β-arrestin2 mediates β-2 adrenergic receptor signaling inducing
prostate cancer cell progression. Oncol Rep. 26:1471–1477.
2011.PubMed/NCBI
|
|
14
|
Stanojkovic TP, Zizak Z,
Mihailovic-Stanojevic N, Petrovic T and Juranic Z: Inhibition of
proliferation on some neoplastic cell lines-act of carvedilol and
captopril. J Exp Clin Cancer Res. 24:387–395. 2005.PubMed/NCBI
|
|
15
|
Miranda CL, Stevens JF, Helmrich A, et al:
Antiproliferative and cytotoxic effects of prenylated flavonoids
from hops (Humulus lupulus) in human cancer cell lines. Food Chem
Toxicol. 37:271–285. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
McDonald PG, Antoni MH, Lutgendorf SK, et
al: A biobehavioral perspective of tumor biology. Discov Med.
5:520–526. 2005.PubMed/NCBI
|
|
17
|
Chida Y, Hamer M, Wardle J and Steptoe A:
Do stress-related psychosocial factors contribute to cancer
incidence and survival? Nat Clin Pract Oncol. 5:466–475. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cole SW and Sood AK: Molecular pathways:
beta-adrenergic signaling in cancer. Clin Cancer Res. 18:1201–1206.
2012. View Article : Google Scholar :
|
|
19
|
Zhang D, Ma Q, Wang Z, Zhang M, Guo K,
Wang F and Wu E: β2-adrenoceptor blockage induces G1/S phase arrest
and apoptosis in pancreatic cancer cells via Ras/Akt/NFκB pathway.
Mol Cancer. 10:1462011. View Article : Google Scholar
|
|
20
|
Chakroborty D, Sarkar C, Basu B, Dasgupta
PS and Basu S: Catecholamines regulate tumor angiogenesis. Cancer
Res. 69:3727–3730. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Perez-Sayans M, Somoza-Martin JM,
Barros-Angueira F, Diz PG, Gandara Rey JM and Garcia-Garcia A:
Beta-adrenergic receptors in cancer: therapeutic implications.
Oncol Res. 19:45–54. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang D, Ma QY, Hu HT and Zhang M:
β2-adrenergic antagonists suppress pancreatic cancer cell invasion
by inhibiting CREB, NFκB and AP-1. Cancer Biol Ther. 10:19–29.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lin Q, Wang F, Yang R, Zheng X, Gao H and
Zhang P: Effect of chronic restraint stress on human colorectal
carcinoma growth in mice. PLoS One. 8:e614352013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rajagopal S, Rajagopal K and Lefkowitz RJ:
Teaching old receptors new tricks: biasing seven-transmembrane
receptors. Nat Rev Drug Discov. 9:373–386. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ji Y, Chen S, Li K, Xiao X, Zheng S and Xu
T: The role of β-adrenergic receptor signaling in the proliferation
of hemangioma-derived endothelial cells. Cell Div. 8:12013.
View Article : Google Scholar
|
|
26
|
Wisler JW, DeWire SM, Whalen EJ, et al: A
unique mechanism of beta-blocker action: carvedilol stimulates
beta-arrestin signaling. Proc Natl Acad Sci USA. 104:16657–16662.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Baker JG, Hill SJ and Summers RJ:
Evolution of β-blockers: from anti-anginal drugs to ligand-directed
signalling. Trends Pharmacol Sci. 32:227–234. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dhillon AS, Hagan S, Rath O and Kolch W:
MAP kinase signalling pathways in cancer. Oncogene. 26:3279–3290.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Barron TI, Sharp L and Visvanathan K:
Beta-adrenergic blocking drugs in breast cancer: a perspective
review. Ther Adv Med Oncol. 4:113–125. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shenoy SK, Drake MT, Nelson CD, et al:
Beta-arrestin-dependent, G protein-independent ERK1/2 activation by
the beta2 adrenergic receptor. J Biol Chem. 281:1261–1273. 2006.
View Article : Google Scholar
|