Introduction
The Kirsten rat sarcoma viral oncogene homolog
(KRAS) protein is an important signaling mediator in the epidermal
growth factor receptor (EGFR) pathway, which regulates cell growth
and is commonly targeted in cancer therapy (1,2).
Mutations in codon 12 and 13 of the KRAS gene are found in
~30% of colorectal cancer (CRC) cases and are associated with an
increased risk of recurrence and mortality (3). Various preclinical and clinical
studies have revealed that the presence of KRAS activating
mutations in CRC correlates with resistance to EGFR monoclonal
antibodies (EGFR mAb), such as cetuximab (Erbitax) and panitumumab
(Vectibix) (4–8). As such, the US Food and Drug
Administration (FDA) has recommended that EGFR mAbs should not be
given to patients with tumors harboring KRAS mutations in codon 12
or 13 (6). This requires the
KRAS mutation status to be assessed prior to treatment, yet
clinicians currently lack the ability to detect these mutations
with a high sensitivity, high-throughput and simple method.
Sanger sequencing is the gold standard for detecting
KRAS mutations; however, it is highly inefficient and has
relatively low sensitivity. In response to increasing demands in
the medical setting, various KRAS mutation detection assays
have been developed and described in the literature and different
providers offer commercial test kits (9,10).
Real-time PCR based methods are the most cost-effective and require
shorter working times than other methods. The Scorpion,
Amplification Refractory Mutation System (ARMS), and TaqMelt
systems are among the widely used real-time PCR-based KRAS
mutation detection systems (11–13).
While these are highly sensitive and reproducible methods, each has
its advantages and disadvantages. The Scorpion and ARMS methods are
based on mutation-matched primers, which amplify mutated loci more
efficiently than primers with wild-type (WT) sequences or other
mutations. Seven reactions are necessary to detect the mutation
status and thus the amount of DNA needed is ~800 ng (12). Alternatively, the TaqMelt method is
based on the melting curve analysis after PCR and is able to detect
a total of 19 mutations in KRAS codons 12, 13 and 61, yet it
requires sophisticated instruments and software. Hence, a more
simple and cost-effective detection assay capable of producing high
quality results may greatly benefit cancer diagnostics.
In addition to mutation sequencing at clinical
laboratories, most of these assays use an amplification-dependent
detection method sometimes combined with melting curve analysis of
the amplicon. The use of high-throughput sequencing for detecting
KRAS mutations has been described and some providers have
started to offer dedicated cancer sequencing panels (14). While high-throughput sequencing has
been used as a reference for testing the performance of new
KRAS detection assays, this approach could be extended to
wider applications. Assuming that PCR-based detection assays are
able to achieve a mutation sensitivity greater than or equal to
that of a Sanger sequencing-based approach, PCR-based detection
assays should be sufficient to confirm mutations found by
high-throughput sequencing.
In the present study, we aimed at developing a new
PCR-based detection assay using Eprobe, or ‘Eprobe-mediated PCR’.
Eprobe is a fluorescence probe enabling quantification analysis and
melting curve analysis using real-time PCR machines (15–17).
Eprobe binds complementary DNA with higher affinity than normal
oligonucleotides by using cationic dye moieties (18) and thus leads to a competitive effect
observed in primer annealing and extension. This characteristic
enables specific sequence enrichment-similar to using peptide and
locked nucleic acids (PNA and LNA, respectively) as a clamping
probe (19–27), to allow for the detection of even
miniscule amounts of mutated DNA. Notably, this method can detect
somatic mutations with high accuracy in simple steps that employ
commonly used laboratory equipment and a small amount of DNA,
unlike other methods. Thus, Eprobe-mediated PCR enables efficient
detection of mutation status and is a major advance in cancer
diagnostics. Here, we demonstrate a novel method to detect
KRAS mutations at codon 12 and 13 by Eprobe-mediated PCR
with a higher sensitivity than conventional Sanger sequencing,
which is more time and cost-effective than other technologies.
Materials and methods
Reagents and control DNA
DNA oligonucleotides were purchased from
Sigma-Aldrich (Ishikari, Japan), and stored as 100 mM stock
solutions in 10 mM Tris-HCl with 1 mM EDTA (pH 8.0). WT human
genomic DNA was purchased from Promega (Tokyo, Japan). Heterozygote
human genomic DNA with seven mutations (G12A, G12C, G12D, G12R,
G12S, G12V and G13D) in the KRAS gene codon 12 and 13 were
obtained from Horizon Diagnostics (Cambridge, UK). Eprobes (shown
in Table I) were obtained from K.K.
DNAFORM (Yokohama, Japan). Point mutations in codon 12 of the
KRAS gene (GAT, GCT, GTT, AGT, TGT) were cloned into the
plasmid pGEM-T (Promega) as previously described (26) and stored in glycerol at −80°C for
long-term conservation. Point mutation of codon 13 (GAC) in the
KRAS gene was prepared using the same protocol. The
KRAS codon 12 (CGT) point mutation was prepared using the
synthetic DNA ordered and inserted into a pUC57 plasmid (GenScript,
Tokyo, Japan). The sequences of WT and all seven mutant clones were
verified on ABI PRISM 3100 Avant (Applied Biosystems, Tokyo,
Japan), diluted in TE buffer and stored at −20°C until use.
 | Table ISequences of Eprobe for KRAS
mutation detection and genotyping. |
Table I
Sequences of Eprobe for KRAS
mutation detection and genotyping.
| Eprobe name | Genotype | Length (bases) | Sequence |
|---|
| Rkw19d16 | Wild-type | 19 |
5′-CTCzTGCCTACGCCACCAG-3′ |
| K12GCTe3 | G12A | 14 |
5′-AGCTGCTGGCGzAG-3′ |
| K12TGTe3 | G12C | 14 |
5′-AGCTTGTGGCGzAG-3′ |
| K12GATe3 | G12D | 14 |
5′-AGCTGATGGCGzAG-3′ |
| K12CGTe3 | G12R | 14 |
5′-AGCTCGTGGCGzAG-3′ |
| K12AGTe3 | G12S | 14 |
5′-AGCTAGTGGCGzAG-3′ |
| K12GTTe3 | G12V | 14 |
5′-AGCTGTTGGCGzAG-3′ |
| K13GACe3 | G13D | 14 |
5′-AGCTGGTGACGzAG-3′ |
Design of Eprobe and real-time PCR
Human KRAS specific primers for KRAS
codon 12 and 13 (KRAS-F, 5′-TTATAAGG CCTGCTGAAAATGACTGAA-3′
and KRAS-R, 5′-TGAATTAGCTGTATCGTCAAGGCACT-3′) were based on
literature (28) and amplified a 92
bp DNA fragment. Different PCR reagents and enzyme master mixes
were used with specific Eprobes during PCR. Eprobes were designed
complementary to the reverse strand for mutation detection by the
WT probe and to the forward strand for genotyping by the mutant
(MT) Eprobe (Table I).
Highly sensitive mutation detection by
high resolution melting analysis
PCR assays were performed using 1.5 μl of 5X
LightCycler 480 Genotyping Master, 0.5 μl of DMSO, 2.5
μl of diluted template DNA (2.5 ng/reaction), 0.5 μM
of forward primer, 0.1 μM of reverse primer and 0.4
μM Eprobe in a total volume of 10 μl. Amplification
reactions and melting curve analysis were from real-time PCR
experiments run on a Rotor-Gene Q (Qiagen K.K., Tokyo, Japan) after
activation of the hot-start enzyme for 10 min at 95°C, followed by
50 cycles of 15 sec at 95°C, 15 sec at 63°C and 12 sec at 72°C.
Amplification signals were detected during the annealing step of
each cycle at 63°C, using a SYBR Green I (483 nm) filter. Melting
curve analysis and high resolution melting analysis were performed
from 40 to 75°C with a temperature increase of 0.5°C/sec. All PCR
reactions and melting curve experiments were performed in
triplicate.
PCR standard curves were generated by analyzing Ct
values acquired from the Rotor-Gene Q instrument with Rotor-Gene
Q-Pure Detection (version 2.0.2; Qiagen K.K.). The data was
transferred to Microsoft Excel (Microsoft, Redmond, VA, USA) and Ct
values were plotted against the template DNA concentrations.
Logarithmic trend lines were added, and equations and R-squared
values obtained by using Microsoft Excel are shown in the graphs.
Microsoft Excel provides slopes as log10(x) values, which were
transformed into ln(x) by multiplying the slope value by 2.303
[ln(x) = 2.303 × log10(x)]. The PCR efficiency to evaluate the
competing effect is based on seven different concentrations of WT
plasmid DNA templates. The slope values were used to calculate PCR
efficiencies with a Thermo Fisher online web tool at: http://www.thermoscientificbio.com/webtools/qpcref-ficiency/.
Amplification, melting and derivative melting curves were obtained
with the Rotor-Gene Q instruments as indicated above, and the data
points were transferred to Microsoft Excel for plotting.
Mutation genotyping by melting curve
analysis
Amplification reactions using AmpliTaq Gold were set
up in 96-well plates using 5 μl template DNA (50
ng/reaction), 12.5 μl of AmpliTaq Gold PCR Master Mix, 0.1
μM of forward primer, 0.5 μM of reverse primer, and
0.2 μM Eprobe in a total volume of 25 μl. Real-time
PCR experiments were run on a Rotor-Gene Q or LightCycler 480
(Roche Diagnostics, Mannheim, Germany) after activation of the
hot-start enzyme for 10 min at 95°C, followed by 50 cycles of 15
sec at 95°C, 15 sec at 60°C, and 12 sec at 72°C. Amplification
signals were detected during the annealing step of each cycle at
60°C, using a SYBR-Green I (Rotor-Gene Q, 483 nm; LightCycler 480,
483–533 nm) filter. For melting curve analysis, the PCR was
followed by heating the reaction mixture to 95°C for 15 sec,
cooling to 37°C, holding at 37°C for 7 min, and then slowly heating
again to 95°C at a ramp rate of 2.2°C/sec with continuous
fluorescence acquisition at the indicated wavelengths. Samples were
analyzed in triplicate. Genotyping was performed by the ‘Melting
Curve Genotyping’ mode of LightCycler 480 software (version
1.5.1.62; Roche Diagnostics). After setting of the parameters
(temperature range, 55–70°C; score threshold, 0.70; and resolution
threshold, 0.20), ‘auto grouping’ was carried out for the mutation
genotyping by comparison with standard samples for each mutation
ratio.
Amplification, melting and derivative melting curves
were obtained from the LightCycler 480 instrument as indicated
above, and the data points were transferred to Microsoft Excel for
plotting.
Clinical samples
Tumor samples were obtained from patients with CRC
who were surgically treated at Gunma University Hospital (Gunma,
Japan) between 2002 and 2006. All the samples were immediately
frozen after surgical resection and stored at −80°C until DNA
extraction. Tumor samples were also preserved in a formalin-fixed
paraffin-embedded (FFPE) form. Previous examination with a
microscope confirmed that each FFPE tissue sample contained a
sufficient number of tumor cells for analysis. In the present
study, 92 frozen and 35 FFPE tissue samples were available for gene
analysis. An institutional approval and an informed consent from
all the patients were obtained in writing.
DNA extraction
DNA was extracted from a 3- to 5-mm cube of frozen
tissues that were collected using a DNA Mini kit (Qiagen K.K.)
according to the manufacturer’s instructions. For FFPE samples, two
to three 5-μm thick sections were sliced from each block
with the maximum number of tumor-rich areas. To suppress normal
tissue contamination, the tumor area of the section was macro
dissected. DNA was extracted using a QIAamp DNA FFPE tissue kit
(Qiagen K.K.) according to the manufacturer’s instructions. After
extraction, all the DNA templates were diluted in TE buffer to 10
ng/μl and stored at −20°C. Stored templates were diluted to
1 ng/μl with H2O just before Eprobe-PCR with the
WT Eprobe assay.
Sanger sequencing
Mutation screening in clinical samples for
KRAS codon 12 and 13 was performed using PCR conditions and
direct sequencing following previously described protocols
(25). The PCR reactions were
performed in a final volume of 25 μl containing GeneAmp 10X
PCR Gold buffer (Life Technologies, Carlsbad, CA, USA), 1.5 mM of
MgCl2 solution, 200 μM of dNTPs, 500 nM of each
primer (forward primer, 5′-TGAAGTACAGTTCATTACGATACACG-3′ and
reverse primer, 5′-GGAAAGTAAAGTTCCCATATTAATGGT-3′), 1 unit of
AmpliTaq Gold DNA polymerase (Life Technologies), and 20 ng of
genomic DNA. Thermal cycling conditions included preincubation at
94°C for 5 min, followed by 35 cycles at 94°C for 15 sec, 60°C for
30 sec, 72°C for 1 min and extension at 72°C for 5 min. The PCR
products were purified using the QIAquick PCR purification kit
(Qiagen K.K.) and processed for DNA sequencing reaction using ABI
PRISM BigDye Terminator version 3.1 (Applied Biosystems) with a
forward primer. Sequence data were generated using the ABI PRISM
3100 DNA Analyzer (Applied Biosystems).
Next-generation sequencing
Target sequencing libraries for Illumina HiSeq
sequencing were prepared using an Illumina TruSeq DNA Library Prep
kit (Illumina, San Diego, CA, USA) with multiplexed sample
barcoding and amplicon tagging following the manufacturer’s
instructions. The Illumina sequencing libraries were run on the
Illumina HiSeq 2000 utilizing a 100 bp single end sequencing read
protocol.
FASTQ files generated from either the Illumina HiSeq
sequencing runs were trimmed with the adapter sequence from sample
data with FASTX-toolkit (v0.0.13.2). The trimmed data were mapped
to references with the Burrows-Wheeler aligner (v0.6.2-r126). SAM
tools (v0.1.18) were utilized to convert the aligned sequence data
from a sequence alignment/map (SAM) to a binary sequence alignment
(BAM) format. The pileup files from BAM files were created with SAM
tools. Single nucleotide polymorphism (SNP) was examined using
VarScan2 (v2.3.5jar). SNPs were characterized as being
significantly different from the reference sequence if the variant
to reference base frequency was >1%.
Results
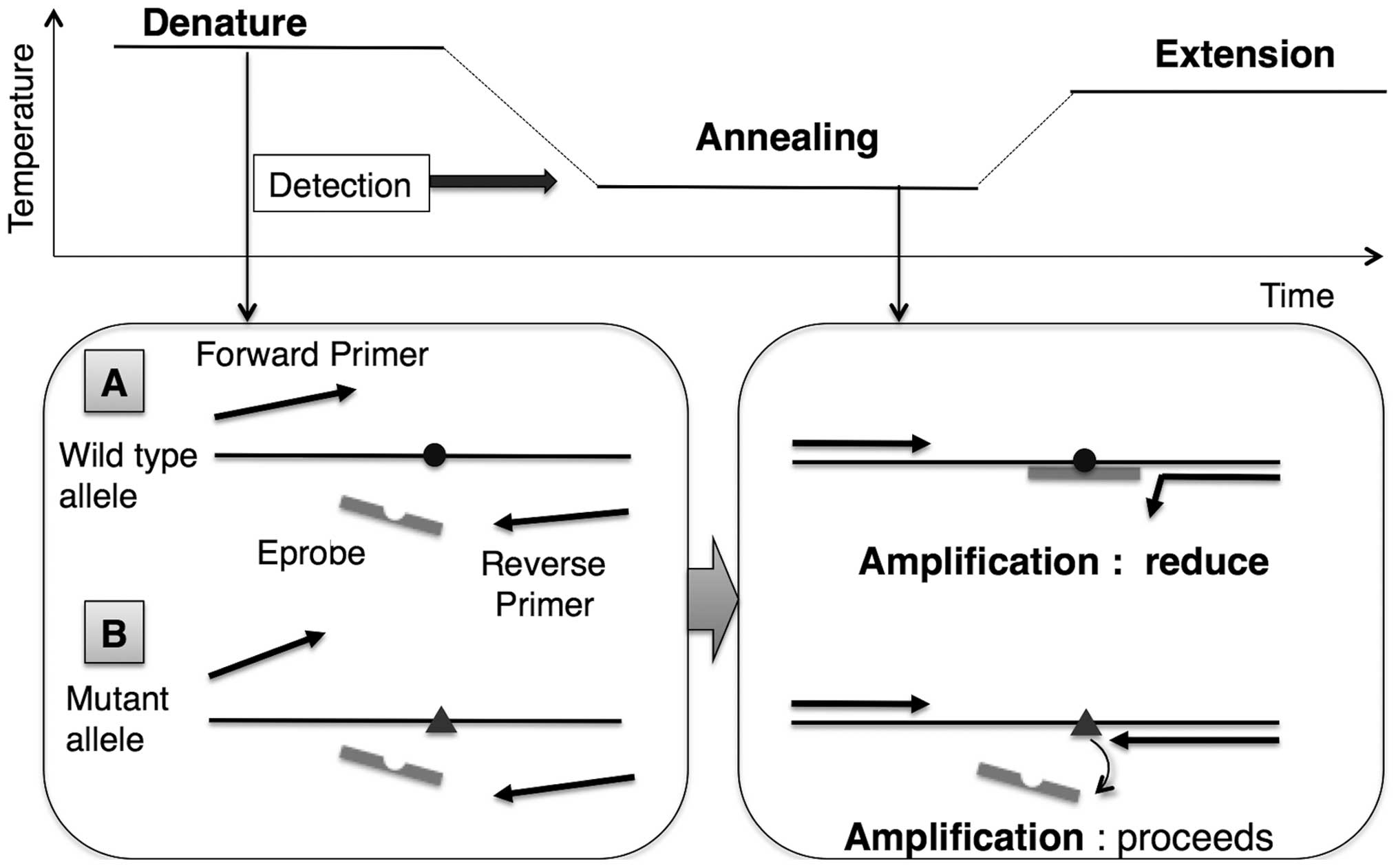
Working principle of mutation enrichment
by competitive, allele-specific Eprobe-mediated real-time PCR
Eprobe-mediated PCR is able to detect the targeted
mutation via enrichment of the mutant amplicon by a simple design
(Fig. 1). During the reaction, the
WT Eprobe binds the WT template with high affinity in the annealing
and extension steps, thus preventing generation of the PCR
amplicon. In contrast, a mismatch base pair between the WT Eprobe
and the mutant template reduces hybridization, resulting in
enrichment of the mutant amplicon. The WT Eprobe is designed
complementary to the reverse strand for mutation detection
(Table I). Standard PCR protocols
are used with the exception of an asymmetric primer ratio
(KRAS-F:KRAS-R=5:1) to improve Eprobe binding to the
forward strand. The annealing temperature was set to 63°C, which is
slightly lower than the Tm value of the full-complementary match
between the WT Eprobe and WT template. Taq DNA polymerase lacking
5′-3′ exonuclease activity in LightCycler 480 Genotyping Master is
utilized to avoid degradation of WT Eprobe, which binds the WT
template during the extension reaction.
Confirmation of Eprobe-mediated
competitive real-time PCR
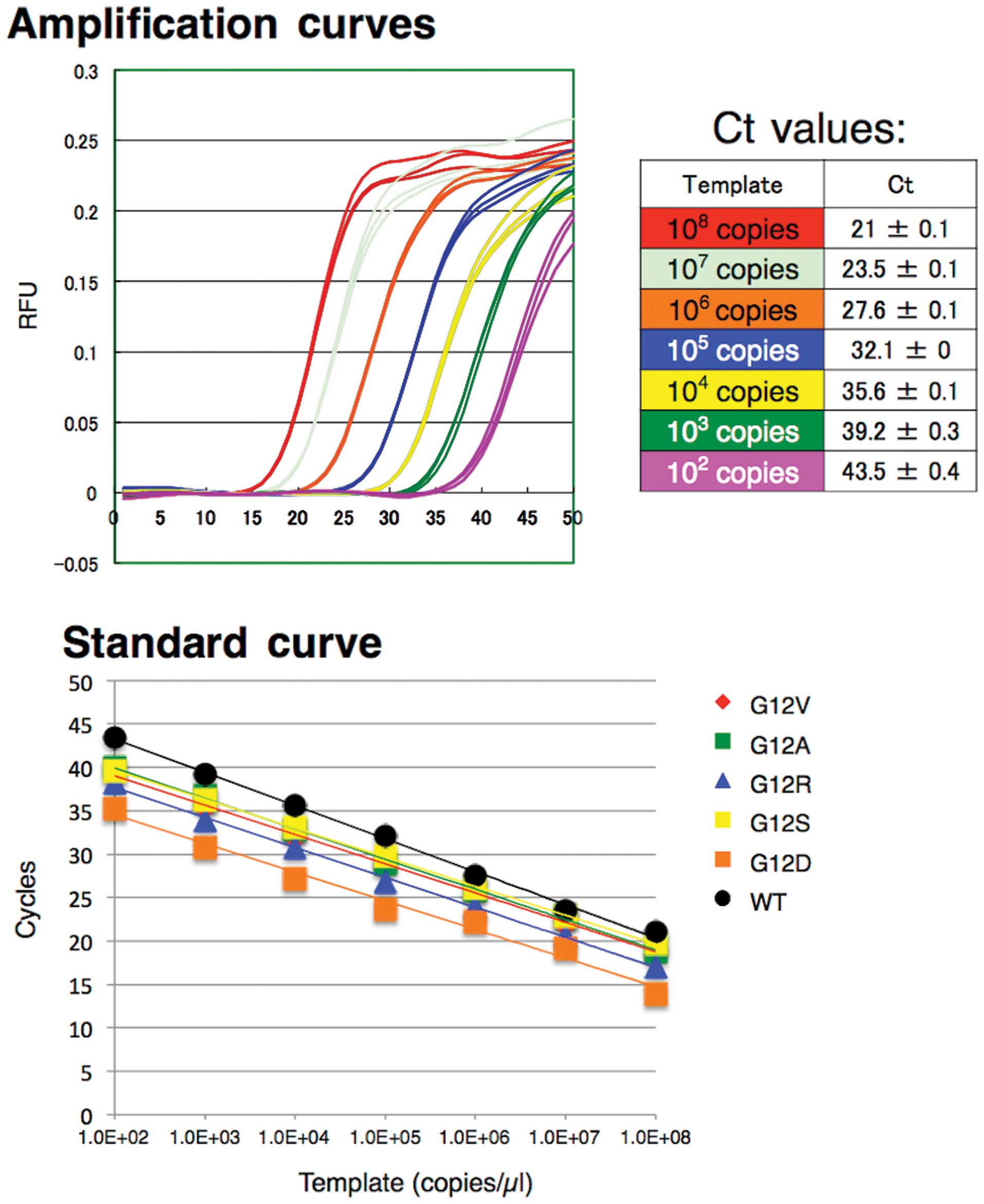
We tested a serial dilution range from
102 to 108 copies of KRAS WT and MT
plasmid DNA (Fig. 2). Each dilution
was assayed in triplicate. The WT of Eprobe detected amplification
of the WT template in real-time and demonstrated a good linear
range for detection in the dilution series. Standard curves of
triplicate Ct values for each dilution resulted in an 83%
amplification efficiency, as calculated from the slope (Fig. 2 and Table II). In contrast, G12C and G13D
plasmid DNA templates failed to show amplification curves; however,
a mismatch peak was detected on the melting curve analysis (data
not shown). These results suggest that the amplifications were
successful, but the lower affinity hybridization of the WT Eprobe
to the mutant DNA limits the amplification signal. Other MT plasmid
templates exhibit good amplification curves and a linear detection
range in the dilution series with efficiency values of 93–100%
(Table II); thus demonstrating
that the competitive effect is inefficient for the MT template, yet
present between the WT Eprobe and KRAS-R.
| Table IIEfficiency for each mutation
type. |
Table II
Efficiency for each mutation
type.
| Genotype | Sequence
(12) | Sequence
(13) |
Efficiency
(%) |
|---|
| WT | GGT | GGC | 83.0 |
| G12S | AGT | | 99.6 |
| G12C | TGT | | NDa |
| G12R | CGT | | 94.7 |
| G12D | GAT | | 101.3 |
| G12V | GTT | | 97.6 |
| G12A | GCT | | 93.1 |
| G13D | | GAC | NDa |
Melting curve analysis and high
resolution melting (HRM) analysis by competition with WT
Eprobe
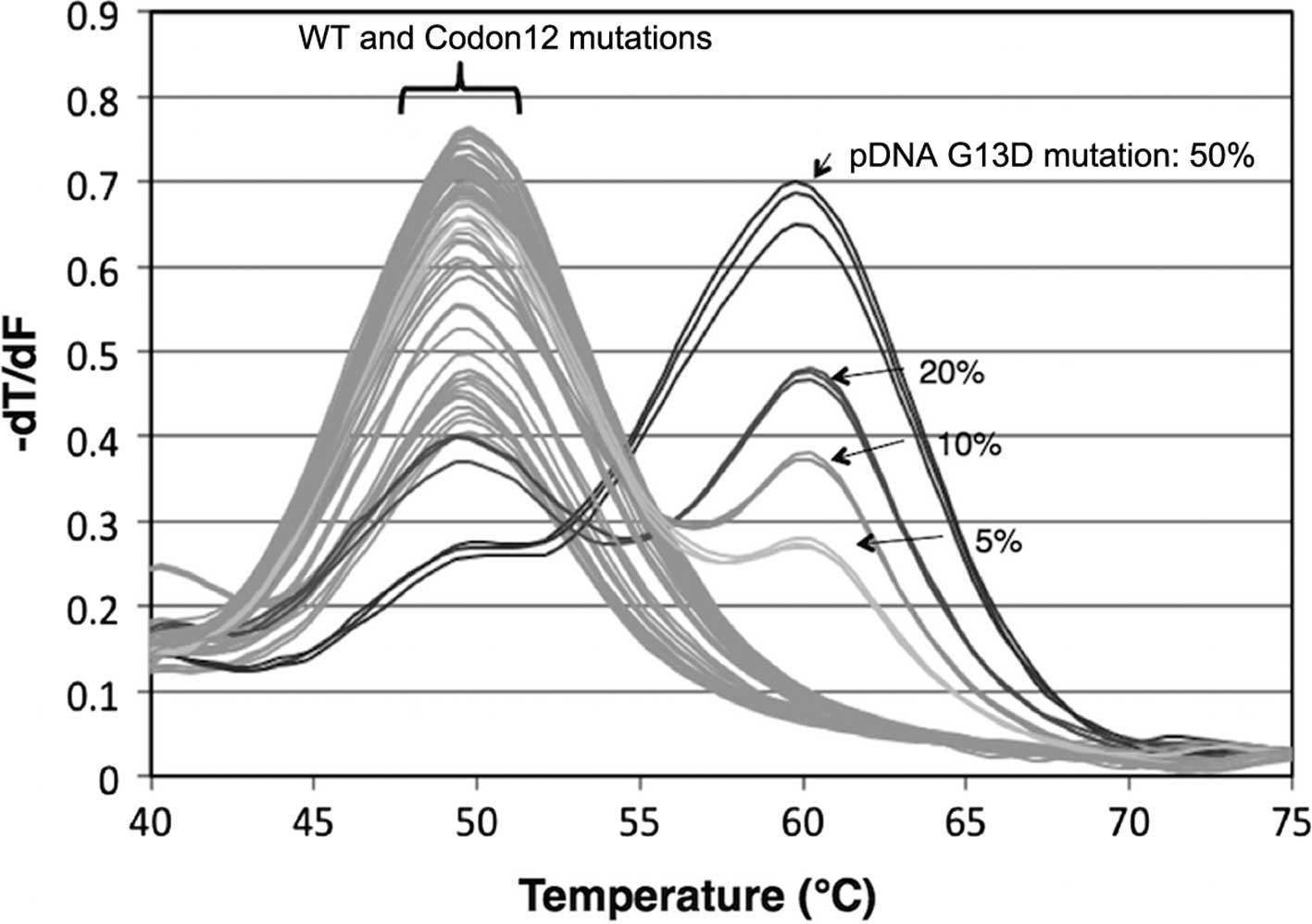
We evaluated the ability of KRAS low-level
mutation detection using 25,000 copies/reaction of plasmid DNA and
2.5 ng/reaction of human genomic DNA. Samples of each mutation
ratio were prepared by diluting mutant plasmid DNA with WT plasmid
DNA, and then analyzing mutant detection by real-time PCR. Melting
curve analysis and high resolution melting (HRM) analysis provided
Tm values for the indicated genotypes. The Tm of KRAS WT
(64.2°C) was higher than the Tm values of all seven KRAS
mutant types (MTs) in codon 12 and 13 (57.3–54.5°C). For each
mutant, a >1% mutation ratio exhibited peaks in the lower region
of the Tm values; however, it was difficult to determine values
<1% since the curve shapes failed to show distinct peaks.
On HRM analysis, we detected small changes of curves
at <1% mutation ratio by comparing them to the standard curve
derived from the WT amplicon (Fig.
3). We evaluated the numerical value from the confidence scores
of each sample assigned by the comparison of the melting curves
with the WT template. The threshold of KRAS mutation
detection was set at 98%. Using plasmid DNA, we achieved mutation
detection as low as a 0.05–0.1% ratio for each genotype (Table III), yet the human genomic DNA
showed an amplification efficiency lower than that of the plasmid
DNA. Notably, Eprobe-PCR achieved 1–2.5% mutation detection for
each genotype using 2.5 ng of genomic DNA (Table III). These results indicate that
Eprobe-PCR with WT Eprobe can detect KRAS mutations with
high sensitivity even when present at low levels.
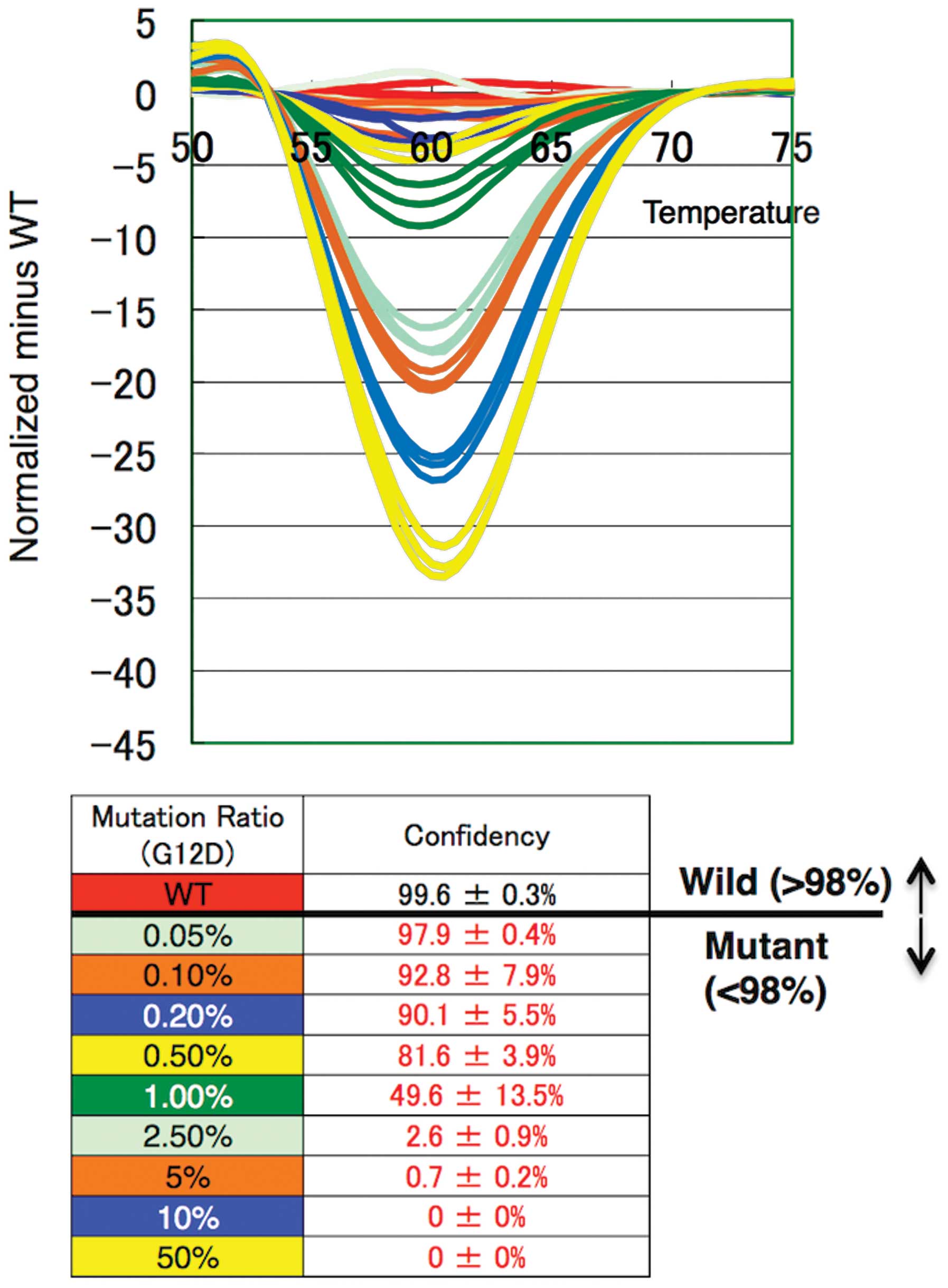 | Figure 3High resolution melting curve
analysis and differential plot of each G12D mutation ratio, 0,
0.05, 0.1, 0.2, 0.5, 1.0, 2.5, 5.0, 10 and 50%. Each color
represents three replicates. The threshold of KRAS mutation
detection was set at 98%. KRAS, Kirsten rat sarcoma viral
oncogene homolog. |
| Table IIIKRAS mutation detection. |
Table III
KRAS mutation detection.
| Mutation | Codon
12 | Codon
13 | Tm
(Obs)
(°C) | Mutation
ratio(pDNA)
(%) | Mutation ratio
(gDNA)
(%) |
|---|
| Wild | GGT | GGC | 64.2 | | |
| G12A | GCT | | 54.8 | 0.05 | 1.0 |
| G12C | TGT | | 57.0 | 0.05 | 1.0 |
| G12D | GAT | | 55.5 | 0.05 | 1.0 |
| G12R | CGT | | 57.3 | 0.05 | 1.0 |
| G12S | AGT | | 57.2 | 0.05 | 1.0 |
| G12V | GTT | | 56.2 | 0.05 | 2.5 |
| G13D | | GAC | 54.5 | 0.1 | 1.0 |
Melting curve analysis by Eprobe for
mutation genotyping
We tested mutation genotyping by MT Eprobes as
presented in Table I. The lengths
of Eprobes were shorter than that of the WT Eprobe for the
low-level mutation detection to avoid competition between the
primers and Eprobes. PCR reactions using AmpliTaq Gold Master Mix
were run on LightCycler 480 to generate a melting genotyping for
each MT template (Fig. 4). Some
combinations, such as G12R mutation and G12A MT Eprobe, exhibited a
slightly higher Tm value than other mutations and the WT template
on melting curve analysis. To confirm this, we used a positive
control for each mutation genotype and the highest Tm value to
identify the genotype. Importantly, Eprobe-based PCR yielded a
genotyping sensitivity as low as a 5 to 10% mutation ratio for each
mutant.
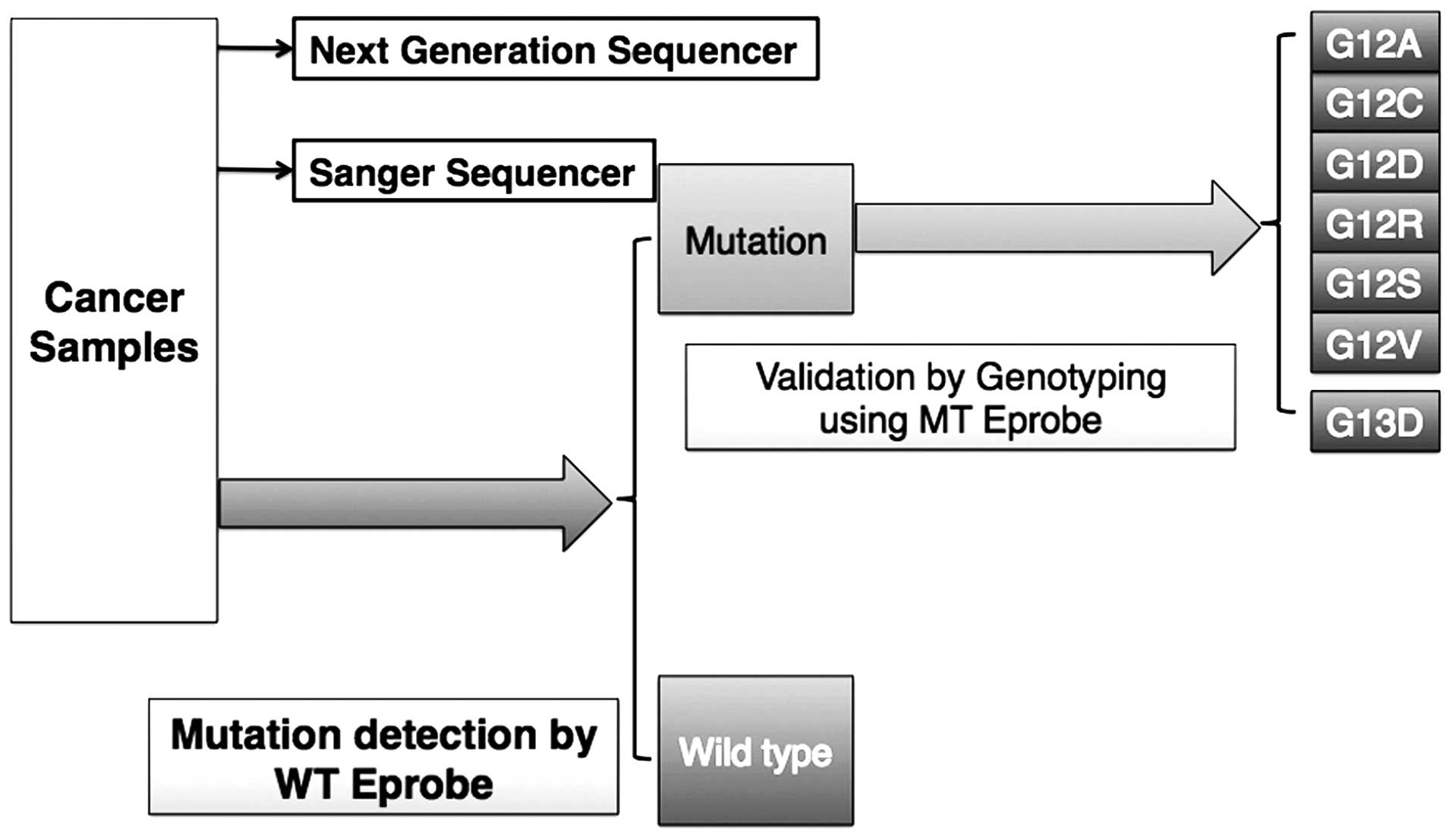
Assay of clinical samples
Eprobe-based PCR with WT Eprobe only requires a
small amount of DNA sample to KRAS mutations; this may then
be followed with MT Eprobe PCR to both verify these results as well
as genotype the mutation (Fig. 5).
To demonstrate the feasibility of Eprobe-based PCR in a clinical
setting, we performed the assay on DNA extracted from 92 frozen and
35 FFPE CRC tissue samples. To determine the mutation status with
the WT Eprobe, 2.5 ng of genomic DNA from each clinical sample was
used per reaction. In the DNA extracted from the frozen tissue,
Eprobe mediated PCR by WT Eprobe detected 33/92 mutated samples
(36%), whereas Sanger sequencing identified mutation in 20 samples
(22%) that were all detected by the Eprobe method (Table IV). These results demonstrate that
the Eprobe-PCR method is sufficient to detect KRAS mutations
using a small amount of clinical sample. Genotyping results with
the MT Eprobe identified 20 mutant DNA samples also detected by
Sanger sequencing, and 13 that went undetected (Table IV).
| Table IVSummary of KRAS mutation
detection on frozen clinical sample assay. |
Table IV
Summary of KRAS mutation
detection on frozen clinical sample assay.
| Sanger Sequencing
(SS) | Eprobe-PCR |
|---|
| No. of samples | 92 | 92 |
| Wild-type
status | 71 | 59 |
| Mutated | 20 | 33a (20 +13) |
| Unclear | 1 | 0 |
We performed a next-generation sequencing (NGS) on
these 13 genomic DNA samples that gave differing results with the
Eprobe-PCR and Sanger sequencing methods and one that yielded an
unclear result with Sanger sequencing (Table V). NGS analyses showed that the 13
DNA samples contained KRAS mutations with a 1–10% mutation
ratio and were consistent with the Eprobe-PCR results. Furthermore,
of the 35 DNA samples extracted from the FFPE tissue, 10 (29%) were
positive for KRAS mutations that were consistent with the
results from the Sanger sequencing (Table VI). These findings demonstrate that
Eprobe-PCR with the WT Eprobe allows for the detection of
KRAS mutations with high sensitivity, consistent to that
observed with NGS and more efficient than Sanger sequencing.
| Table VDiscrepancy of the results for each
method. |
Table V
Discrepancy of the results for each
method.
| Sample name | Sanger
sequencing | Eprobe mediated PCR
| Next-generation
sequencing
|
|---|
| Mutation
detection (WT Eprobe) | Genotyping (MT
Eprobe) |
Genotype | Mutation ratio
(%) |
|---|
| 62 | WT | MT | G12V | G12V | 7.4 |
| 202 | WT | MT | G12V | G12V | 3.3 |
| 207 | WT | MT | G12C | G12C | 1.7 |
| 212 | WT | MT | G12V | G12V | 2.7 |
| 215 | WT | MT | G12V | G12V | 3.3 |
| 217 | WT | MT | G12C | G12C | 1.4 |
| 224 | WT | MT | G12D | G12D | 6.7 |
| 227 | WT | MT | G13D | G13D | 6.5 |
| 230 | WT | MT | G12V | G12V | 3.0 |
| 244D | WT | MT | G13D | G13D | 9.5 |
| 247T | WT | MT | G13D | G13D | 8.9 |
| 305 | WT | MT | G13D | G13D | 6.3 |
| 321 | Unclear | MT | G13D | G13D | 1.1 |
| Table VIResults of FFPE samples. |
Table VI
Results of FFPE samples.
| Eprobe-PCR | Sanger Sequencing
(SS) |
|---|
| No. of samples | 35 | 35 |
| Wild-type
status | 25 | 25 |
| Mutated | 10 | 10 |
| Unclear | 0 | 0 |
Discussion
In the present study, we have presented a method to
detect and genotype somatic KRAS mutations in CRC samples
using Eprobe-mediated PCR. Eprobes can be used directly for
amplification analysis and melting curve analysis for mutation
detection and genotyping, respectively. It can also be used as a
competitive probe to enrich for mutant amplicon.
The guidelines detailing the design of Eprobes to
enrich for mutations via competition-mediated PCR are simple and
have been previously published (17). Our group designed and optimized the
Eprobes for the present study using the tool ‘ECHO/DNA
Thermodynamics’ (18). The WT
Eprobe overlaps with the reverse primer at two bases, causing
competition during the annealing and extension steps. An annealing
temperature slightly lower than the Tm value of the WT
Eprobe/template combination (63 vs. 64.2°C) yields a good
competition effect. In addition, it is essential to use Taq DNA
polymerase that lacks 5′-3′ exonuclease activity, since this
activity is able to digest the WT Eprobe during the extension
reaction. These optimization techniques effectively prevent
amplification of the WT template and lead to enrichment of the
mutant amplicons. The Eprobe method exhibits a lower competition
effect than PNA and LNA (19–27);
however, it also works as a detection probe. The relatively low
efficiency value of the WT amplification (83% compared to 93–100%,
Table II) allows the WT Eprobe to
be used as both a detection probe and a competition probe. This
competitive effect enables the detection of mutations present at
low ratios (0.05–0.1% mutant in WT background). The results
demonstrated the detection of the target sequence by fluorescence
and primer competition to reduce WT template amplification with one
WT Eprobe. Furthermore, this system simplifies the process to
detect KRAS mutations in codons 12 and 13, and reduces costs
since it only requires a standard primer set, PCR reaction mix and
an Eprobe.
To assess Eprobe-mediated PCR performance in a
clinical setting, we assayed 92 samples extracted from frozen CRC
tissues and evaluated the accuracy of the assay in comparison with
the Sanger sequencing. Significantly, out of 92 samples, our WT
Eprobe detected the presence of mutations in 33 samples (36%),
including 20 (27%) identified as mutants by the Sanger sequencing,
and 13 samples (14%) not detected by Sanger but confirmed by NGS.
We evaluated this discrepancy in KRAS mutation genotyping by
Eprobe-mediated PCR with MT Eprobes and NGS (Table V). Eprobes are designed to not
overlap with primers and have Tm values below the annealing
temperatures of the PCR primers. The MT Eprobes accurately
genotyped each of the 33 samples identified as mutants by the WT
Eprobe. This demonstrates that Eprobe-mediated PCR is able to
detect even a small amount of mutated KRAS DNA, which the
conventional sequencing method cannot. Sanger sequencing is the
gold standard for identification of KRAS mutations in codons
12 and 13, yet it has a detection threshold of at least 20%
(29). Thus, in a clinical setting,
Eprobe-mediated PCR may provide more accurate information for
determining which anti-EGFR mAb to use for the CRC patients. NGS
may become the primary method to detect somatic mutations in order
to ensure the necessary sensitivity and accuracy required by
clinical standards. Our Eprobe-mediated PCR method is independent
of the NGS method, and offers a more time and cost-effective method
with equivalent sensitivity and accuracy; and thus may be a
suitable alternative to NGS or a second method to confirm data
acquired by NGS.
We also assayed clinical samples from FFPE tissue
and found no discrepancy between the results from Eprobe-mediated
PCR and Sanger sequencing (Table
VI). In general, most clinical samples are fixed and preserved
for pathological assessment in hospitals or institutes. However,
the DNA extracted from the FFPE tissues is fragmented and
chemically modified, making it difficult to use this DNA in
molecular studies. The target base length is important for
successful PCR when assaying DNA isolated from FFPE samples
(32). As the Eprobe and primers in
the present study are designed to generate a 92 bp amplicon, the
PCR reaction is more likely to be successful even with short
fragmented DNA obtained from FFPE tissues. Nevertheless, further
investigation with larger sample sizes is required to verify the
usefulness of Eprobe-mediated PCR in DNA isolated from FFPE
samples.
Conventional real-time PCR-based KRAS
mutation detection kits have a detection sensitivity of 95–99% and
a specificity of 100% (30). Our
Eprobe-PCR method satisfies these requirements as it detects
mutated DNA (via the WT Eprobe) present at 0.05–0.1% in plasmid DNA
and 1–2.5% in human genomic DNA as determined by HRM analysis. This
raises the specificity to 100% for detecting mutations using these
templates.
The FDA recommends that CRC patient tumor biopsies
be assessed for KRAS mutation status prior to treatment with
anti-EGFR mAbs. Our method has the potential to quickly provide
necessary and sufficient information to physicians. It is also
likely to reduce the cost of gene sequencing not only for
KRAS, but also other common oncogenic mutations, including
those BRAF, PIK3CA and PTEN (31).
Eprobe-mediated PCR is superior to other methods,
such as Scorpion, ARMS and TaqMelt, in terms of the amount of DNA
required. Notably, only 2.5 ng of template DNA samples is needed
for detection by Eprobe-PCR, compared to ~100 ng/μl for
other methods (11–13). The need for only a small amount of
template is beneficial since DNA extraction is laborious and
time-consuming. Furthermore, excess template can be used for future
analyses with other methods; this is particularly useful in cases
where only a small amount of tissue is obtained.
In conclusion, we have developed a novel detection
system capable of detecting KRAS mutations with high
sensitivity using Eprobe-mediated PCR and compared its performance
to Sanger sequencing and NGS. The simplicity and high sensitivity
of Eprobe PCR may be suitable in a clinical setting that requires
swift and accurate KRAS gene mutation detection.
Acknowledgments
We acknowledge Dr Matthias Harbers for his
contributions to the manuscript preparation. This study was
supported by i) research grants for RIKEN Omics Science Center from
the Japanese Ministry of Education, Culture, Sports, Science and
Technology (MEXT) to Y.H., ii) a research grant from the Japanese
Ministry of Education, Culture, Sports, Science and Technology
(MEXT) through RIKEN Preventive Medicine and Diagnosis Innovation
Program to Y.H., iii) a Research Grant from the Japanese Ministry
of Education, Culture, Sports, Science and Technology (MEXT) to the
RIKEN Center for Life Science Technologies, iv) FY2013 subsidy of
expenses by METI (Minister of Economy, Trade and Industry) for
measures for small business support, and v) a research grant and
operational grant for the Division of Thoracic and Visceral Organ
Surgery, Gunma University Graduate School of Medicine from the
Ministry of Education, Culture, Sports, Science and Technology
(MEXT) of the Japanese Government to K.S.
References
|
1
|
Mendelsohn J and Baselga J: Epidermal
growth factor receptor targeting in cancer. Semin Oncol.
33:369–385. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Downward J: Targeting RAS signalling
pathways in cancer therapy. Nat Rev Cancer. 3:11–22. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Andreyev HJ, Norman AR, Cunningham D,
Oates JR and Clarke PA: Kirsten ras mutations in patients with
colorectal cancer: The multicenter ‘RASCAL’ study. J Natl Cancer
Inst. 90:675–684. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Allegra CJ, Jessup JM, Somerfield MR,
Hamilton SR, Hammond EH, Hayes DF, McAllister PK, Morton RF and
Schilsky RL: American Society of Clinical Oncology provisional
clinical opinion: Testing for KRASgene mutations in patients with
metastatic colorectal carcinoma to predict response to
anti-epidermal growth factor receptor monoclonal antibody therapy.
J Clin Oncol. 27:2091–2096. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lièvre A, Bachet JB, Le Corre D, Boige V,
Landi B, Emile JF, Côté JF, Tomasic G, Penna C, Ducreux M, et al:
KRASmutation status is predictive of response to cetuximab therapy
in colorectal cancer. Cancer Res. 66:3992–3995. 2006. View Article : Google Scholar
|
|
6
|
Normanno N, Tejpar S, Morgillo F, De Luca
A, Van Cutsem E and Ciardiello F: Implications for KRAS status and
EGFR-targeted therapies in metastatic CRC. Nat Rev Clin Oncol.
6:519–527. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bardelli A and Siena S: Molecular
mechanisms of resistance to cetuximab and panitumumab in colorectal
cancer. J Clin Oncol. 28:1254–1261. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Amado RG, Wolf M, Peeters M, Van Cutsem E,
Siena S, Freeman DJ, Juan T, Sikorski R, Suggs S, Radinsky R, et
al: Wild-type KRAS is required for panitumumab efficacy in patients
with metastatic colorectal cancer. J Clin Oncol. 26:1626–1634.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cavallini A, Valentini AM, Lippolis C,
Campanella D, Guerra V and Caruso ML: KRASgenotyping as biomarker
in colorectal cancer: A comparison of three commercial kits on
histologic material. Anticancer Res. 30:5251–5256. 2010.PubMed/NCBI
|
|
10
|
Malapelle U, Carlomagno C, de Luca C,
Bellevicine C and Troncone G: KRAS testing in metastatic colorectal
carcinoma: Challenges, controversies, breakthroughs and beyond. J
Clin Pathol. 67:1–9. 2014. View Article : Google Scholar
|
|
11
|
Lee S, Brophy VH, Cao J, Velez M, Hoeppner
C, Soviero S and Lawrence HJ: Analytical performance of a PCR assay
for the detection of KRAS mutations (codons 12/13 and 61) in
formalin-fixed paraffin-embedded tissue samples of colorectal
carcinoma. Virchows Arch. 460:141–149. 2012. View Article : Google Scholar :
|
|
12
|
Gonzalez de Castro D, Angulo B, Gomez B,
Mair D, Martinez R, Suarez-Gauthier A, Shieh F, Velez M, Brophy VH,
Lawrence HJ, et al: A comparison of three methods for detecting
KRAS mutations in formalin-fixed colorectal cancer specimens. Br J
Cancer. 107:345–351. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tol J, Dijkstra JR, Vink-Börger ME,
Nagtegaal ID, Punt CJ, Van Krieken JH and Ligtenberg MJ: High
sensitivity of both sequencing and real-time PCR analysis of
KRASmutations in colorectal cancer tissue. J Cell Mol Med.
14:2122–2131. 2010. View Article : Google Scholar
|
|
14
|
Chambers PA, Stead LF, Morgan JE, Carr IM,
Sutton KM, Watson CM, Crowe V, Dickinson H, Roberts P, Mulatero C,
et al: Mutation detection by clonal sequencing of PCR amplicons and
grouped read typing is applicable to clinical diagnostics. Hum
Mutat. 34:248–254. 2013. View Article : Google Scholar
|
|
15
|
Ikeda S, Kubota T, Yuki M and Okamoto A:
Exciton-controlled hybridization-sensitive fluorescent probes:
Multicolor detection of nucleic acids. Angew Chem Int Ed Engl.
48:6480–6484. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lezhava A, Ishidao T, Ishizu Y, Naito K,
Hanami T, Katayama A, Kogo Y, Soma T, Ikeda S, Murakami K, et al:
Exciton Primer-mediated SNP detection in SmartAmp2 reactions. Hum
Mutat. 31:208–217. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hanami T, Delobel D, Kanamori H, Tanaka Y,
Kimura Y, Nakasone A, Soma T, Hayashizaki Y, Usui K and Harbers M:
Eprobe mediated real-time PCR monitoring and melting curve
analysis. PLoS One. 8:e709422013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kimura Y, Hanami T, Tanaka Y, de Hoon MJ,
Soma T, Harbers M, Lezhava A, Hayashizaki Y and Usui K: Effect of
thiazole orange doubly labeled thymidine on DNA duplex formation.
Biochemistry. 51:6056–6067. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nagai Y, Miyazawa H, Huqun, Tanaka T,
Udagawa K, Kato M, Fukuyama S, Yokote A, Kobayashi K, Kanazawa M,
et al: Genetic heterogeneity of the epidermal growth factor
receptor in non-small cell lung cancer cell lines revealed by a
rapid and sensitive detection system, the peptide nucleic
acid-locked nucleic acid PCR clamp. Cancer Res. 65:7276–7282. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Arcila M, Lau C, Nafa K and Ladanyi M:
Detection of KRAS and BRAFmutations in colorectal carcinoma roles
for high-sensitivity locked nucleic acid-PCR sequencing and
broad-spectrum mass spectrometry genotyping. J Mol Diagn. 13:64–73.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dono M, Massucco C, Chiara S, Sonaglio C,
Mora M, Truini A, Cerruti G, Zoppoli G, Ballestrero A, Truini M, et
al: Low percentage of KRASmutations revealed by locked nucleic acid
polymerase chain reaction: Implications for treatment of metastatic
colorectal cancer. Mol Med. 18:1519–1526. 2012. View Article : Google Scholar
|
|
22
|
Oh JE, Lim HS, An CH, Jeong EG, Han JY,
Lee SH and Yoo NJ: Detection of low-level KRASmutations using
PNA-mediated asymmetric PCR clamping and melting curve analysis
with unlabeled probes. J Mol Diagn. 12:418–424. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Beau-Faller M, Legrain M, Voegeli A-C,
Guérin E, Lavaux T, Ruppert AM, Neuville A, Massard G, Wihlm JM,
Quoix E, et al: Detection of K-Rasmutations in tumour samples of
patients with non-small cell lung cancer using PNA-mediated PCR
clamping. Br J Cancer. 100:985–992. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Däbritz J, Hänfler J, Preston R, Stieler J
and Oettle H: Detection of Ki-ras mutations in tissue and plasma
samples of patients with pancreatic cancer using PNA-mediated PCR
clamping and hybridisation probes. Br J Cancer. 92:405–412.
2005.PubMed/NCBI
|
|
25
|
Araki T, Shimizu K, Nakamura K, Nakamura
T, Mitani Y, Obayashi K, Fujita Y, Kakegawa S, Miyamae Y, Kaira K,
et al: Usefulness of peptide nucleic acid (PNA)-clamp smart
amplification process version 2 (SmartAmp2) for clinical diagnosis
of KRAS codon 12 mutations in lung adenocarcinoma: Comparison of
PNA-clamp SmartAmp2 and PCR-related methods. J Mol Diagn.
12:118–124. 2010. View Article : Google Scholar
|
|
26
|
Tatsumi K, Mitani Y, Watanabe J, Takakura
H, Hoshi K, Kawai Y, Kikuchi T, Kogo Y, Oguchi-Katayama A, Tomaru
Y, et al: Rapid screening assay for KRASmutations by the modified
smart amplification process. J Mol Diagn. 10:520–526. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Miyamae Y, Shimizu K, Mitani Y, Araki T,
Kawai Y, Baba M, Kakegawa S, Sugano M, Kaira K, Lezhava A, et al:
Mutation detection of epidermal growth factor receptor and
KRASgenes using the smart amplification process version 2 from
formalin-fixed, paraffin-embedded lung cancer tissue. J Mol Diagn.
12:257–264. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Krypuy M, Newnham GM, Thomas DM, Conron M
and Dobrovic A: High resolution melting analysis for the rapid and
sensitive detection of mutations in clinical samples: KRAS codon 12
and 13 mutations in non-small cell lung cancer. BMC Cancer.
6:2952006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tsiatis AC, Norris-Kirby A, Rich RG, Hafez
MJ, Gocke CD, Eshleman JR and Murphy KM: Comparison of Sanger
sequencing, pyrosequencing, and melting curve analysis for the
detection of KRASmutations: Diagnostic and clinical implications. J
Mol Diagn. 12:425–432. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Aubin F, Gill S, Burkes R, Colwell B,
Kamel-Reid S, Koski S, Pollett A, Samson B, Tehfe M, Wong R, et al:
Canadian Expert Group consensus recommendations: KRAS testing in
colorectal cancer. Curr Oncol. 18:e180–e184. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Blons H, Rouleau E, Charrier N, Chatellier
G, Côté JF, Pages JC, de Fraipont F, Boyer JC, Merlio JP, Morel A,
et al MOKAECM Collaborative Group: Performance and cost efficiency
of KRAS mutation testing for metastatic colorectal cancer in
routine diagnosis: The MOKAECM study, a nationwide experience. PLoS
One. 8:e689452013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ludyga N, Grünwald B, Azimzadeh O, Englert
S, Höfler H, Tapio S and Aubele M: Nucleic acids from long-term
preserved FFPE tissues are suitable for downstream analyses.
Virchows Arch. 460:131–140. 2012. View Article : Google Scholar : PubMed/NCBI
|