Introduction
Antiangiogenic therapy is considered significant for
the treatment of cancer (1).
However, accumulating evidence suggests that antiangiogenic therapy
shows transient antitumor activity and enhances tumor invasiveness
and metastasis (2,3). The complex changes in the tumor
microenvironment elicited by antiangiogenic therapy, which result
in a hypoxic and inflammatory microenvironment, are the main cause
of tumor refractoriness. Notably, numerous tumor-associated
inflammatory cells and their cytokines, e.g.,
CD11b+Gr1+ myeloid-derived suppressor cells
(MDSCs), tumor-associated macrophages (TAMs), granulocyte
colony-stimulating factor (G-CSF) and tumor necrosis factor-α
(TNF-α), are recruited into the tumor microenvironment (4,5).
Therefore, remission or resolution of the inflammatory tumor
microenvironment is crucial for improving the efficacy of
antiangiogenic therapies.
Versican, a versatile extracellular matrix
proteoglycan present in a variety of tissues and commonly
overexpressed in tumor stroma and cancer cells (6), plays a key role in cancer development
and progression by contributing to cell adhesion, migration,
angiogenesis and the formation of an inflammatory tumor
microenvironment (7). By activating
multiple types of inflammatory cells through the Toll-like receptor
2 (TLR-2) and then eliciting the production of many proinflammatory
cytokines, versican strongly enhances tumor progression (8). Additionally, the inflammatory and
tumor cells enhance versican expression, which in turn induces the
secretion cascade of inflammatory cytokines to generate an
inflammatory microenvironment that provides permissive conditions
for tumor progression and metastases (9).
Endostatin, a broad-spectrum endogenous
angio-genesis inhibitor, suppresses tumor growth mainly by
selectively blocking the binding of vascular endothelial growth
factor (VEGF) to endothelial cells (ECs) to inhibit the
VEGF-stimulated proliferation, migration and tube formation of ECs
(10,11). Several lines of direct and indirect
evidence indicate that endostatin has caused a significant
reduction in microvessel density and resulted in the inhibition of
tumor growth. However, the antitumor efficacy of endostatin alone
is of short duration, and confers no significant survival benefit
to tumor bearing mice (12–14). Findings of a recent study showed
that versican, NF-κB and HIF-1α, which are associated with
inflammatory and hypoxic changes in the tumor microenvironment,
were found to be overexpressed in tumor tissues from animal models
refractory to endostatin treatment. Additionally, MDSCs and
inflammatory cytokines were largely recruited into the peripheral
blood and the tumor microenvironment (15). However, the role of versican and
tumor-associated inflammatory cells in the tumor microenvironment
changes caused by endostatin administration is not entirely clear,
and the connection of versican with tumor-associated inflammatory
cells, and with other inflammatory and immune regulators in the
tumor microenvironment, remains to be elucidated. Accordingly, we
hypothesized that versican, a ‘bridge’ connecting inflammation with
tumor progression as well as playing a central role in the
generation of inflammatory tumor microenvironment is a promising
candidate for the intervention of inflammatory changes in the tumor
microenvironment elicited by antiangiogenic therapy.
Thus, we designed a short-hairpin (sh) RNA targeting
versican and assessed the effects of versican silencing on the
bioactivity of B16F1 and Lewis lung carcinoma (LLC) cell lines. At
the same time, the established B16F1 and LLC tumor models were used
to investigate the effect of versican silencing combined with
endostatin on the tumor burden of mice. Furthermore, we studied the
effect of versican on the changes in the tumor microenvironment
elicited by endostatin, and examined the related mechanisms.
Materials and methods
Cell culture
LLC and B16F1 melanoma cell lines were obtained from
the American Type Culture Collection (ATCC; Manassas, VA, USA). The
cells were cultured in Dulbecco’s modified Eagle’s medium
(Gibco-BRL, Grand Island, NY, USA) supplemented with 10% calf serum
and 1% penicillin/streptomycin, and maintained in a humidified 5%
CO2 atmosphere incubator at 37°C.
Knockdown of versican in LLC and B16F1
cells
Short-hairpin RNAs (shRNA) targeting mouse versican
(V1 isoform) were designed and cloned into the
pcDNA6.2-GW/EmGFP-miR vector (Invitrogen, Shanghai, China), and
transfected into LLC and B16F1 cells by Lipofectamine 2000
(Invitrogen) according to the manufacturer’s instructions. The
cells were then selected in 8 μg/ml Blasticidin S HCl
(Invitrogen), sorted for green fluorescent protein (GFP) expression
and cloned. The targeting sequence included
5′-GTACACAGTTGATGAAATAC-3′. The pcDNA6.2-GW/EmGFP-miR-neg (shC)
plasmid served as a negative control (Invitrogen).
Groups
Six groups of each model were analyzed in the
present study: normal saline (B16F1-NS and LLC-NS), normal saline +
pcDNA6.2-GW/EmGFP-miR-neg (B16F1/shC-NS, LLC/shC-NS), normal saline
+ pcDNA6.2-GW/EmGFP-miR-versican (B16F1/shVCAN-NS, LLC/shVCAN-NS),
endostatin (B16F1-ES, LLC-ES), endostatin +
pcDNA6.2-GW/EmGFP-miR-neg (B16F1/shC-ES, LLC/shC-ES) and endostatin
+ pcDNA6.2-GW/EmGFP-miR-versican (B16 F1/shVCAN-ES,
LLC/shVCAN-ES).
Western blot analysis
Equal amounts of protein were separated by 8–12%
SDS-PAGE and transferred to polyvinylidene fluoride (PVDF)
membranes (Millipore, Shanghai, China) by electroblotting. The
membranes were probed with specific antibodies including HIF-1α
(1:100; ab113642), versican (1:200; ab19345) (both from Abcam,
Cambridge, UK) or NF-κB (1:100; no. 8242S; CST, Boston, MA, USA).
Blots were developed with horseradish peroxidase (HRP)-conjugated
secondary antibodies and chemiluminescent substrate on Kodak X-ray
film.
Cell proliferation, Transwell migration
and invasion assays
Cells were seeded at a density of
1×103−3×103 cells/well on 96-well plates,
cultured for 72 h, and subjected to a CCK-8 colorimetric assay
(Dojindo, Shanghai, China), according to the manufacturer’s
instructions. The migration and invasive ability of cells was
determined using the Matrigel (BD Biosciences)-coated 24-well
Transwell chambers (Corning Costar, Beijing, China). The cells
(1×105) were seeded in the top chamber and incubated for
24 or 48 h. The migrating or invading cells were counted using a
light microscope (Olympus UIS2; magnification, ×20, three random
fields/well were analyzed by ImageJ).
Tumor models and treatment protocol
C57BL/6 mice were subcutaneously implanted with 100
ml solution containing 3–5×105 cells in the right
mid-dorsal flank. The tumor volume (mm3) was calculated
as length × width2/2 (16). The time when the tumors reached
volumes of 10–50 mm3 was designated as day 0, after
which normal saline or endostatin [3 mg/kg (15); Simcere-Medgenn Bio-Pharmaceutical
Co., Ltd. Shandong, China] was administered daily i.v. (by caudal
vein injection) for 9 days to the tumor-bearing mice. On days 0, 3,
6 and 9 after initiation of treatment, the tumor volume was
estimated and mice (three from each group) were sacrificed to
harvest tumor tissues and blood samples for subsequent analyses
(15). Experimental procedures and
protocols were approved by the Animal Ethics Committee of Sichuan
University.
Flow cytometry
Analyses of the MDSCs and TAMs in the tumor tissue
and peripheral blood were conducted as previously described
(15). Single-cell suspensions were
stained with fluorochrome-labeled antibody-targeting murine CD11b
(PE-CY5-labeled), Gr1 (PE-labeled), F4/80 (APC-labeled) or an
appropriate isotype control antibody (all from Tianjin Sungene
Biotech Co., Ltd.) and were analyzed by flow cytometry (BD
FACSCalibur; BD Biosciences).
Luminex xMAP assays
A commercially available mouse cytokine magnetic
bead panel kit (Millipore, Billerica, MA, USA) was used to evaluate
the cytokine levels in collected mouse serum and tumor tissue. On
days 0, 3, 6 and 9 after initiation of treatment, the serum was
collected from non-anticoagulated blood from all mice, and the
protein lysate of tumor tissue was collected. The cytokines
comprising G-CSF, TNF-α, IL-6, VEGF and IL-10 were measured
according to the manufacturer’s instructions (MILLIPLEX®
MAP) based on Luminex xMAP technology (Millipore).
Immunohistochemistry
As previously described (15), paraffin-embedded tumor tissues were
sectioned (4–5 μm), and incubated with the following
antibodies: HIF-1α, versican (both from Abcam, Cambridge, UK),
NF-κB (CST) and CD31 (BD Biosciences). Randomly chosen fields were
photographed at a magnification of ×400 with a microscope (Leica
DM2500, Germany). Immunopositive cells and staining intensities
were quantified by measuring the pixel area of the positive-stained
tissue using Image-Pro Plus 6 software. At least three random
fields were evaluated for each section and the averages were
compared.
Statistical analysis
Data were presented as means ± SD. The Statistical
Package for the Social Sciences (SPSS) version 19.0 (Chicago, IL,
USA) was used for statistical analysis. Statistical significance
between the groups was determined using one-way ANOVA, and the
Bonferroni method was used to compare multiple means. Differences
were considered significant at P<0.05.
Results
Construction of shVCAN stable-transfected
B16F1 and LLC cell lines
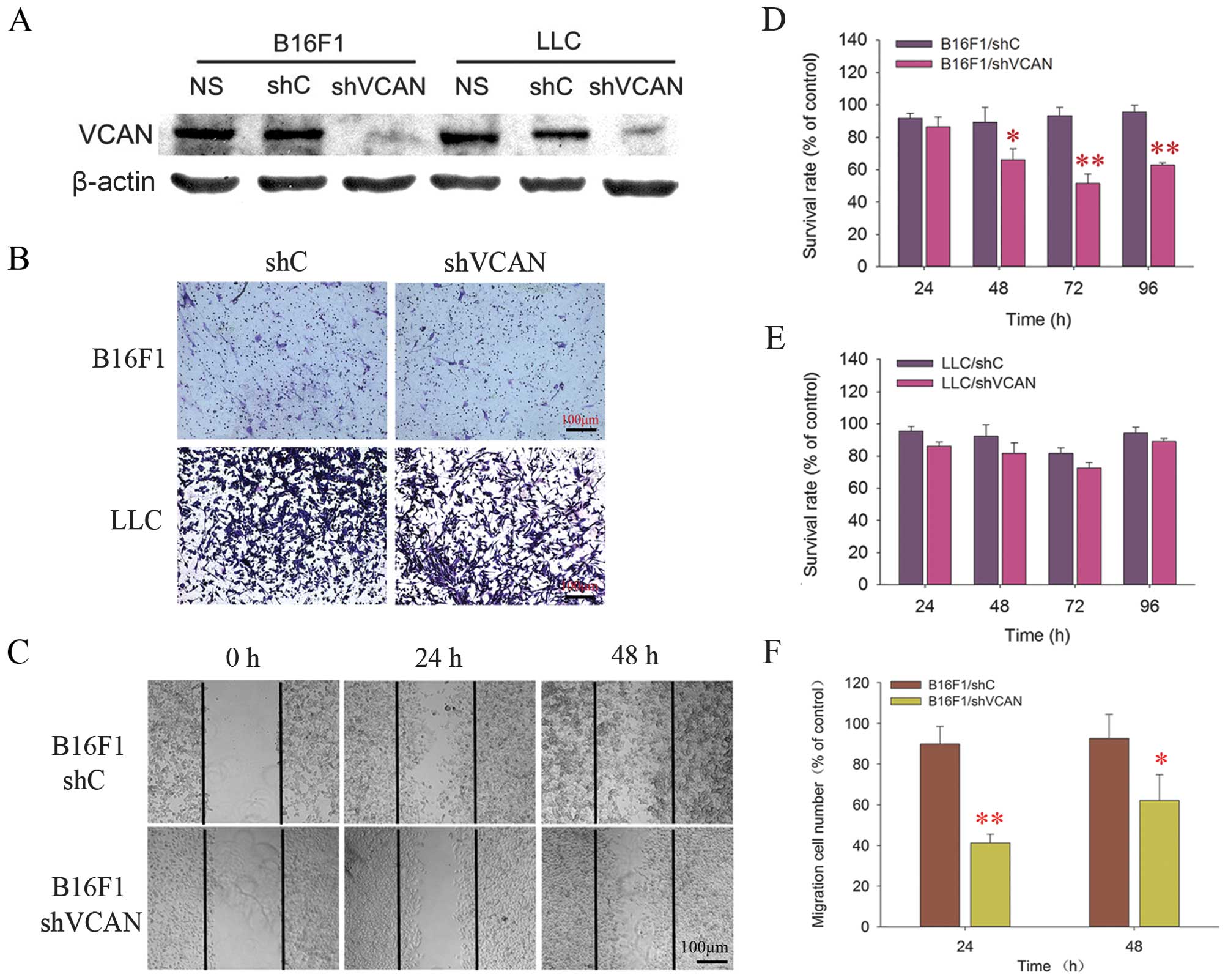
We first performed versican knockdown in B16F1 and
LLC cells in vitro through the construction of shVCAN
stable-transfected cell lines as described in ‘Materials and
methods’. The overall transfection rates of the stable transfected
cell lines were estimated to be >70%, as confirmed by flow
cytometric analysis of GFP (data not shown). Successful silencing
of versican expression was confirmed by western blot analysis,
which revealed that versican expression was markedly downregulated
in shVCAN stable-transfected cell lines (Fig. 1A).
Effects of silencing of versican on the
bioactivity of B16F1 and LLC cells in vitro
To assess the potential effects of versican
silencing on the bioactivity of shVCAN stable-transfected cell
lines. CCK-8 analysis, wound-healing and Transwell invasion assays
were performed. The results showed that versican silencing had an
inhibitory effect on the proliferative and migratory ability of
B16F1 cells, but did not affect the invasion properties of B16F1
cells. However, the versican silencing had no effect on LLC cells
in terms of the aforementioned bioactivity (Fig. 1B–F).
Antitumor efficacy of versican silencing
and endostatin in vivo
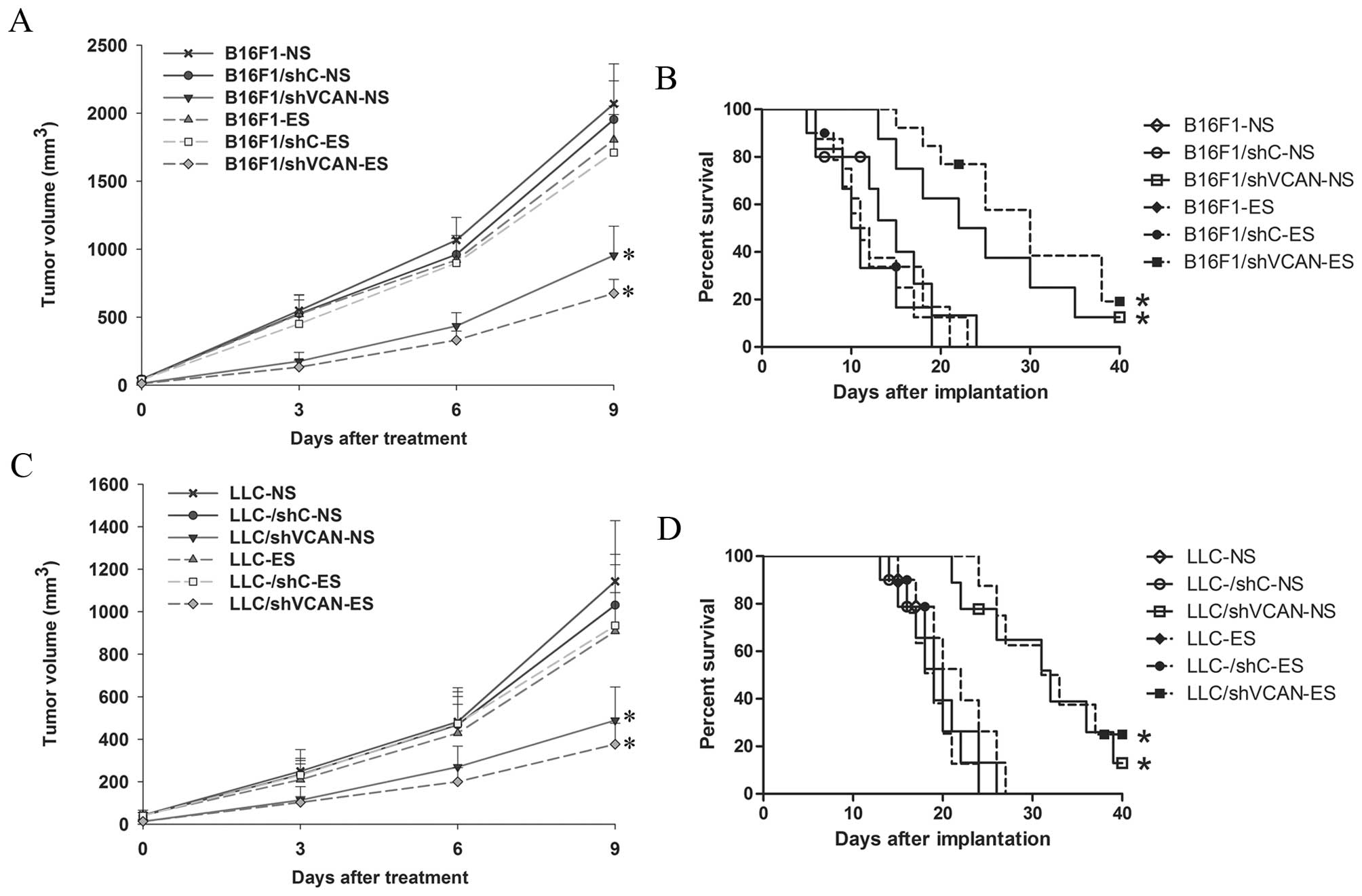
The established B16F1 and LLC tumor models were used
to investigate the effect of combining versican silencing with
endostatin on the tumor burden of mice. In the first experiment,
the mice were observed for primary tumor growth (Fig. 2A and C). The monotherapy with
endostatin showed modest inhibition of tumor growth compared with
the NS-treated groups. The shVCAN stable-transfected groups showed
significant inhibition of tumor growth compared with the NS-treated
and shC stable-transfected control groups (more effectively in
shVCAN-ES groups). The second experiment was conducted to examine
the life-prolonging effect of the treatments (Fig. 2B and D). All the groups of mice that
received NS-treated, shC stable-transfected or endostatin treatment
alone died of tumor burden within 24 days (LLC, 27 days) of B16F1
implantation. The shVCAN-transfected groups showed a significantly
prolonged survival time. These results indicated that the antitumor
efficacy of endostatin was modest. However, versican silencing had
a significant antitumor effect that was more effective when
combined with endostatin.
Versican silencing reduces the
accumulation of MDSCs and TAMs elicited by endostatin
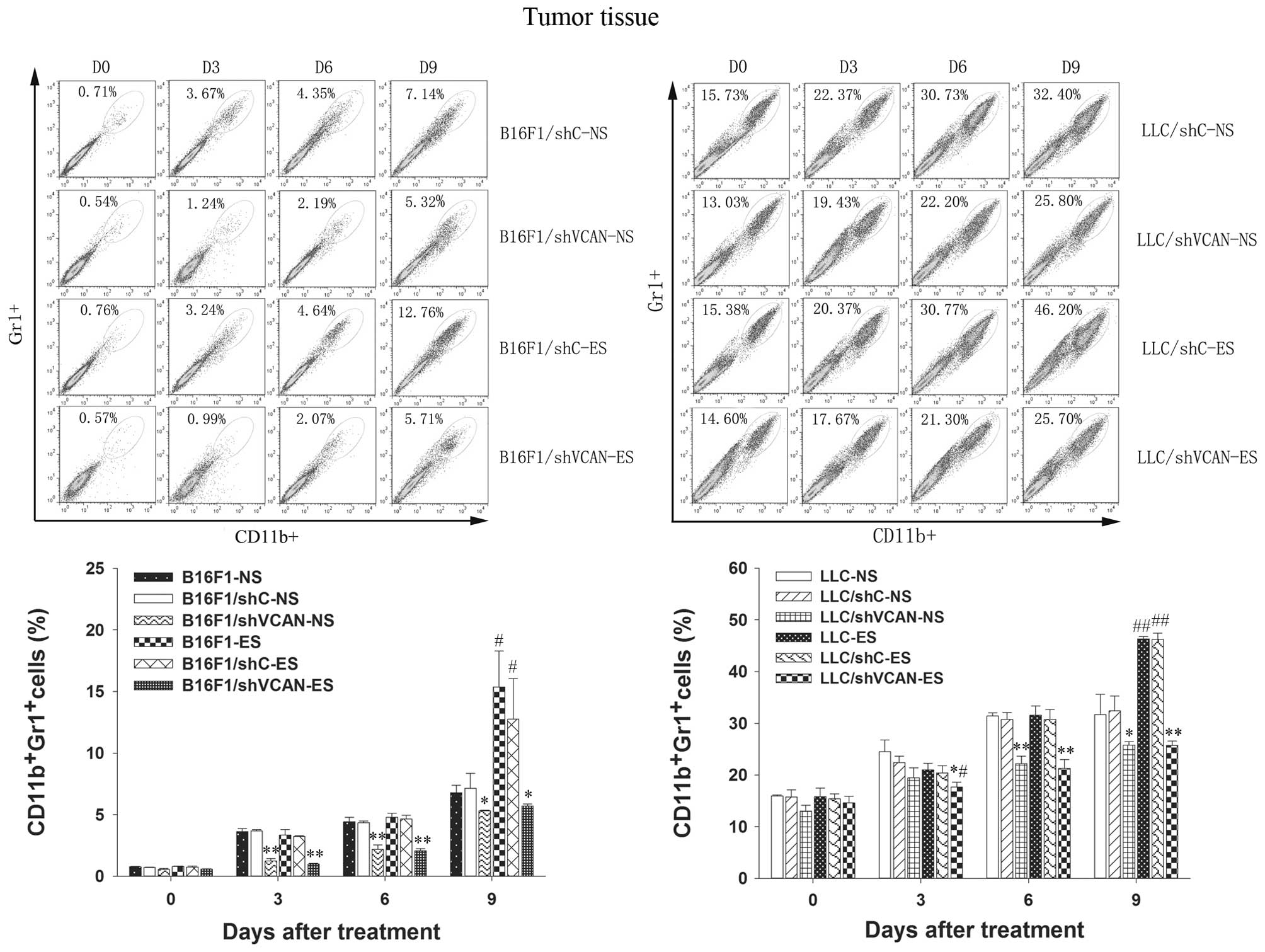
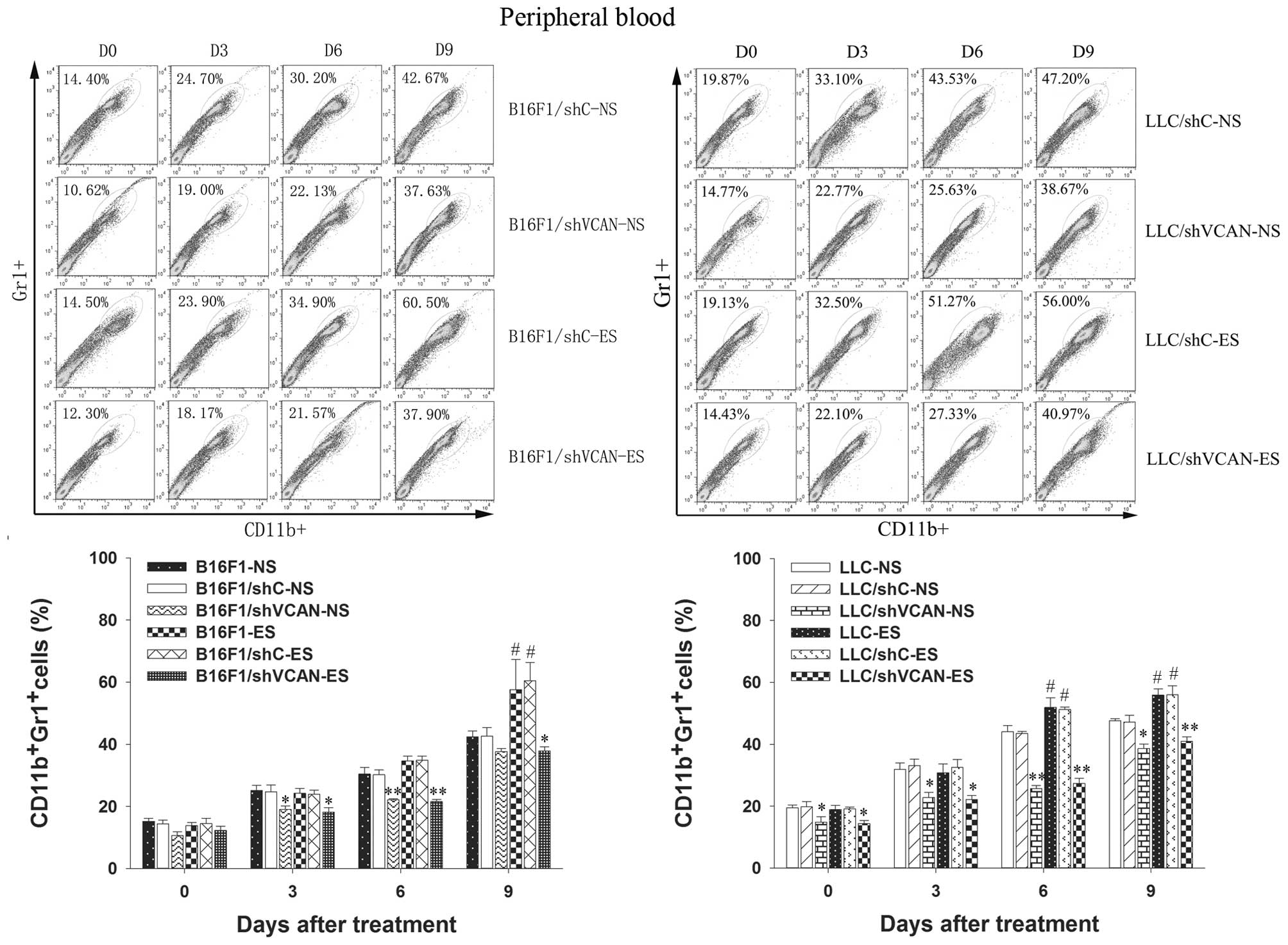
To test the hypothesis that versican silencing in
the tumor microenvironment reduces the tumor refractoriness to
endostatin by reducing the recruitment of tumor-associated
inflammatory cells, we examined the presence of MDSCs in the tumor
tissue and peripheral blood, and TAMs in the tumor tissue, by flow
cytometry. Figs. 3 and 4 show that MDSCs in the tumor tissue and
peripheral blood increased gradually with the duration of
endostatin treatment (on day 9 after treatment, P<0.05).
However, a clear reduction in MDSCs was found in all shVCAN
stable-transfected groups, as compared with shC stable-transfected
and untransfected models (on day 3–9 after treatment, P<0.05).
Notably, on day 3 after treatment, a reduction in MDSCs in the
tumor tissue was observed in the LLC/shVCAN-ES group, as compared
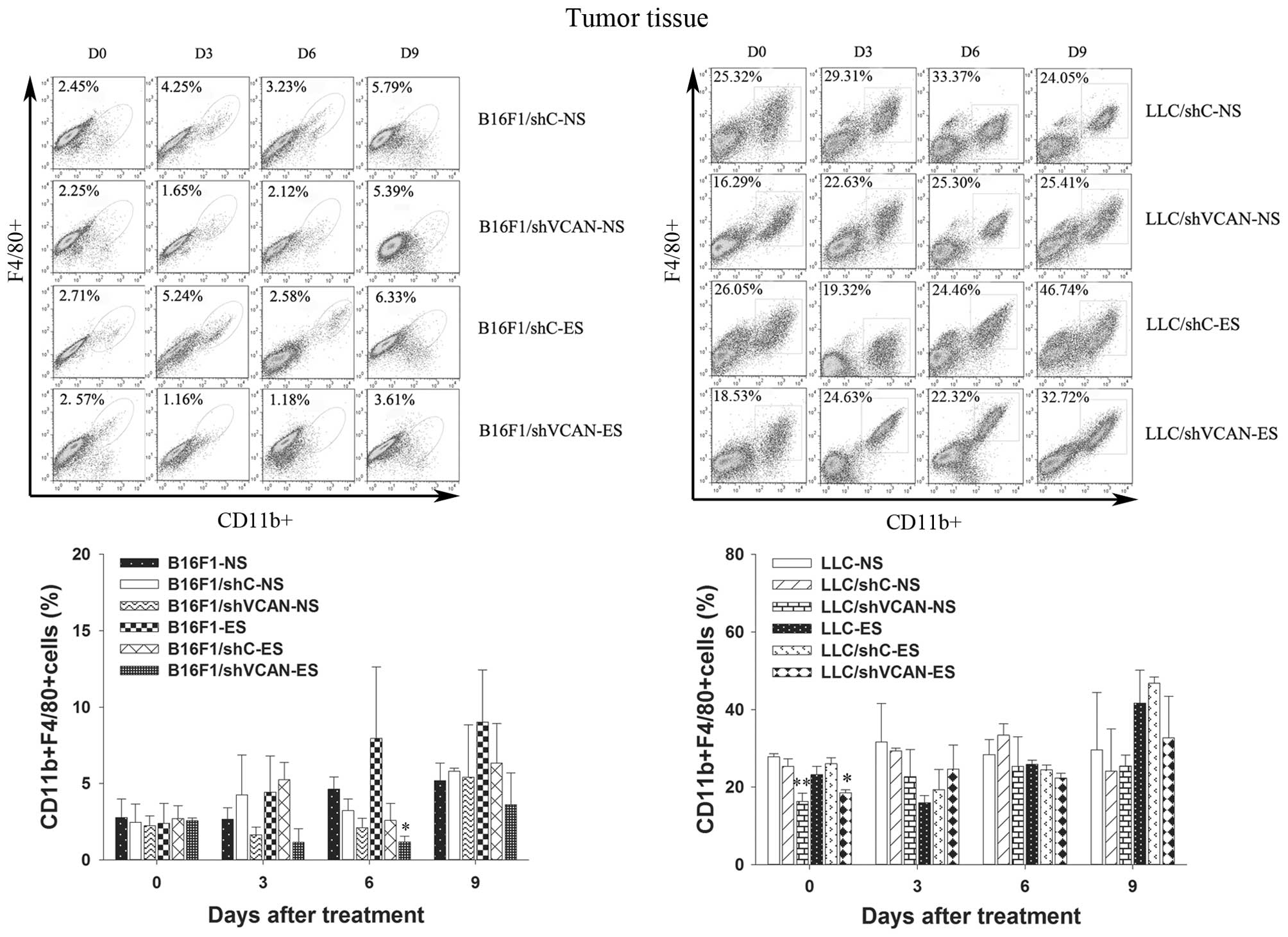
with the LLC/shVCAN-NS group. As shown in Fig. 5, a modest reduction in TAMs in the
tumor tissue of B16F1/shVCAN-ES group was found, as compared with
the B16F1-ES group (on day 6 after treatment, P<0.05). However,
a reduction in TAMs in the tumor tissue of LLC tumor models was
found in all shVCAN stable-transfected groups only on day 0 after
treatment, as compared with the shC stable-transfected and
untransfected groups (P<0.05). Taken together, these results
indicated that MDSCs were largely recruited into the peripheral
blood and the tumor microenvironment of tumor models after
endostatin treatment; however, versican silencing reduced the MDSCs
accumulation. The recruitment of TAMs in tumor tissue after
endostatin treatment was found only on day 9 after treatment.
Versican silencing reduced the number of TAMs in the tumor tissue
on day 6 after treatment in B16F1 tumor models (and on day 0 in LLC
tumor models).
Versican silencing reduces the
accumulation of tumor-associated inflammatory cytokines elicited by
endostatin
To determine the inflammatory changes in the tumor
microenvironment elicited by endostatin, Luminex xMAP assays were
conducted to examine the level of inflammatory cytokines, including
G-CSF, TNF-α, IL-6, VEGF and IL-10. Fig. 6 shows the levels of these cytokines
in the tumor tissue. On day 9 after treatment, a clear increase in
G-CSF, TNF-α, IL-6, VEGF and IL-10 was found in the endostatin
monotherapy groups, as compared with the NS-treated models.
However, a clear reduction in these cytokines was observed in all
the shVCAN-ES groups, as compared with the endostatin monotherapy
groups. Fig. 7 shows the levels of
the aforementioned cytokines in the serum. On day 9 after
treatment, a clear increase in G-CSF and VEGF was found in the
endostatin monotherapy groups, as compared with the NS-treated
groups. However, a clear reduction was observed in all the
shVCAN-ES groups, as compared with the endostatin monotherapy
groups (the same change in TNF-α was found on day 6 after
treatment, in IL-6 the change was identified on day 6–9 after
treatment, and in IL-10 on day 3 and 9 after treatment).
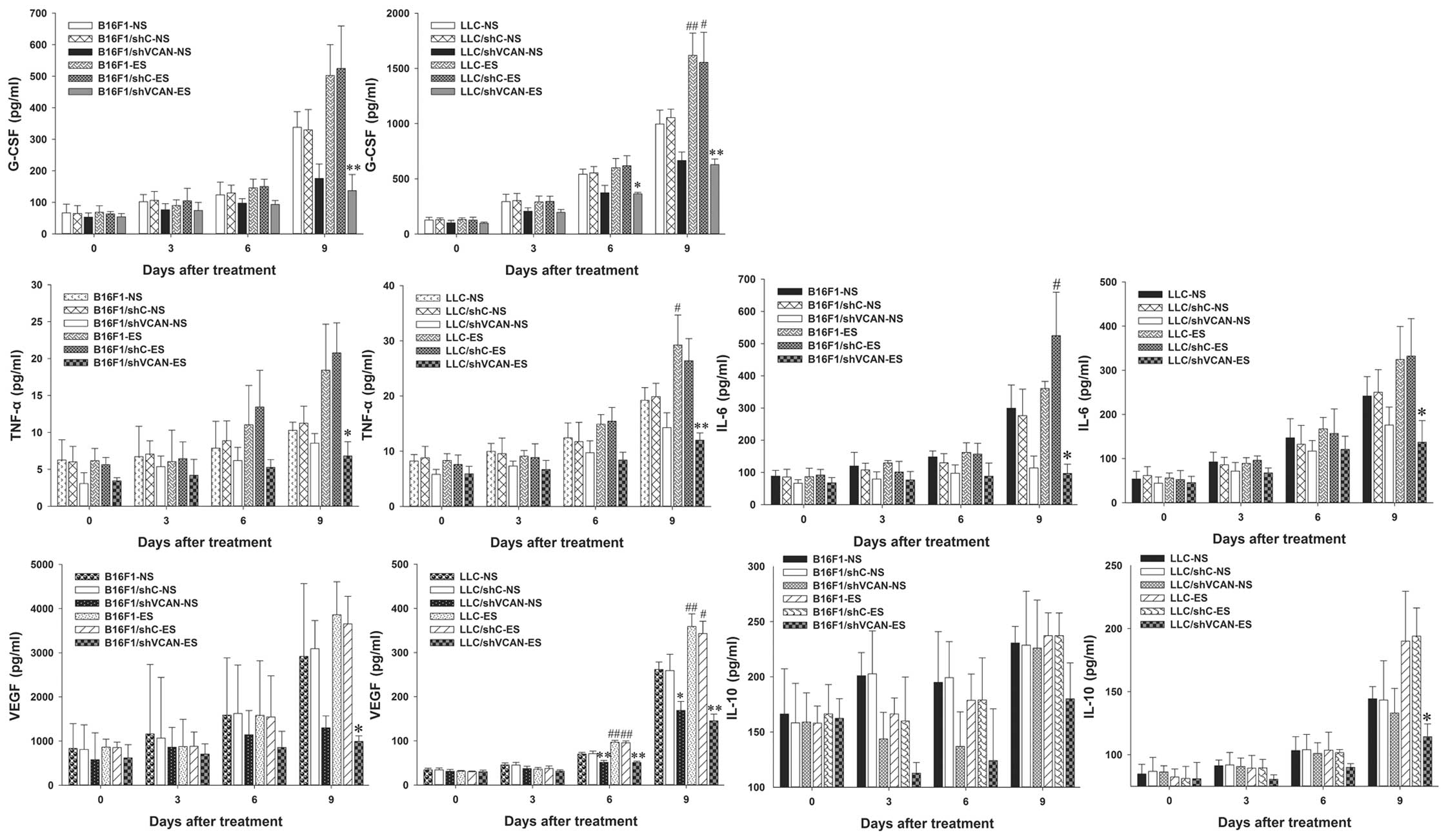 | Figure 6Versican silencing reduces the
accumulation of tumor-associated inflammatory cytokines elicited by
endostatin in tumor tissue. On days 0, 3, 6 and 9 after the
initiation of endostatin treatment, Luminex xMAP assays were
conducted to determine the level of G-CSF, TNF-α, IL-6, VEGF and
IL-10 in tumor tissue of B16F1 and LLC tumor-bearing mice. n=3.
shVCAN vs. shC, *P<0.05, **P<0.01. ES vs. NS,
#P<0.05, ##P<0.01. G-CSF, granulocyte
colony-stimulating factor; TNF-α, tumor necrosis factor-α; VEGF,
vascular endothelial growth factor; LLC, Lewis lung carcinoma;
shVCAN, short-hairpin RNA targeting versican. |
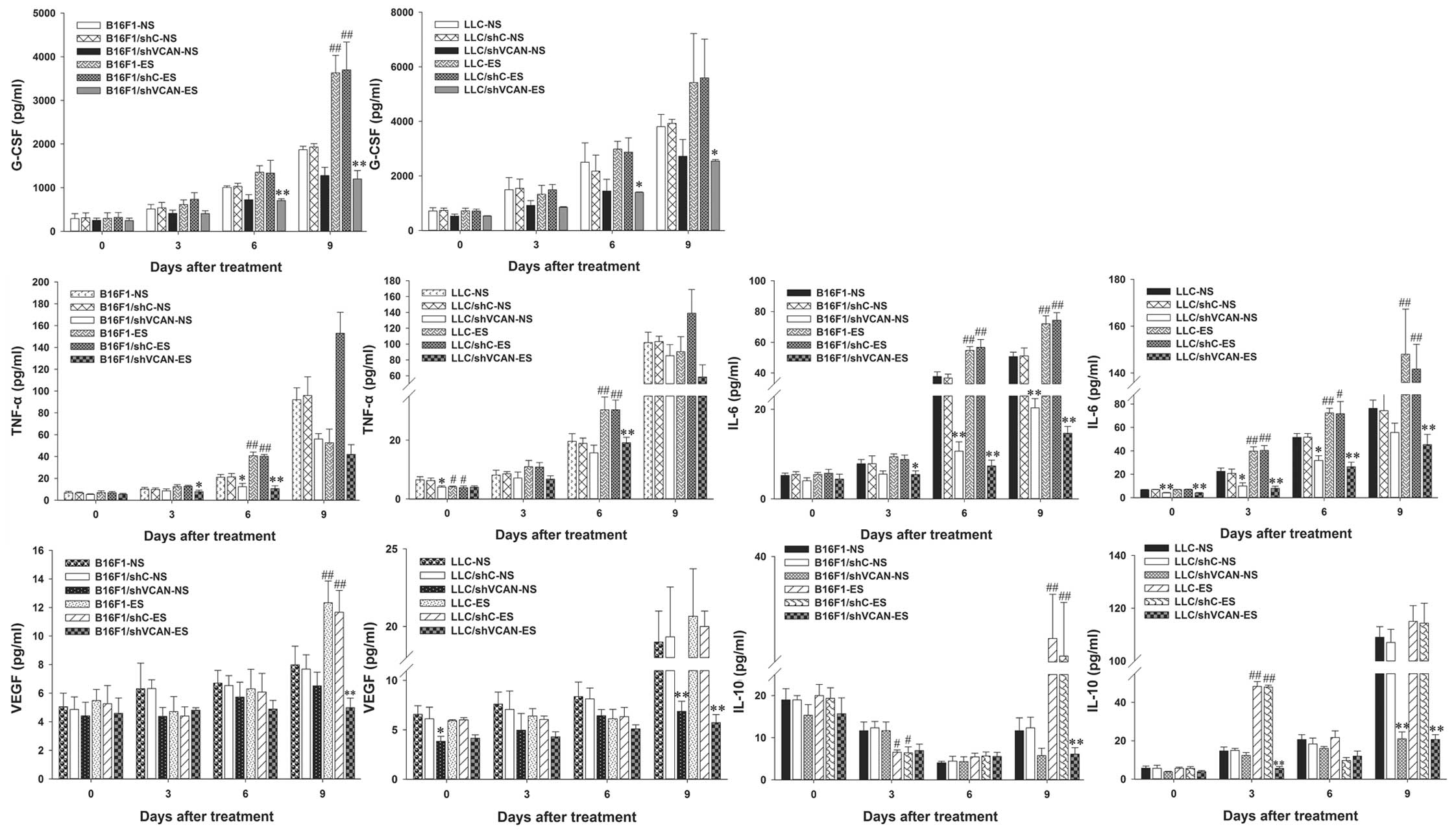 | Figure 7Versican silencing reduces the
accumulation of tumor-associated inflammatory cytokines elicited by
endostatin in serum. On days 0, 3, 6 and 9 after the initiation of
endostatin treatment, Luminex xMAP assays were conducted to
determine the level of G-CSF, TNF-α, IL-6, VEGF and IL-10 in the
serum of B16F1 and LLC tumor-bearing mice. n=3. shVCAN vs. shC,
*P<0.05, **P<0.01. ES vs. NS, #P<0.05,
##P<0.01. G-CSF, granulocyte colony-stimulating
factor; TNF-α, tumor necrosis factor-α; VEGF, vascular endothelial
growth factor; LLC, Lewis lung carcinoma; shVCAN, short-hairpin RNA
targeting versican. |
The results indicated that endostatin elicited an
increase in G-CSF, TNF-α and IL-6 in serum and tumor tissue (more
significant on day 9 after treatment), and to some extent versican
silencing reversed this tendency. The level of VEGF decreased at an
early stage of endostatin treatment (on day 3–6 after treatment),
but reverted quickly on day 9 after treatment. The level of IL-10
in the serum decreased slightly at an early stage of endostatin
treatment (on day 3–6 after treatment); however, it increased
significantly on day 9 after treatment. Versican silencing
alleviated the increase in IL-10 elicited by endostatin.
Versican silencing alleviated the
endostatin-elicited tumor inflammatory microenvironment by leading
to downregulation of NF-κB and HIF-1α
Versican and NF-κB are generally acknowledged to be
proinflammatory factors (8,17). In order to explore the related
mechanisms of how versican silencing alleviated the
endostatin-elicited tumor inflammatory microenvironment, western
blot analysis and immunohistochemical assays were conducted to
determine the expression of versican, NF-κB and HIF-1α, and
immunohistochemical assays were used to evaluate the expression of
CD31 in tumor tissue.
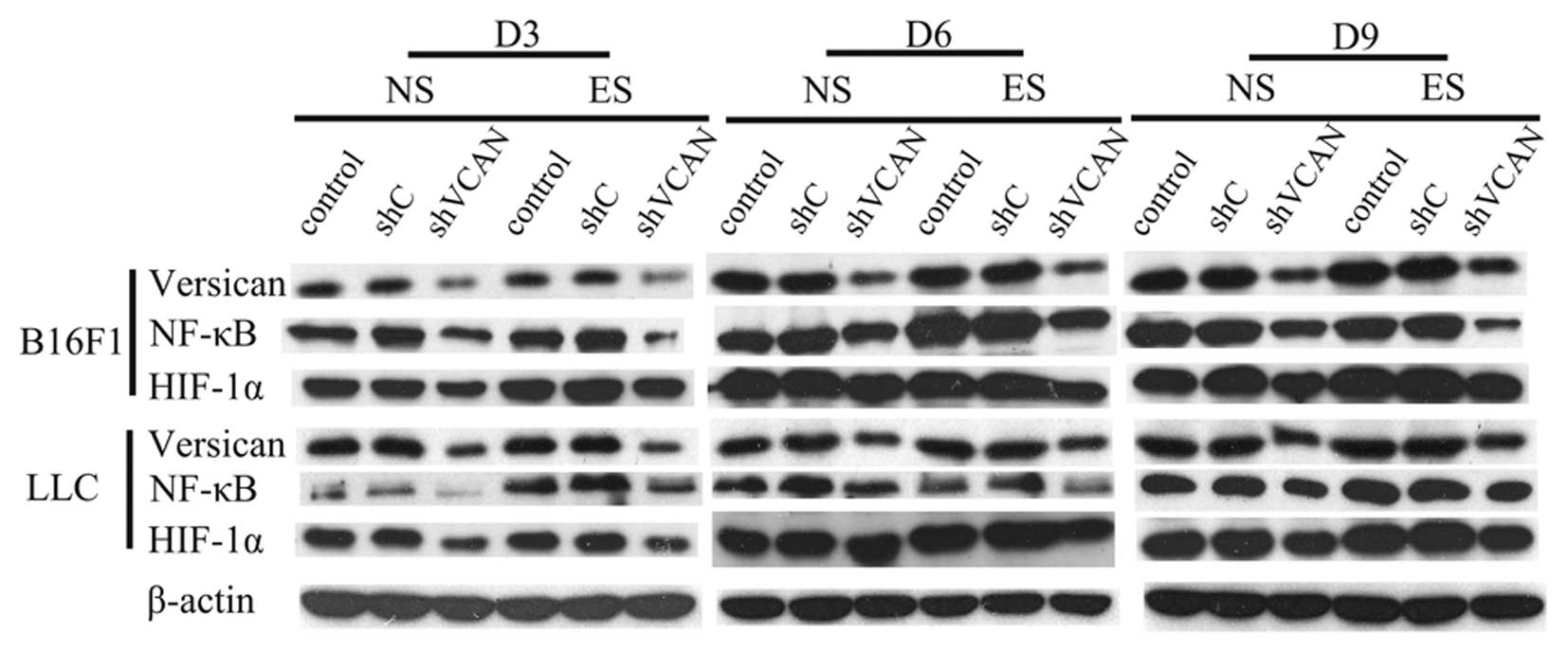
The results showed that the expression of versican
was attenuated in the shVCAN-transfected groups, as compared with
the shC stable transfected and untransfected groups (Figs. 8A and 10), and that endostatin has the ability
to increase the expression level of versican (Fig. 8A). With the duration of endostatin
treatment, NF-κB increased gradually, but was reduced with versican
silencing (Fig. 8B and 10). Notably, on day 9 after treatment, a
lower expression level of NF-κB was detected in the shVCAN+ES group
compared with the shVCAN+NS group in the B16F1 tumors (Fig. 8B). With the duration of endostatin
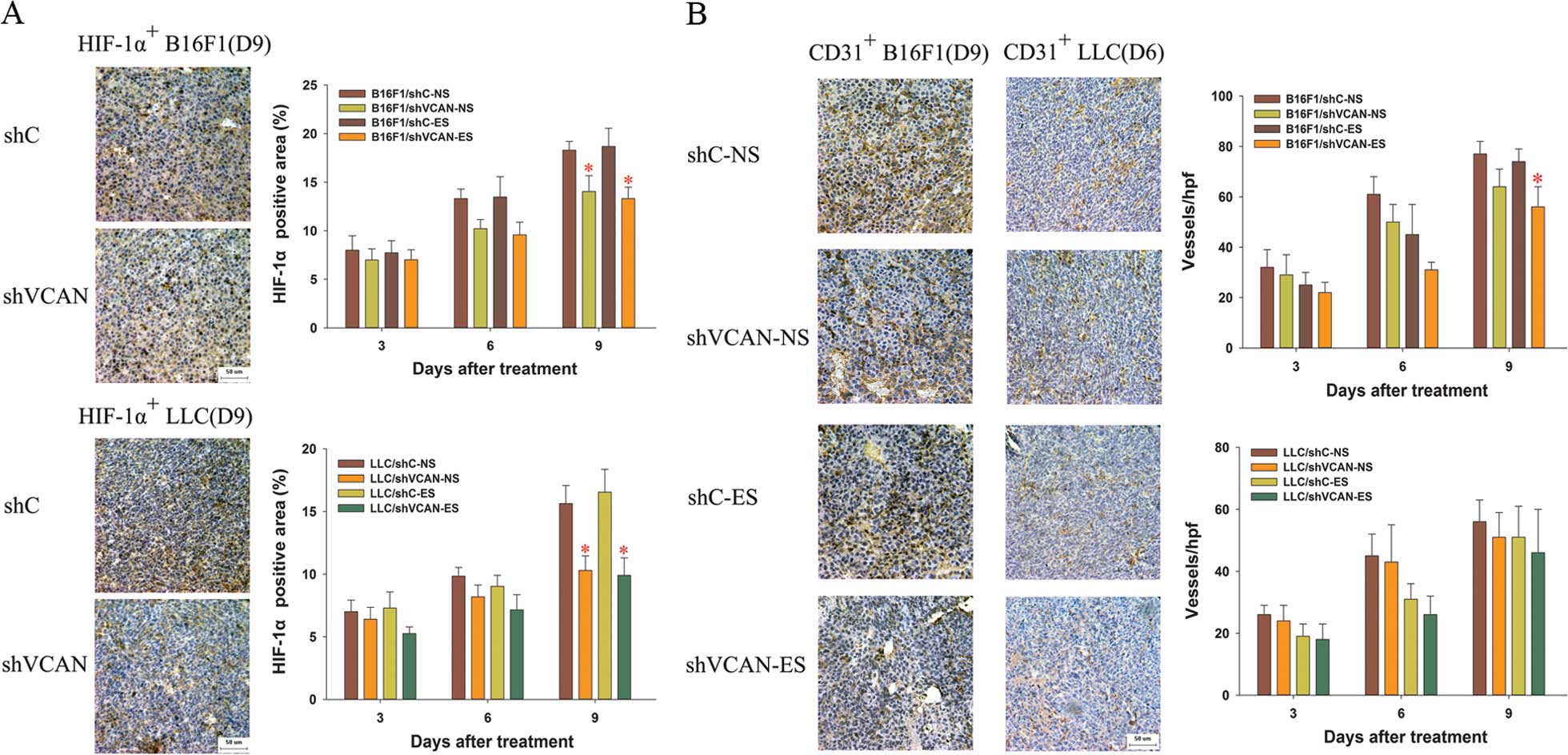
treatment, HIF-1α increased gradually, but was reduced with
versican silencing (Figs. 9A and
10). At an early stage of
endostatin treatment (on day 3–6 after treatment), microvessel
density was lower in the endostatin-treated group than in the
NS-treated group (P>0.05) (Fig.
9B). Compared with the endostatin monotherapy groups, the
microvessel density decreased in the shVCAN+ES groups (in B16F1
tumor models, P<0.05) (Fig.
9B).
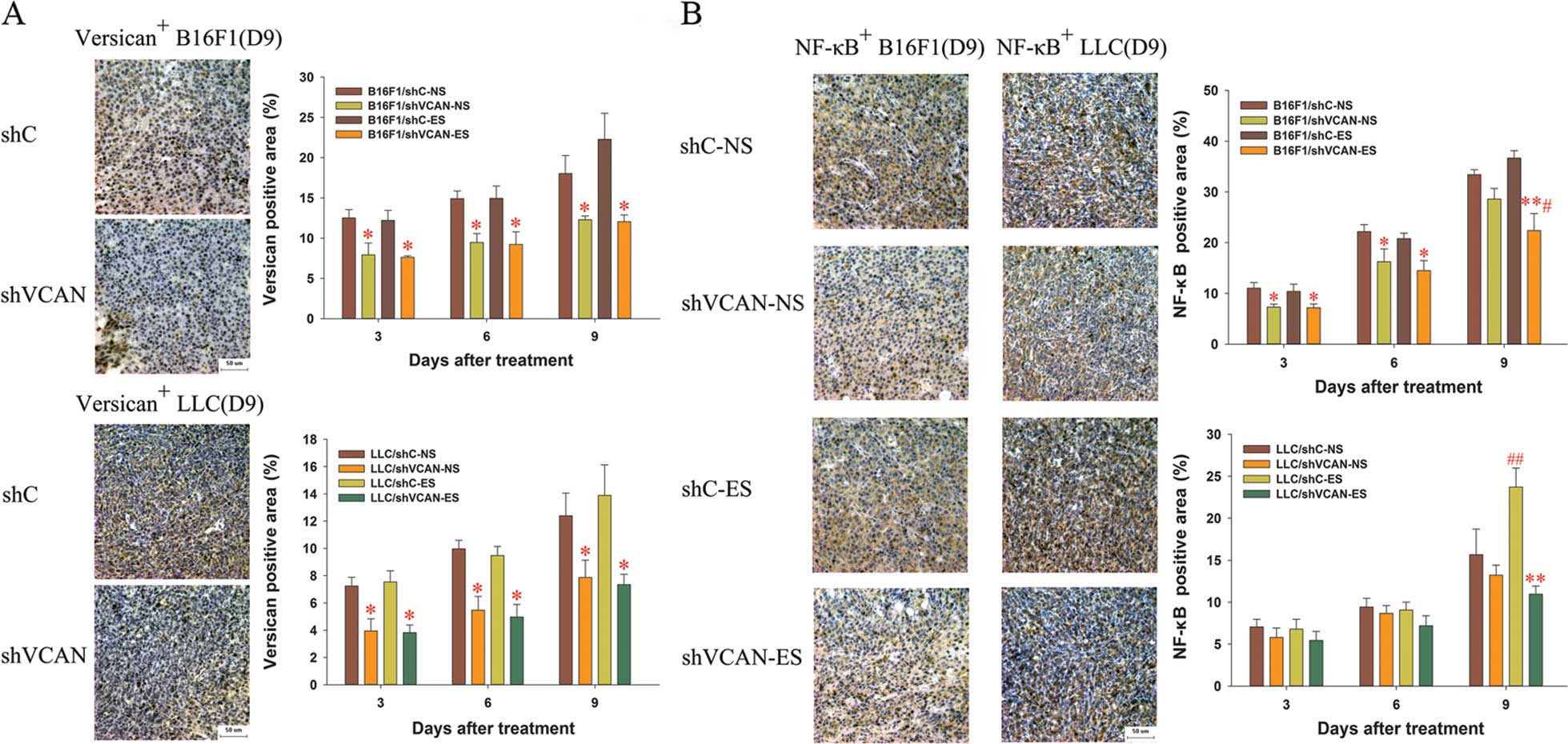 | Figure 8Immunohistochemical assays of the
expression of versican and NF-κB in B16F1 and LLC tumor tissue. On
days 3, 6 and 9 after the initiation of endostatin treatment, the
expression of (A) versican and (B) NF-κB was detected by
immunohistochemical assays. Scale bar, 50 μm. Original
magnification, ×400 for all panels with a microscope. At least
three random fields were evaluated for each section. shVCAN vs.
shC, *P<0.05, **P<0.01. ES vs. NS,
#P<0.05, ##P<0.01. LLC, Lewis lung
carcinoma; shVCAN, short-hairpin RNA targeting versican. |
Discussion
In the present study, B16F1 and LLC cell lines with
stably silenced expression of versican were initially established,
and the effects of versican silencing on the bioactivity of shVCAN
stable-transfected cell lines were assessed to eliminate its
influence on exploring the role of versican in the development of
th einflammatory tumor microenvironment in vivo. We found
that versican silencing exerted an inhibitory effect on the
proliferative and migratory ability of B16F1 cells, but did not
affect LLC cells. Consistent with our findings, a previous study
showed that versican V0/V1 silencing caused a reduction in the
proliferation and migratory ability in human melanoma SK-mel-131
cell lines (18). Notably, the
knockdown of versican expression in A549 lung cancer cells by RNA
interference significantly inhibited tumor growth in vivo
but not in vitro (19).
Although versican has different effects on biological behavior in
different cell lines its mechanisms remain to be elucidated.
Several observations have been made in the present
study concerning combination therapy based on versican silencing
and endostatin anti-angiogenesis. Endostatin showed modest
inhibition of tumor growth, and had no effect on the survival time
of tumor-bearing mice. Versican silencing alone effectively
suppressed orthotopic tumor growth and significantly prolonged
survival time in the B16F1 and LLC tumor models. This effect was
enhanced with combined versican silencing and endostatin treatment.
The major cause of the tumor refractoriness to the antiangiogenic
therapy may be associated with the inflammatory and
immunosuppressive changes of the tumor microenvironment elicited by
endostatin administration. By reducing the recruitment of
tumor-associated inflammatory and immunosuppressive cells, and
achieving remission of tumor-inflammatory cytokines, versican
silencing of cancer cells in the tumor microenvironment alleviated
the tumor refractoriness to antiangiogenic therapy with endostain.
These suggestions were supported by our own results.
Myeloid-derived suppressor cells (MDSCs) and tumor-associated
macrophages (TAMs) in the tumor tissue and peripheral blood of
B16F1 and LLC tumor-bearing mice were examined by flow cytometry,
and the inflammatory cytokines (including G-CSF, TNF-α, IL-6, VEGF
and IL-10) were assessed by Luminex xMAP assays. A clear increase
in MDSCs in the blood and tumor tissue was identified in all the
endostatin-treated groups of tumor-bearing models, as compared with
NS-treated ones (more significant on day 9 after treatment).
However, the versican silencing of cancer cells reversed this
tendency. On day 9 after treatment, an increase in TAMs was found
in the tumor tissue in all the endostatin-treated groups of
tumor-bearing models (P>0.05). Versican silencing reduced the
number of TAMs in the tumor tissue on day 6 after treatment in
B16F1 tumors, and on day 0 after treatment in LLC tumors. In
addition, endostatin caused an increase in G-CSF, TNF-α and IL-6 in
serum and tumor tissue (more significant on day 9 after treatment),
and to some extent, versican silencing reversed this tendency.
There was a reduced level of VEGF and IL-10 in serum and tumor
tissue at the early stage of endostatin treatment (on days 3–6
after treatment). However, the level of VEGF and IL-10 increased
quickly on day 9 after the treatment. Versican silencing
attenutated the increase in IL-10 elicited by endostatin.
CD11b+Gr1+ MDSCs, a class of
immature myeloid cells, also described as a subset of
tumor-associated inflammatory cells, are recruited by inflammation
cytokines secreted by tumor and stromal cells (20). It is a commonly held view that MDSCs
act as ‘bridges’ linking inflammation and cancer (20). MDSCs are induced by tumor-secreted
and host-secreted factors, many of which are proinflammatory
molecules, such as TGF-β, TNF-α, IL-6, IL-10, G-CSF, GM-CSF, CCL2
and CXCL12 (20,21). The induction of MDSCs by
proinflammatory mediators led to the hypothesis that inflammation
promotes the accumulation of MDSCs which in turn, downregulate
immune surveillance and antitumor immunity, thereby facilitating
tumor growth (20). In addition,
MDSCs play an important role in tumor growth through the induction
of angiogenesis (21). Previous
findings have suggested that MDSCs contribute to refractoriness to
antiangiogenic therapy, and that the recruitment of MDSCs
stimulated by overexpressed proinflammatory factors after anti-VEGF
treatment is a major cause of the resistance of tumors to
antiangiogenic therapies (22,23).
In the present study, we found that antiangiogenic therapy with
endostatin elicited inflammatory and immunosuppressive changes in
the tumor microenvironment, including the accumulation of MDSCs and
TAMs, and an increase in G-CSF, TNF-α, IL-6, VEGF and IL-10. The
versican silencing alleviated the inflammatory and
immunosuppressive changes in the tumor microenvironment, and
improved the antitumor efficacy of endostatin. Our findings may
contribute in elucidating the inflammation promoting the
accumulation of MDSCs, which in turn, facilitate tumor
progression.
TAMs comprise a heterogeneous cell population
originating from mononuclear phagocytic lineage (24,25).
TAMs are the second well-described population of myeloid cells that
have been shown to exert a negative effect on antitumor immune
responses. The relationship between TAMs and MDSCs has not been
completely defined, although it has been suggested that TAMs may in
part be derived from or be associated with MDSCs (26). Diversity and plasticity are
characteristics of TAMs, which can be polarized to different
phenotypic subgroups under different microenvironmental conditions
(27). The classically activated
(M1-like) TAMs reduce angiogenesis, increase inflammation and
express antitumor activity. The mediators (IL-12, TNF-α and IL-6)
of M1 macrophage-mediated inflammation maintain a high level by M1
macrophage (28). However, the
alternatively activated (M2-like) TAMs usually promote tumor growth
and stimulate angiogenesis (29,30).
M2 macrophages exhibit immune suppression through the production of
immunosuppressive cytokines (e.g., IL-10 and TGF-β) and the
recruitment of regulatory T cells through the secretion of CCL22
(28). In the tumor
microenvironment, many cytokines skew the polarization of TAMs from
an M1-like phenotype to an M2-like phenotype (e.g., IL-4, IL-6,
IL-13, IL-10 and TGF-β) (31). In
general, most TAMs in the tumor microenvironment obtain M2-like
properties and resemble ‘tolerant’ macrophages, and an increased
number of TAMs correlate with vessel density and poor prognosis
(30). In the present study, we
found that TAMs in the tumor microenvironment increased modestly on
day 9 after endostatin treatment (P>0.05). Versican silencing
reduced the number of TAMs in the tumor tissue on day 6 after
treatment in B16F1 tumors, and on day 0 after treatment in LLC
tumors. However, versican silencing had no effect on TAMs on day 9
after endostatin treatment, possibly caused by the polarization of
TAMs from an M2-like phenotype to an M1-like phenotype. We also
found that IL-6 and IL-10 in the tumor microenvironment and serum,
which skewed the polarization of TAMs from an M1-like to an M2-like
phenotype, increased with endostatin treatment but were reduced by
versican silencing. Moreover, the level of TNF-α and VEGF, which
can be secreted by M2 macrophages, were also down-regulated by
versican silencing. These findings indicate that versican silencing
can skew the polarization of TAMs from an M2-like phenotype to an
M1-like phenotype through the regulation of the cytokines described
above.
Among the many different mediators of the
recruitment of inflammatory cells into the tumor microenvironment,
versican and NF-κB are generally acknowledged to be proinflammatory
factors (8,17). Versican strongly enhances tumor
progression by activating multiple types of inflammatory cells by
combining with the cell surface of TLR-2, and eliciting the
production of proinflammatory cytokines (8). The inflammatory cytokines (e.g.,
TNF-α, IL-1β, IL-6 and IL-8) are involved in the TLR-2-mediated
NF-κB pathway, which regulates the transcription genes associated
with the immune and inflammatory responses (32). Recently, investigators reported that
hemiterpene rotundarpene (4-caffeoyl-3-methyl-but-2-ene-1,4-diol,
an extract from the bark of the Ilex rotunda Thunb.)
attenuated the production of inflammatory mediators by suppressing
activation of the TLR-2-mediated NF-κB pathway (32). Although versican and NF-κB are
proinflammatory factors, the direct relationship between them
remains elusive. In the present study, as previously described, we
found that versican silencing alleviated the inflammatory and
immunosuppressive changes elicited by endostatin, and disrupted the
positive feedback for the extension of inflammation, and thus
improved the antitumor efficacy of endostatin. In order to explore
the associated mechanisms, we conducted immunohisto chemical and
western blot analyses to determine the expression of versican,
NF-κB and HIF-1α. We found that the expression of NF-κB and HIF-1α
was greater in the endostatin monotherapy group compared with the
NS-treated ones, but decreased in the shVCAN-transfected groups.
This finding suggested that versican silencing resulted in the
downregulation of NF-κB and HIF-1α. Versican affected the
expression of NF-κB via many inflammatory cytokines, including
G-CSF, TNF-α and IL-6, which were involved in the TLR-2-mediated
NF-κB pathway. Using Luminex xMAP assays, we found that there was a
clear increase in G-CSF, TNF-α and IL-6 in endostatin monotherapy
groups in the B16F1 and LLC tumor models on day 9 after endostatin
treatment, as compared with NS-treated ones. However, a clear
reduction in G-CSF, TNF-α and IL-6 was observed in the
shVCAN-transfected groups, as compared with the shC-transfected and
untransfected ones. These findings indicate that versican silencing
reduced the accumulation of G-CSF, TNF-α and IL-6 elicited by
endostatin in serum and tumor tissue. Based on the results, we
hypothesize that the silencing of versican may suppress activation
of the TLR-2-mediated NF-κB pathway by reducing the production of
inflammatory cytokines (e.g., TNF-α and IL-6) involved in this
pathway. However, additional studies are required to clarify the
exact mechanisms of the interaction of versican and NF-κB.
In summary, our findings indicate that the leading
cause of tumor refractoriness to antiangiogenic therapy is
associated with inflammatory and immunosuppressive changes in the
tumor microenvironment elicited by endostatin administration.
Versican silencing improved the antitumor efficacy of endostatin by
alleviating its induced alterations in the tumor microenvironment.
Versican silencing in the tumor micro-environment may offer a
promising approach to reverse the tumor refractoriness to
antiangiogenic therapies.
Acknowledgments
The present study funding was supported by the
National Natural Science Foundation of China (nos. 81071864 and
81372506).
References
|
1
|
Verheul HM, Hammers H, van Erp K, Wei Y,
Sanni T, Salumbides B, Qian DZ, Yancopoulos GD and Pili R: Vascular
endothelial growth factor trap blocks tumor growth, metastasis
formation, and vascular leakage in an orthotopic murine renal cell
cancer model. Clin Cancer Res. 13:4201–4208. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Paez-Ribes M, Allen E, Hudock J, Takeda T,
Okuyama H, Vinals F, Inoue M, Bergers G, Hanahan D and Casanovas O:
Antiangiogenic therapy elicits malignant progression of tumors to
increased local invasion and distant metastasis. Cancer Cell.
15:220–231. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zuniga RM, Torcuator R, Jain R, Anderson
J, Doyle T, Ellika S, Schultz L and Mikkelsen T: Efficacy, safety
and patterns of response and recurrence in patients with recurrent
highgrade gliomas treated with bevacizumab plus irinotecan. J
Neurooncol. 91:329–336. 2009. View Article : Google Scholar
|
|
4
|
Shojaei F, Wu X, Malik AK, Zhong C,
Baldwin ME, Schanz S, Fuh G, Gerber HP and Ferrara N: Tumor
refractoriness to anti-VEGF treatment is mediated by
CD11b+Gr1+ myeloid cells. Nat Biotechnol.
25:911–920. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Soeda S, Nakamura N, Ozeki T, Nishiyama H,
Hojo H, Yamada H, Abe M and Sato A: Tumor-associated macrophages
correlate with vascular space invasion and myometrial invasion in
endometrial carcinoma. Gynecol Oncol. 109:122–128. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kischel P, Waltregny D, Dumont B, Turtoi
A, Greffe Y, Kirsch S, De Pauw E and Castronovo V: Versican
overexpression in human breast cancer lesions: Known and new
isoforms for stromal tumor targeting. Int J Cancer. 126:640–650.
2010. View Article : Google Scholar
|
|
7
|
Wang W, Xu GL, Jia WD, Ma JL, Li JS, Ge
YS, Ren WH, Yu JH and Liu WB: Ligation of TLR2 by versican: A link
between inflammation and metastasis. Arch Med Res. 40:321–323.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kim S, Takahashi H, Lin WW, Descargues P,
Grivennikov S, Kim Y, Luo JL and Karin M: Carcinoma-produced
factors activate myeloid cells through TLR2 to stimulate
metastasis. Nature. 457:102–106. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gao D, Joshi N, Choi H, Ryu S, Hahn M,
Catena R, Sadik H, Argani P, Wagner P, Vahdat LT, et al: Myeloid
progenitor cells in the premetastatic lung promote metastases by
inducing mesenchymal to epithelial transition. Cancer Res.
72:1384–1394. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ling Y, Yang Y, Lu N, You QD, Wang S, Gao
Y, Chen Y and Guo QL: Endostar, a novel recombinant human
endostatin, exerts antiangiogenic effect via blocking VEGF-induced
tyrosine phosphorylation of KDR/Flk-1 of endothelial cells. Biochem
Biophys Res Commun. 361:79–84. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang HL, Ning T, Li M, Lu ZJ, Yan X, Peng
Q, Lei N, Zhang H and Luo F: Effect of endostatin on preventing
postoperative progression of distant metastasis in a murine lung
cancer model. Tumori. 97:787–793. 2011.
|
|
12
|
Ning T, Yan X, Lu ZJ, Wang GP, Zhang NG,
Yang JL, Jiang SS, Wu Y, Yang L, Guan YS, et al: Gene therapy with
the angiogenesis inhibitor endostatin in an orthotopic lung cancer
murine model. Hum Gene Ther. 20:103–111. 2009. View Article : Google Scholar
|
|
13
|
Ning T, Jiang M, Peng Q, Xi Y, Lu ZJ, Peng
YL, Wang HL, Lei N, Zhang H, Lin HJ, et al: Low-dose endostatin
normalizes the structure and function of tumor vasculature and
improves the delivery and anti-tumor efficacy of cytotoxic drugs in
a lung cancer xenograft murine model. Thorac Cancer. 3:229–238.
2012. View Article : Google Scholar
|
|
14
|
Huang G and Chen L: Discrepancies between
antiangiogenic and antitumor effects of recombinant human
endostatin. Cancer Biother Radiopharm. 24:589–596. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang H, Wang Z, Peng Q, Liu YY, Zhang W,
Wu L, Wang X and Luo F: Tumor refractoriness to endostatin
anti-angiogenesis is associated with the recruitment of CD11b+Gr1+
myeloid cells and inflammatory cytokines. Tumori. 99:723–733.
2013.
|
|
16
|
Li XQ, Shang BY, Wang DC, Zhang SH, Wu SY
and Zhen YS: Endostar, a modified recombinant human endostatin,
exhibits synergistic effects with dexamethasone on angiogenesis and
hepatoma growth. Cancer Lett. 301:212–220. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ben-Neriah Y and Karin M: Inflammation
meets cancer, with NF-κB as the matchmaker. Nat Immunol.
12:715–723. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hernández D, Miquel-Serra L, Docampo MJ,
Marco-Ramell A and Bassols A: Role of versican V0/V1 and CD44 in
the regulation of human melanoma cell behavior. Int J Mol Med.
27:269–275. 2011.
|
|
19
|
Zheng PS, Wen J, Ang LC, Sheng W,
Viloria-Petit A, Wang Y, Wu Y, Kerbel RS and Yang BB: Versican/PG-M
G3 domain promotes tumor growth and angiogenesis. FASEB J.
18:754–756. 2004.PubMed/NCBI
|
|
20
|
Ostrand-Rosenberg S and Sinha P:
Myeloid-derived suppressor cells: Linking inflammation and cancer.
J Immunol. 182:4499–4506. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gabrilovich DI and Nagaraj S:
Myeloid-derived suppressor cells as regulators of the immune
system. Nat Rev Immunol. 9:162–174. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shojaei F, Wu X, Qu X, Kowanetz M, Yu L,
Tan M, Meng YG and Ferrara N: G-CSF-initiated myeloid cell
mobilization and angiogenesis mediate tumor refractoriness to
anti-VEGF therapy in mouse models. Proc Natl Acad Sci USA.
106:6742–6747. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Carbone C, Moccia T, Zhu C, Paradiso G,
Budillon A, Chiao PJ, Abbruzzese JL and Melisi D: Anti-VEGF
treatment-resistant pancreatic cancers secrete proinflammatory
factors that contribute to malignant progression by inducing an EMT
cell phenotype. Clin Cancer Res. 17:5822–5832. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gordon S and Taylor PR: Monocyte and
macrophage heterogeneity. Nat Rev Immunol. 5:953–964. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pollard JW: Trophic macrophages in
development and disease. Nat Rev Immunol. 9:259–270. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sica A and Bronte V: Altered macrophage
differentiation and immune dysfunction in tumor development. J Clin
Invest. 117:1155–1166. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Martinez FO, Helming L and Gordon S:
Alternative activation of macrophages: An immunologic functional
perspective. Annu Rev Immunol. 27:451–483. 2009. View Article : Google Scholar
|
|
28
|
Mantovani A, Bottazzi B, Colotta F,
Sozzani S and Ruco L: The origin and function of tumor-associated
macrophages. Immunol Today. 13:265–270. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mantovani A and Sica A: Macrophages,
innate immunity and cancer: Balance, tolerance, and diversity. Curr
Opin Immunol. 22:231–237. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Qian BZ and Pollard JW: Macrophage
diversity enhances tumor progression and metastasis. Cell.
141:39–51. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sica A, Saccani A and Mantovani A:
Tumor-associated macrophages: A molecular perspective. Int
Immunopharmacol. 2:1045–1054. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kim YJ, Jung EB, Lee MS, Seo SJ, Kim MH,
Lee MW and Lee CS: Rotundarpene inhibits toll-like receptor 2
activation-induced production of inflammatory mediators in
keratinocytes by suppressing the Akt and NF-κB pathways. Int
Immunopharmacol. 18:325–332. 2014. View Article : Google Scholar : PubMed/NCBI
|