Introduction
Nuclear factor-κB (NF-κB) is an important
tumor-related nuclear transcription factor and is overexpressed and
continuously activated in malignant glioma cells, thereby playing a
role in promoting the survival of tumor cells (1,2).
Sulfasalazine (SAS) has been widely used as an anti-inflammatory
drug in the clinic. Because of its significant antitumor effect on
human glioma, colon cancer, breast cancer, lymphoma and other
malignant cells (3–7), it has been used in a phase 1/2
prospective, randomized study for the treatment of progressive
malignant glioma (8). At present,
most studies suggest that the antitumor mechanism of SAS is related
to its effective inhibition of NF-κB signaling (9–11).
Autophagy is an evolutionarily conserved cellular
metabolic process. It can promote cell survival and may also
promote cell death via different mechanisms. Autophagy plays
different roles depending on the drug, cell type or time of drug
action and its mechanism is not fully understood (12–15).
Therefore, study of the dual role of autophagy may provide new
clues for tumor treatment.
Research has shown that NF-κB regulates autophagy by
upregulating the mRNA and protein levels of the autophagy promoter
Beclin 1; it can also inhibit autophagy by activating autophagy
inhibitor mTOR (16). Other
research has shown that autophagy can also inactivate NF-κB
activity by degrading its upstream IκB kinases (IKKs) (17). Recent research found that SAS
induced autophagy at the same time as causing the growth inhibition
of human small cell lung cancer cell lines NCI-H69 and NCI-H82
(18). Yet, whether SAS induces
autophagy in other types of tumor cells and the related mechanism
of autophagy in the antitumor effect of SAS still require further
investigation.
Our previous research demonstrated that the
multifunctional protein p62/sequestosome 1 (encoded by
SQSTM1) is involved in the autophagic degradation process of
ubiquitinated proteins as an adaptor protein and therefore it plays
an important role in the mechanism by which autophagy promotes
human ovarian cancer cells to survive (19). p62 is a multiple domain protein,
which can combine with ubiquitin through the UBA domain and can
bind the autophagy regulatory protein LC3 via the LIR domain,
resulting in these proteins performing autophagic degradation.
Several studies have shown that autophagy can suppress the
development and progression of tumors by downregulating the levels
of p62 protein (20,21). In addition, p62 can also act with an
apoptosis-regulating molecular switch RIP1 by zinc-finger domains
to form a signaling complex, which functions as a scaffold protein
in the activation of IKKs, leading to degradation of the inhibitory
molecule IκB and resulting in the activation of NF-κB signaling
(22–25). A study demonstrated that knockdown
of p62 with an antisense construct severely impaired the activation
of NF-κB in response to TNFα (26).
Duran et al also proved that p62 is necessary for the
survival of human lung adenocarcinoma cells (27); therefore, high expression of p62 can
be used as a marker of NF-κB signaling activation (28).
SAS can inhibit glioma growth, but whether it does
this by influencing autophagy to inhibit the activity of NF-κB is
not clear. Furthermore, it is important to elucidate whether the
multifunctional protein p62 participates in the regulation of NF-κB
signaling by SAS. Therefore, we inhibited autophagy by 3-MA and
inhibited the mRNA and protein expression of p62 by RNA
interference (RNAi) technology, to investigate whether both
autophagy and p62 participate in the anti-glioma mechanism of SAS,
in order to provide insight into a potential new target for the
antitumor therapy of SAS.
Materials and methods
Cell lines
The human glioma U251 cell line was obtained from
the Department of Pathophysiology, Jilin University Norman Bethune
Medical College. The cells were cultured in Iscove’s modified
Dulbecco’s medium (IMDM; Gibco-BRL, Carlsbad, CA, USA) supplemented
with 10% fetal bovine serum (FBS; Invitrogen Life Technologies,
Carlsbad, CA, USA) and cultured at 37°C in 5% CO2 with
high humidity.
Cell viability assays
Cells were plated at a density of 0.7×104
cells/well in 96-well plates. The next day, different
concentrations of SAS were added to the wells and incubated for 4,
8 and 24 h. Each treatment was repeated in 5 wells. To each well,
we added 20 µl MTT (Sigma-Aldrich, St. Louis, MO, USA) and
incubated the plates for 4 h; 150 µl dimethyl-sulphoxide was
then added to dissolve the formazan crystals. Absorbance was
measured with a VMax microplate reader (Molecular Devices,
Sunnyvale, CA, USA) at a wavelength of 570 nm.
Flow cytometry
After exposure to different experimental conditions,
cells were trypsinized and incubated with propidium iodide (PI; 1
µg/ml) and Annexin V-FITC (1 µg/ml; Invitrogen) for
15 min at room temperature. Samples were then analyzed for
apoptosis by a FACScan flow cytometer (Becton-Dickinson, Franklin
Lakes, NJ, USA) within 1 h.
Western blot analysis
Lysate proteins (30–50 µg) were separated by
12% w/v SDS-polyacrylamide gel electrophoresis and transferred onto
PVDF membranes (Millipore, Bedford, MA, USA). Membranes were
blocked with 5% nonfat dry milk in buffer [10 µM Tris-HCl
(pH 7.6), 100 µM NaCl and 0.1% Tween-20] for 1 h at room
temperature, incubated with the desired primary antibody overnight
at 4°C and then incubated with the horseradish
peroxidase-conjugated secondary antibody (Thermo Scientific,
Waltham, MA, USA) at a 1:2,000 dilution for 1 h at room
temperature. Immunoreactive bands were visualized using the DAB
(Sigma-Aldrich) coloration method. Protein levels were quantified
by densitometry using Quantity One software (Bio-Rad).
Indirect immunofluorescence staining and
confocal laser microscopy
Cells were fixed with 4% paraformaldehyde, stained
with Hoechst 33258 (2 µg/ml, Sigma-Aldrich) for 30 min and
examined using confocal laser microscopy to observe apoptotic
nuclei. For indirect immunofluorescence staining, cells were fixed
with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100 and
blocked with bovine serum albumin. They were then incubated with
primary antibodies against p65 and p50 (1:50 dilution) overnight at
4°C, and then in FITC/Rhodamine Red-conjugated secondary antibodies
(1:400 dilution) (all antibodies, Santa Cruz Biotechnology, Santa
Cruz, CA, USA) for 0.5 h and stained with Hoechst 33258 (2
µg/ml) for 2 min and examined by confocal fluorescence
microscopy.
p62 knockdown by small interfering RNA
(siRNA)
siRNA sequences targeting human p62/SQSTM1
(GenBankAccession NM_003900) and a non-target sequence were
constructed by Genechem (Shanghai, China). The p62 siRNA (si-p62)
sequence was GAC-ATC-TTC-CGAATC-TAC-A and that of non-target siRNA
(scramble) was TTC-TCC-GAA-CGT-GTC-ACG-T. Briefly, cells were
transfected with si-p62 or si-Scramble using Lipofectamine 2000
(Invitrogen), according to the manufacturer’s protocol.
NF-κB luciferase reporter gene
analysis
Cells were plated at a density of
4×104/well in 24-well plates. When the cells had reached
~80% confluency, the cells were transfected with firefly luciferase
gene reporter plasmid encoding pNF-κBLuc (Beyotime Institute of
Biotechnology, Shanghai, China) using Lipofectamine 2000.
Transfection efficacy was controlled by cotransfection with the
Renilla luciferase (pRL-null) plasmid (Beyotime Institute of
Biotechnology). Cells were lysed after stimulation, and luciferase
activity was measured following the manufacturer’s instructions
(Promega). The ratio of firefly luciferase activity to
Renilla luciferase activity represented the NF-κB
transcriptional activity.
RNA extraction and reverse
transcription-PCR
We used TRIzol reagent (Invitrogen) to extract total
cellular RNA. Reverse transcription was performed to generate cDNA,
which was then amplified by PCR. The sequences of the primers were
as follows: p62: forward, 5′-GAA-CTC-CAG-TCC-CTA-CAG-AT-3′ and
reverse, 5′-CGA-TGT-CAT-AGT-TCT-TG-TC-3′; GAPDH: forward,
5′-GGG-TGA-TGC-TGG-TGC-TGA-GTA-TGT-3′ and reverse
5′-AAG-AAT-GGG-AGT-TGC-TGT-TGA-AGT-3′. PCR products were run on 1%
agarose gel electrophoresis containing ethidium bromide, visualized
by figure gel image processing system and analyzed by GIS 1D gel
image system software (Tanon, Shanghai, China). The ratio of p62
and GAPDH reflcted the changes in p62 levels.
Statistical analysis
Experiments were performed at least 3 times and data
are presented as mean ± SD. Statistical analysis of the data was
performed using one-way ANOVA; differences between treatment means
were examined with Dunnett’s tests. P<0.05 was considered to
indicate a statistically significant difference.
Results
Apoptosis and autophagy are induced by
SAS accompanied by inhibition of NF-κB signaling in U251 cells
U251 cells were treated with different doses of SAS
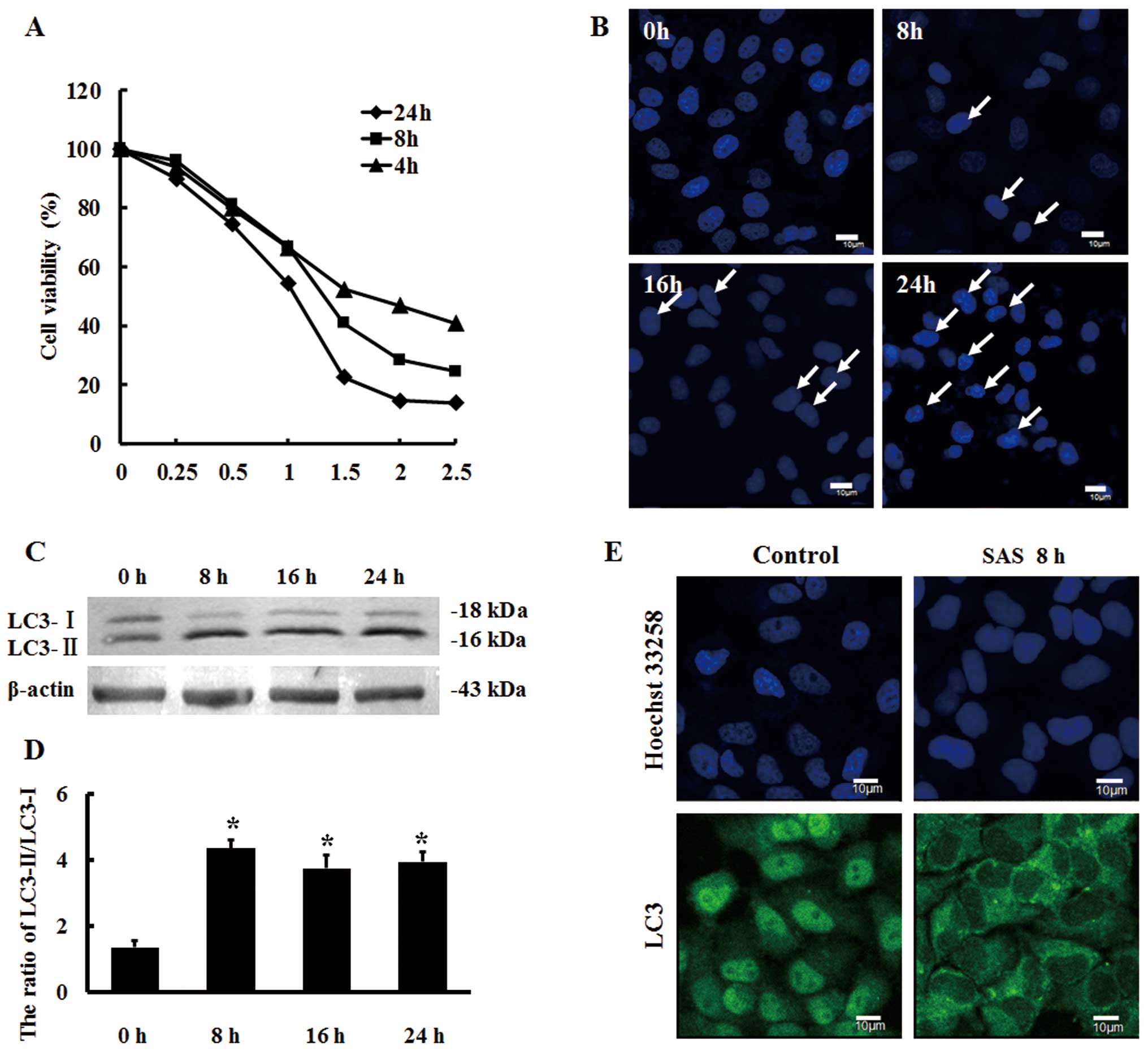
for different time intervals, and the MTT assay results showed that
SAS effectively inhibited the proliferation of U251 cells in a
dose- and time-dependent manner (Fig.
1A). From the resulting IC50 values, we selected 1.5
µM SAS for the treatment of U251 cells at different time
intervals.
We observed cell nuclei stained with Hoechst 33258
using confocal laser scanning microscopy. The results revealed that
SAS induced apoptosis in the U251 cells (Fig. 1B). Research suggests that autophagy
may be involved in the antitumor effect of SAS (18). Therefore, we analyzed the protein
expression of the autophagy marker protein LC3 in response to 1.5
µM SAS by western blot analysis and showed that the protein
expression ratio of LC3-II to LC3-I was significantly increased by
SAS treatment for 8 h (Fig. 1C and
D). Furthermore, indirect immunofluorescence showed that LC3
had translocated to the cytoplasm, forming punctate aggregates and
the fluorescence intensity of LC3 was also enhanced (Fig. 1E), suggesting that SAS can induce
autophagy in U251 cells.
Most studies suggest that SAS has an antitumor
effect by inhibiting NF-κB signaling (9–11).
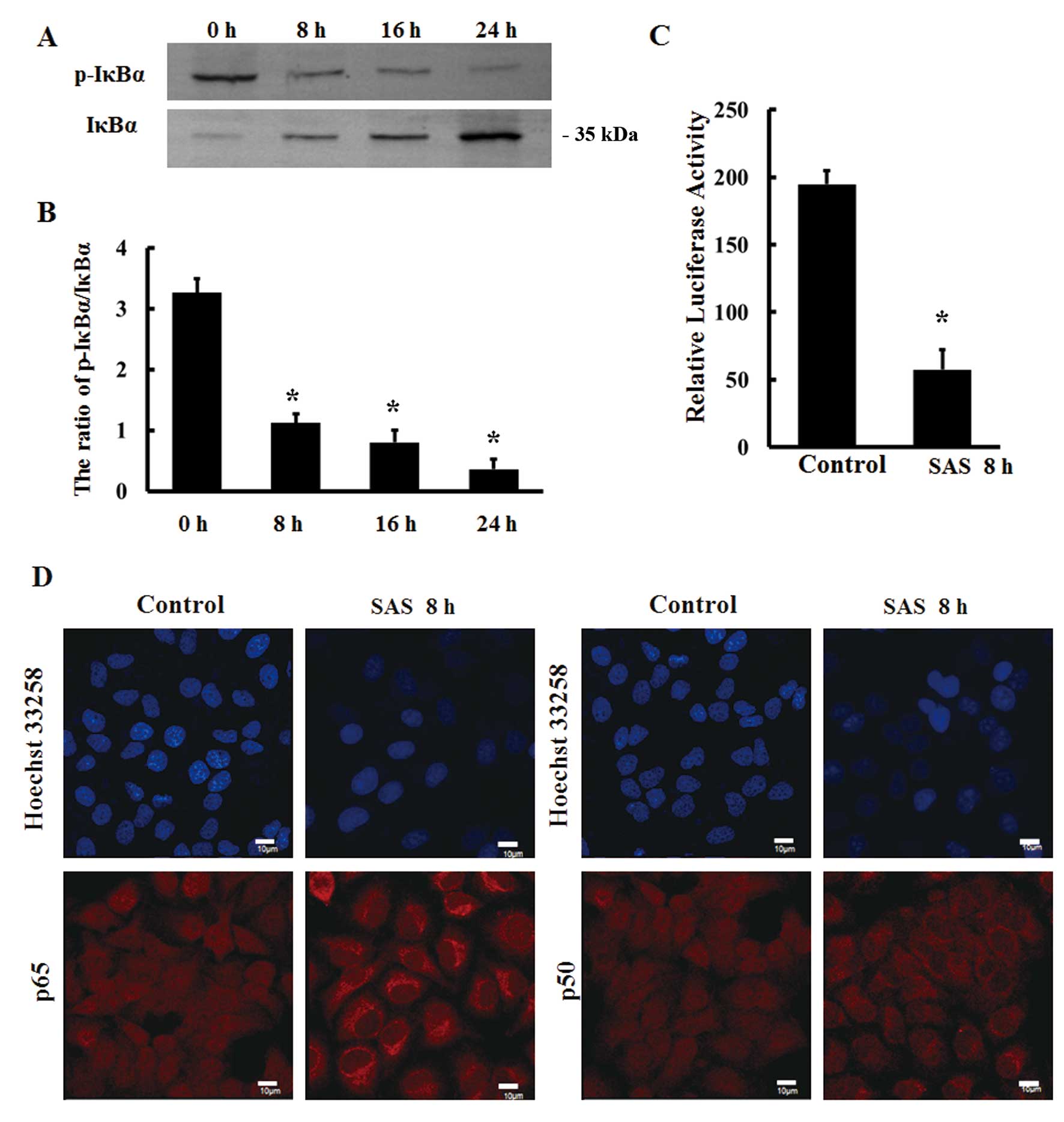
Therefore, we investigated the expression of IκBα in response to
1.5 µM SAS by western blot analysis. The results showed that
the ratio of p-IκBα to IκBα protein expression was significantly
decreased following treatment with SAS for 8 h (Fig. 2A and B). In addition, the
transcriptional activity of NF-κB was reduced by 1.5 µM SAS
in U251 cells as detected by dual luciferase reporter assays
(Fig. 2C). The intracellular
localizations of the NF-κB subunits p65 and p50 were detected by
indirect immunofluorescence, and the results showed that p65 and
p50 were mainly distributed in the cytoplasm following SAS
treatment for 8 h (Fig. 2D). This
finding suggests that SAS can effectively inhibit NF-κB
signaling.
Inhibition of autophagy by 3-MA
suppresses the effects of SAS on NF-κB signaling and apoptosis in
U251 cells
3-MA as a classic autophagy inhibitor has been
widely used in related research (29,30).
Previous research used 3-MA to inhibit autophagy and prove that
autophagy is involved in the growth inhibition of hepatoma cells
induced by SAS (31). Therefore, we
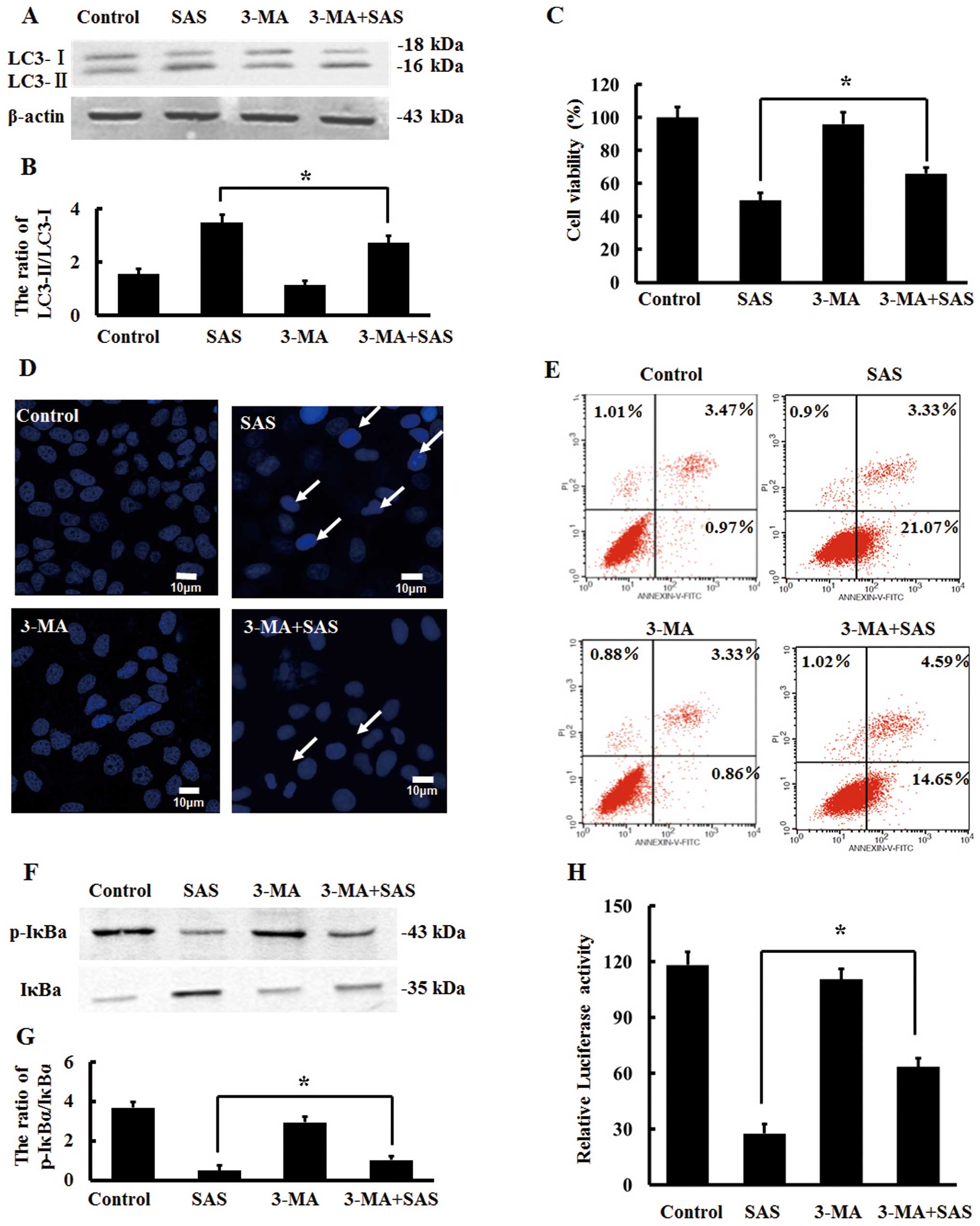
treated U251 cells with a combination of 5 µM 3-MA and 1.5
µM SAS for 8 h and detected the protein expression of LC3 by
western blot analysis; SAS combined with 3-MA resulted in a
reduction in the protein expression ratio of LC3-II to LC3-I
compared with this ratio following SAS alone (Fig. 3A and B). This suggests that 3-MA
effectively inhibits SAS-induced autophagy. The results of MTT
assays showed that the viability of U251 cells was significantly
increased by SAS combined with 3-MA when compared with the
viability of cells treated with SAS alone (Fig. 3C). Compared with the cells treated
with SAS alone, apoptotic nuclei and apoptotic cell ratio were
reduced in the cells treated with SAS combined with 3-MA (Fig. 3D and E). Therefore, inhibition of
autophagy by 3-MA reduced the apoptosis induced by SAS in U251
cells.
These findings motivated us to ascertain whether
inhibition of autophagy by 3-MA can also impact NF-κB signaling at
the same time. Compared with the cells treated with SAS alone, the
protein expression ratio of p-IκBα to IκBα was slightly increased
in the cells treated with SAS combined with 3-MA (Fig. 3F and G). Moreover, dual luciferase
assays demonstrated that the transcriptional activity of NF-κB was
significantly enhanced (Fig. 3H),
suggesting that inhibition of autophagy can weaken SAS-induced
inhibition of NF-κB signaling.
Inhibition of autophagy by 3-MA weakens
p62 reduction by SAS in U251 cells
Multifunctional scaffold protein p62 is well known
as an autophagy marker protein and provides crosstalk for important
signaling pathways, including NF-κB signaling (24,32).
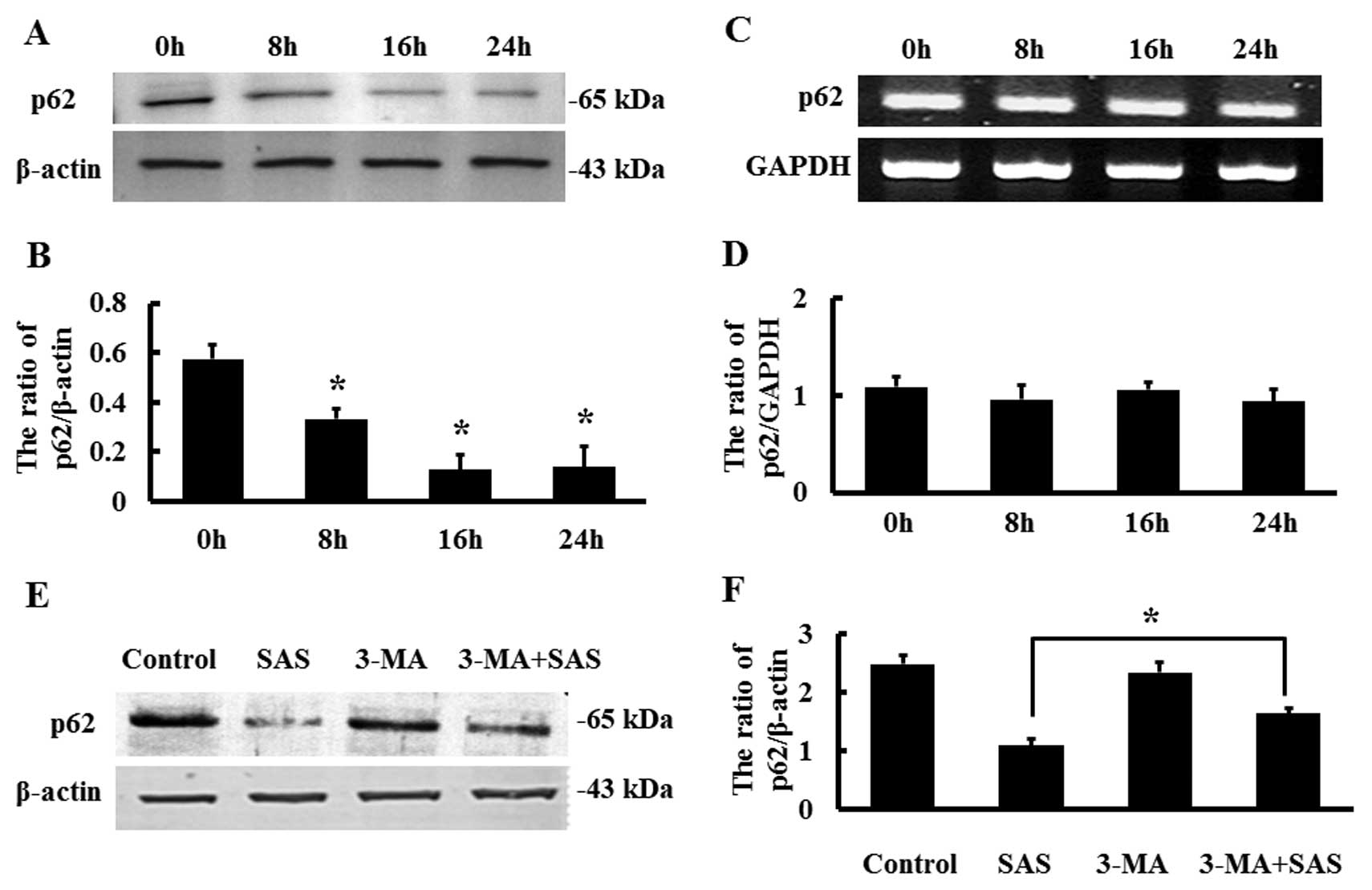
We detected the expression of p62 in response to 1.5 µM SAS
by western blot analysis and found that p62 expression was
decreased in a time-dependent manner (Fig. 4A and B). However, RT-PCR results
showed that the mRNA levels of p62 remained basically unchanged
(Fig. 4C and D). Therefore, we
speculated that the SAS-induced decrease in p62 protein levels may
not be the result of reduced p62 transcript levels, but may be
caused by other processes, such as post-translational
modifications. Compared with the cells treated with SAS alone, the
protein expression of p62 was increased in the cells treated with
SAS combined with 3-MA (Fig. 4E and
F). These results indicate that inhibition of autophagy can
result in increased p62 protein expression.
SAS induces NF-κB signaling inhibition
and apoptosis via a p62-dependent effect in U251 cells
Since 3-MA can enhance the protein expression of p62
by inhibiting autophagy, the changing trend was similar to the
transcriptional activity of NFκB. We applied RNAi technology to
inhibit p62 expression (12). After
the si-p62 recombinant plasmids were transfected into U251 cells
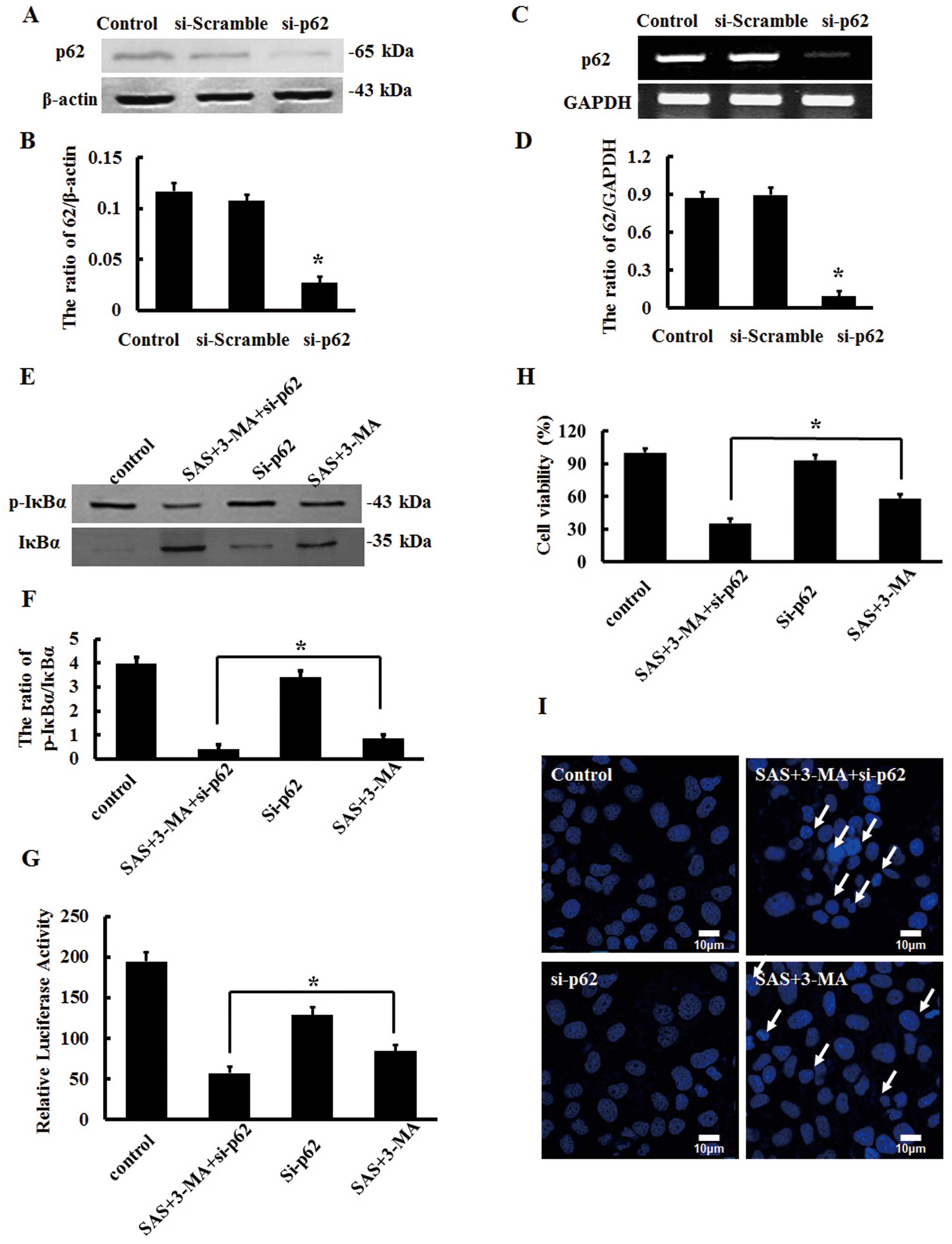
for 24 h, we found that p62 protein expression was significantly
decreased as detected by western blot analysis (Fig. 5A and B). RT-PCR analysis showed that
the mRNA levels of p62 were also significantly decreased (Fig. 5C and D).
In order to identify whether autophagy is involved
in the regulation of NF-κB signaling in a p62-dependent manner, we
applied RNAi technology to inhibit p62 expression, 5 µM 3-MA
to inhibit autophagy and then 1.5 µM SAS to treat U251 cells
for 8 h and detected the protein expression of IκBα by western blot
analysis.
Knockdown of p62 did not influence the expression
ratio of p-IκBα and IκBα. Yet, p62 inhibition inverted the
enhancement of the ratio of p-IκBα to IκBα by 3-MA in the
SAS-treated U251 cells (Fig. 5E and
F). In addition, p62 suppression further decreased the
transcriptional activity of NF-κB in the U251 cells treated with
SAS and 3-MA (Fig. 5G). Similarly,
we detected the viability of U251 cells by MTT assays and found
that knockdown of p62 reversed the enhancement of cell viability by
3-MA in the SAS-treated U251 cells (Fig. 5H), concomitant with an increase in
apoptotic nuclei, as visualized by Hoechst 33258 staining and
confocal laser scanning microscopy (Fig. 5I). These results demonstrate that
the effects of 3-MA on SAS-treated U251 cells are dependent on
p62.
Discussion
NF-κB is one of the most important nuclear
transcription factors related to inflammation and tumors.
Activation of the NF-κB signaling pathway is involved in cancer
progression and chemotherapy drug resistance (33). NF-κB is constitutively activated in
most malignant gliomas (1,2,9,34,35).
Zanotto-Filho et al used siRNA to knockdown NF-κB-p65 and
proved that inhibition of NF-κB induces the apoptosis of glioma
U138MG cells (34). Furthermore,
Robe et al showed that SAS effectively inhibited NF-κB and
induced apoptosis of glial stromal tumor U87 and LN18 and human
glioma U251 cell lines (9). Thus,
in-depth study concerning the regulatory mechanism of the NF-κB
signaling pathway is of significance to further understand glioma
biological characteristics and to develop new glioma therapeutic
strategies. In the present study, we utilized SAS, a drug that can
effectively inhibit the activity of NF-κB, and has recently been
considered as a potential antitumor drug (36,37).
The results showed that SAS effectively inhibited NF-κB signaling.
SAS also induced apoptosis and autophagy in the U251 cells.
As an evolutionarily conserved cellular metabolic
process, autophagy has conflicting roles in cell death according to
the context in different research. Therefore, we were interested in
the role of autophagy induced by SAS in U251 cells. Based on
previous studies, we utilized classic autophagy inhibitor 3-MA in
our research. We found that inhibition of autophagy by 3-MA
suppressed the apoptosis induced by SAS in U251 cells. We also
detected the activation of NF-κB signaling. The inhibition of the
transcriptional activity of NF-κB by SAS was weakened by 3-MA.
Thus, we aimed to ascertain how suppression of autophagy affects
the NF-κB signaling pathway.
Multifunctional scaffold protein p62 is well known
as an autophagy marker protein and provides crosstalk for important
signaling pathways, including NF-κB signaling (24,32).
Accumulating research has confirmed that p62 participates in the
regulation of NF-κB signaling, yet the mechanism is not fully
understood (27,38). Our previous study indicated that p62
is involved in the mechanism of cisplatin-resistance through
autophagy in ovarian cancer cells (19). Thus, we focused on the role of p62
in the suppression of NF-κB signaling induced by SAS. We detected
the protein expression of p62 in response to SAS and found that p62
expression was decreased in a time-dependent manner. However,
RT-PCR results showed that the mRNA levels of p62 remained
basically unchanged. Therefore, we speculated that SAS-induced
decrease in p62 protein levels may not be the result of reduced
transcript levels, but may be caused by other processes, such as
degradation. Inhibition of autophagy by 3-MA reversed the decrease
in p62 induced by SAS. Furthermore, the altered tendency of p62 is
in keeping with the transcriptional activity of NFκB. Thus, we
hypothesized that maintaining the activity of NF-κB signaling is at
least partly dependent on the protein level of p62.
To test our hypothesis, we utilized RNAi technology
to inhibit p62 expression. The results showed that knockdown of p62
weakened the effects of 3-MA on NF-κB signaling inhibition induced
by SAS in U251 cells. Consistent with this, after inhibition of
p62, the protective effect of 3-MA on SAS-treated U251 cells
disappeared; the viability of the cells was decreased and apoptotic
cells were increased.
This indicates that SAS induces NF-κB signaling
inhibition and apoptosis at least partly via a p62-dependent effect
in U251 cells. The results based on the in vitro study
indicate that p62 is an important regulatory factor of NF-κB
signaling in tumorigenesis (27).
In addition, p62 was required for continuous activation of NF-κB
that promoted cell survival in a study based on p62-knockout mice
(38). At present, there are two
well-known explanations for how p62 regulates NF-κB signaling. One
is that p62 can conjugate with aPKCs (PKCζ and PKCλ/ι) resulting in
the phosphorylation of IκB kinase complex (22). Another is the oligomerization of p62
which can activate K63-E3 ubiquitin ligase TRAF6 leading to
ubiquitination of IκBα (24). Both
theories acknowledge that p62 promotes the phosphorylation of IκBα.
This finding is consistent with our results. Yet, further studies
are needed to confirm whether p62 provides signaling activation
center through aggregation or promotes IκBα degradation through
protein degradation pathways including autophagy.
This study demonstrated that apoptosis and autophagy
were induced by SAS accompanied by inhibition of NF-κB signaling in
U251 cells. Inhibition of autophagy by 3-MA can suppress the
effects of SAS on NF-κB signaling and apoptosis in U251 cells. p62
is involved in the mechanism of NF-κB signaling inhibited by SAS in
U251 cells.
In conclusion, the identification of the association
of p62 and NF-κB signaling sheds light on the link between p62 and
the NF-κB signaling pathway, specifically in tumors. Therefore, p62
can act to nucleate different signaling molecules to ensure the
efficiency and selectivity of the signal transduction process.
Given that the NF-κB signaling pathway is commonly deregulated in
cancer, the recent identification of p62 as a critical step in this
pathway may help in the design of better targeted therapies for the
treatment of tumors in which NF-κB signaling is altered.
Acknowledgments
This research was funded by the National Natural
Science Foundation of China (nos. 81272876, 81202552 and 81372793)
and the ‘211 Project’ of Jilin University.
References
|
1
|
Nagai S, Washiyama K, Kurimoto M, Takaku
A, Endo S and Kumanishi T: Aberrant nuclear factor-kappaB activity
and its participation in the growth of human malignant astrocytoma.
J Neurosurg. 96:909–917. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hayashi S, Yamamoto M, Ueno Y, Ikeda K,
Ohshima K, Soma G and Fukushima T: Expression of nuclear
factor-kappa B, tumor necrosis factor receptor type 1 and c-Myc in
human astrocytomas. Neurol Med Chir (Tokyo). 41:187–195. 2001.
View Article : Google Scholar
|
|
3
|
Bredel M, Bredel C, Juric D, Duran GE, Yu
RX, Harsh GR, Vogel H, Recht LD, Scheck AC and Sikic BI: Tumor
necrosis factor-α-induced protein 3 as a putative regulator of
nuclear factor-κB-mediated resistance to O6-alkylating
agents in human glioblastomas. J Clin Oncol. 24:274–287. 2006.
View Article : Google Scholar
|
|
4
|
Lo M, Wang YZ and Gout PW: The
x(c)-cystine/glutamate antiporter: a potential target for therapy
of cancer and other diseases. J Cell Physiol. 215:593–602. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ryan BM, Russel MG, Langholz E and
Stockbrugger RW: Aminosalicylates and colorectal cancer in IBD: a
not-so bitter pill to swallow. Am J Gastroenterol. 98:1682–1687.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Narang VS, Pauletti GM, Gout PW, Buckley
DJ and Buckley AR: Suppression of cystine uptake by sulfasalazine
inhibits proliferation of human mammary carcinoma cells. Anticancer
Res. 23:4571–4579. 2003.
|
|
7
|
Gout PW, Simms CR and Robertson MC: In
vitro studies on the lymphoma growth-inhibitory activity of
sulfasalazine. Anticancer Drugs. 14:21–29. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Robe PA, Martin DH, Nguyen-Khac MT, Artesi
M, Deprez M, Albert A, Vanbelle S, Califice S, Bredel M and Bours
V: Early termination of ISRCTN45828668, a phase 1/2 prospective,
randomized study of sulfasalazine for the treatment of progressing
malignant gliomas in adults. BMC Cancer. 9:3722009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Robe PA, Bentires-Alj M, Bonif M, Rogister
B, Deprez M, Haddada H, Khac MT, Jolois O, Erkmen K, Merville MP,
et al: In vitro and in vivo activity of the nuclear factor-kappaB
inhibitor sulfasalazine in human glioblastomas. Clin Cancer Res.
10:5595–5603. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Habens F, Srinivasan N, Oakley F, Mann DA,
Ganesan A and Packham G: Novel sulfasalazine analogues with
enhanced NF-κB inhibitory and apoptosis promoting activity.
Apoptosis. 10:481–491. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Müerköster S, Arlt A, Witt M, Gehrz A,
Haye S, March C, Grohmann F, Wegehenkel K, Kalthoff H, Fölsch UR,
et al: Usage of the NF-kappaB inhibitor sulfasalazine as
sensitizing agent in combined chemotherapy of pancreatic cancer.
Int J Cancer. 104:469–476. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ogier-Denis E and Codogno P: Autophagy: a
barrier or an adaptive response to cancer. Biochim Biophys Acta.
1603:113–128. 2003.PubMed/NCBI
|
|
13
|
Shintani T and Klionsky DJ: Autophagy in
health and disease: a double-edged sword. Science. 306:990–995.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Marino G and Lopez-Otin C: Autophagy:
molecular mechanisms, physiological functions and relevance in
human pathology. Cell Mol Life Sci. 61:1439–1454. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Eskelinen EL: Maturation of autophagic
vacuoles in mammalian cells. Autophagy. 1:1–10. 2005. View Article : Google Scholar
|
|
16
|
Djavaheri-Mergny M, Amelotti M, Mathieu J,
Besançon F, Bauvy C, Souquère S, Pierron G and Codogno P: NF-kappaB
activation represses tumor necrosis factor-alpha-induced autophagy.
J Biol Chem. 281:30373–30382. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Trocoli A and Djavaheri-Mergny M: The
complex interplay between autophagy and NF-kappaB signaling
pathways in cancer cells. Am J Cancer Res. 1:629–649. 2011.
|
|
18
|
Guan J, Lo M, Dockery P, Mahon S, Karp CM,
Buckley AR, Lam S, Gout PW and Wang YZ: The
xc-cystine/glutamate antiporter as a potential
therapeutic target for small-cell lung cancer: use of
sulfasalazine. Cancer Chemother Pharmacol. 64:463–472. 2009.
View Article : Google Scholar
|
|
19
|
Yu H, Su J, Xu Y, Kang J, Li H, Zhang L,
Yi H, Xiang X, Liu F and Sun L: p62/SQSTM1 involved in cisplatin
resistance in human ovarian cancer cells by clearing ubiquitinated
proteins. Eur J Cancer. 47:1585–1594. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
White E, Karp C, Strohecker AM, Guo Y and
Mathew R: Role of autophagy in suppression of inflammation and
cancer. Curr Opin Cell Biol. 22:212–217. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rosenfeldt MT and Ryan KM: The multiple
roles of autophagy in cancer. Carcinogenesis. 32:955–963. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sanz L, Sanchez P, Lallena MJ, Diaz-Meco
MT and Moscat J: The interaction of p62 with RIP links the atypical
PKCs to NF-kappaB activation. EMBO J. 18:3044–3053. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wooten MW, Seibenhener ML, Mamidipudi V,
Diaz-Meco MT, Barker PA and Moscat J: The atypical protein kinase
C-interacting protein p62 is a scaffold for NF-kappaB activation by
nerve growth factor. J Biol Chem. 276:7709–7712. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wooten MW, Geetha T, Seibenhener ML, Babu
JR, Diaz-Meco MT and Moscat J: The p62 scaffold regulates nerve
growth factor-induced NF-kappaB activation by influencing TRAF6
polyubiquitination. J Biol Chem. 280:35625–35629. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Moscat J, Diaz-Meco MT and Wooten MW:
Signal integration and diversification through the p62 scaffold
protein. Trends Biochem Sci. 32:95–100. 2007. View Article : Google Scholar
|
|
26
|
Moscat J, Diaz-Meco MT, Albert A and
Campuzano S: Cell signaling and function organized by PB1 domain
interactions. Mol Cell. 23:631–640. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Duran A, Linares JF, Galvez AS,
Wikenheiser K, Flores JM, Diaz-Meco MT and Moscat J: The signaling
adaptor p62 is an important NF-kappaB mediator in tumorigenesis.
Cancer Cell. 13:343–354. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rolland P, Madjd Z, Durrant L, Ellis IO,
Layfield R and Spendlove I: The ubiquitin-binding protein p62 is
expressed in breast cancers showing features of aggressive disease.
Endocr Relat Cancer. 14:73–80. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Su J, Xu Y, Zhou L, Yu HM, Kang JS, Liu N,
Quan CS and Sun LK: Suppression of chloride channel 3 expression
facilitates sensitivity of human glioma U251 cells to cisplatin
through concomitant inhibition of Akt and autophagy. Anat Rec
(Hoboken). 296:595–603. 2013. View
Article : Google Scholar
|
|
30
|
Liu N, Xu Y, Sun JT, Su J, Xiang XY, Yi
HW, Zhang ZC and Sun LK: The BH3 mimetic S1 induces endoplasmic
reticulum stress-associated apoptosis in cisplatin-resistant human
ovarian cancer cells although it activates autophagy. Oncol Rep.
30:2677–2684. 2013.PubMed/NCBI
|
|
31
|
Guo W, Zhao Y, Zhang Z, Tan N, Zhao F, Ge
C, Liang L, Jia D, Chen T, Yao M, et al: Disruption of xCT inhibits
cell growth via the ROS/autophagy pathway in hepatocellular
carcinoma. Cancer Lett. 312:55–61. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Moscat J and Diaz-Meco MT: p62 at the
crossroads of autophagy, apoptosis and cancer. Cell. 137:1001–1004.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pikarsky E, Porat RM, Stein I, Abramovitch
R, Amit S, Kasem S, Gutkovich-Pyest E, Urieli-Shoval S, Galun E and
Ben-Neriah Y: NF-kappaB functions as a tumour promoter in
inflammation-associated cancer. Nature. 431:461–466. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zanotto-Filho A, Braganhol E, Schröder R,
De Souza LH, Dalmolin RJ, Pasquali MA, Gelain DP, Battastini AM and
Moreira JC: NFkappaB inhibitors induce cell death in glioblastomas.
Biochem Pharmacol. 81:412–424. 2011. View Article : Google Scholar
|
|
35
|
Kesanakurti D, Chetty C, Rajasekhar
Maddirela D, Gujrati M and Rao JS: Essential role of cooperative
NF-κB and Stat3 recruitment to ICAM-1 intronic consensus elements
in the regulation of radiation-induced invasion and migration in
glioma. Oncogene. 32:5144–5155. 2013. View Article : Google Scholar
|
|
36
|
Chung WJ, Lyons SA, Nelson GM, Hamza H,
Gladson CL, Gillespie GY and Sontheimer H: Inhibition of cystine
uptake disrupts the growth of primary brain tumors. J Neurosci.
25:7101–7110. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Moriuchi S, Glorioso JC, Maruno M, Izumoto
S, Wolfe D, Huang S, Cohen JB and Yoshimine T: Combination gene
therapy for glioblastoma involving herpes simplex virus
vector-mediated codelivery of mutant IkappaBalpha and HSV thymidine
kinase. Cancer Gene Ther. 12:487–496. 2005.PubMed/NCBI
|
|
38
|
Martin P, Diaz-Meco MT and Moscat J: The
signaling adapter p62 is an important mediator of T helper 2 cell
function and allergic airway inflammation. EMBO J. 25:3524–3533.
2006. View Article : Google Scholar : PubMed/NCBI
|