Introduction
Endometrial cancer (EC) is one of the most common
gynecologic malignancies, with an estimated 49,500 new cases and
8200 deaths in 2013 in the United States (1). ECs are classified into two groups:
those that are estrogen related (type I, endometrioid) and those
that are not estrogen related (type II, non-endometrioid). Type I
ECs are usually linked to obesity, hormone-receptor positivity, and
a favorable outcome. In contrast, type II ECs are more common in
older, non-obese women and have a worse prognosis (2). Current treatment, including
hysterectomy, hormonal therapy, and chemotherapy, are only
effective for early-stage ECs. Effective target therapies are not
available for patients with high-grade diagnoses, especially for
individuals with undifferentiated tumors with deep muscle
infiltration (3). Despite the
prevalence of this cancer, molecular mechanisms accounting for the
development and metastasis of EC remain unclear. To allow for the
possibility of targeted therapies for later-stage EC, further
exploration into the molecular mechanisms underlying EC progression
would be beneficial.
Oncostatin M (OSM) is a member of the IL-6 family of
cytokines produced by macrophages, monocytes, T cells, neutrophils,
and several other cell types (4,5). OSM
inhibits the growth of cells from several types of solid tumor,
including breast tumor, melanoma, and osteosarcoma (6–8). In
contrast, OSM promotes the proliferation of cells from other tumor
types, including those derived from prostate, cervical, and ovarian
cells (9–11). These results suggest that OSM may
have different roles depending on the tumor cell type. The role
that OSM plays in EC has not been clearly defined.
OSM signals through a heterodimeric receptor
composed of gp130 and either OSM receptor-b (OSMR) or the leukemia
inhibitory factor receptor (LIFR). These signal via Janus kinases
(JAKs) to activate the STAT3 (signal transducer and activator of
transcription 3), PI3K (phosphatidylinositol-3-kinase), and MAPK
(mitogen-activated protein kinase) cascades (12–14).
Increased STAT3 activity in tumor cells upregulates VEGF expression
and enhances angiogenesis (15),
which has a major role in tumor growth, progression, and metastasis
(16,17). However, it remains unclear whether
OSM contributes to the development and progression of EC and, if it
does, what the mechanism of its action is, especially with respect
to angiogenesis, which involves the STAT3/VEGF pathway.
In this study, we sought to explore the functional,
mechanistic roles of OSM in EC. We analyzed OSM expression in
paraffin-embedded tissue sections and function in multiple cellular
contexts and assayed its effects in tumor-bearing nude mice. We
showed that OSM promoted EC progression. In particular, OSM
promoted angiogenesis in a STAT3-dependent manner and, therefore,
may serve as a potential marker for antiangiogenic therapy
selection.
Materials and methods
Acquisition of tissue specimens
Paraffin-embedded tissue samples were obtained from
patients who were treated at the Shanghai First People’s Hospital
Affiliated to Shanghai Jiao Tong University, Shanghai, China. The
project was approved by the Human Investigation Ethics Committee of
the Shanghai First People’s Hospital, and informed consent were
obtained from all patients prior to the onset of the study. We
obtained 73 EC tissue specimens from patients who underwent initial
hysterectomy, 26 normal endometrial samples from patients who
underwent hysterectomy to treat other diseases such as myoma or
adenomyosis, and 20 atypical hyperplastic tissue specimens from
patients who underwent hysteroscopic examination due to irregular
bleeding. The stages (I–IV) and histological grades (G1-G3) of
these tumors were established according to the criteria of the
International Federation of Gynecology and Obstetrics (FIGO)
surgical staging system (18). None
of the patients had undergone hormone therapy, radiotherapy, or
chemotherapy before surgery. The clinicopathological
characteristics of the EC patients participating in our study are
presented in Table I.
 | Table ICorrelation of OSM expression with
clinicopathological parameters in endometrial carcinomas. |
Table I
Correlation of OSM expression with
clinicopathological parameters in endometrial carcinomas.
| Characteristic | Case (n) | OSM histoscore
|
|---|
| Mean ± SD | P-value |
|---|
| Diagnostic
categories | | | 0.002a |
| Normal | 26 | 2.58±2.59 | |
| Hyperplasia | 20 | 4.45±2.27 | |
| Carcinoma | 73 | 6.52±4.30 | |
| Age | | | 0.480b |
| <60 | 43 | 6.16±3.91 | |
| ≥60 | 30 | 7.03±4.82 | |
| Histology | | | 0.255b |
| Endometrioid | 58 | 6.79±4.15 | |
| Non-endometrioid
(serous/clear) | 15 | 5.47±4.82 | |
| FIGO stage | | | 0.039b |
| Early (I or
II) | 55 | 5.93±4.09 | |
| Late (III or
IV) | 18 | 8.33±4.52 | |
| Histological
grade | | | 0.038b |
| Low (1 or 2) | 43 | 6.12±3.91 | |
| High (3) | 15 | 8.73±4.33 | |
| Myometrial
invasion | | | 0.014b |
| ≤1/2 | 53 | 5.77±4.36 | |
| >1/2 | 20 | 8.50±3.49 | |
| Lymph node
metastasis | | | 0.042b |
| Positive | 22 | 8.05±3.48 | |
| Negative | 51 | 5.86±4.47 | |
Immunohistochemistry analysis
All samples were prepared and analyzed with the
Histostain-Plus kit (rabbit) (MRBiotech, Emeryville, CA, USA)
according to the manufacturer’s protocol. Staining was performed on
paraffin-embedded specimens using primary antibodies as follows:
anti-OSM (1:100; Boster, Wuhan, China). After 16 h of incubation,
the sections were incubated with a biotinylated secondary antibody
(MRBiotech). We then added a horseradish peroxidase-conjugated
avidin-biotin complex to the sections. OSM expression was detected
by diaminobenzidine as per the manufacturer’s instructions. The
staining percentage was scored in the following manner: 0, 0–5%; 1,
5–25%; 2, 25–50%; 3, 50–75%; and 4, 75–100%. The staining intensity
was scored as follows: 0, negative; 1, weak; 2, moderate; 3,
strong. Then, immunoreactivity scores for each case were obtained
by multiplying the values of the two parameters described above. As
a negative control, phosphate-buffered saline (PBS) was used to
replace the primary antibody.
Microvessel density (MVD) was assessed by
immunostaining for CD34 (Abcam, Cambridge, UK), which highlights
the tumor/tissue vascularity. The microvessels were counted in
three fields per section. The positive-stained blood vessels in the
selected areas were analyzed at x400 magnification.
Cell culture
The human EC cell lines Ishikawa and HEC-1B were
used (American Type Culture Collection, Manassas, VA, USA). The
human umbilical vascular endothelial cells (HUVECs) that we used
were obtained from Key-Gen Biotech (nanjing, China). Ishikawa and
HEC-1B cells were maintained according to the supplier’s
instructions in DMEM/F12 (Gibco, Auckland, new Zealand)
supplemented with 10% fetal bovine serum (FBS) (HyClone, Logan, UT,
USA). HUVECs were grown in RPMI-1640 (Gibco) supplemented with 10%
FBS. Cells were cultured in a humidified atmosphere with 95% air/5%
CO2 at 37°C.
RNA extraction and quantitative real-time
PCR
Total RnA was prepared from the EC cell lines using
TRIzol RnA isolation reagents (Invitrogen, Carlsbad, CA, USA), and
cDnA was then generated with the Prime Script RT reagent kit
(Takara Inc., Otsu, Japan), using the manufacturer’s instructions.
A 10-µl PCR reaction with single-stranded cDnA as the
template was carried out with 40 cycles of denaturation (95°C) for
5 sec, annealing (60°C) for 34 sec, and elongation (60°C) for 30
sec using SYBR Premix Ex Taq (Takara Inc.). The primer sequences
are listed in Table II. For all
qPCR experiments, values on the y axis represent
2(−∆Ct), where ∆Ct is the difference between the Ct for
the gene of interest and the Ct for the gene used for
normalization. All data were obtained in triplicate from three
independent experiments.
| Table IIPrimer sequences for real-time PCR
analysis. |
Table II
Primer sequences for real-time PCR
analysis.
| mRnA | Primer
sequence |
|---|
| OSM | Forward
5′-CGGACAGACAGACAGACACC-3′ |
| Reverse
5′-GAGAACAGCCCAGAAGTTGG-3′ |
| VEGFA | Forward
5′-CCTTGCCTTGCTGCTCTACCTC-3′ |
| Reverse
5′-TTCTGCCCTCCTCCTTCTGC-3′ |
| β-actin | Forward
5′-CTGGGACGACATGGAGAAAA-3′ |
| Reverse
5′-AAGGAAGGCTGGAAGAGTGC-3′ |
Enzyme-linked immunosorbent assay
(ELISA)
VEGF and OSM protein levels were detected in the
culture medium using solid-phase sandwich ELISA assays as per the
manufacturer’s instructions (DVE00, R&D Systems, Minneapolis,
Mn, USA and EK0478, Boster). According to the manufacturer, the
VEGF assay sensitivity was 0.7 pg/ml, and the assay range was
15.6–1000 pg/ml. The OSM assay sensitivity was 0.7 pg/ml, and the
assay range was 15.6–1000 pg/ml. Culture medium was collected three
times independently for statistical analysis.
Western blot analysis
Total protein was extracted using a RIPA kit
(Beyotime, Shanghai, China) containing a 1% dilution of the
protease inhibitor phenylmethylsulfonyl fluoride (Beyotime) as per
the manufacturer’s instructions. Total protein concentration was
estimated using the BCA method (Pierce, Rockford, IL, USA) as per
the manufacturer’s instructions. Similar amounts of protein were
analyzed in each lane of a 10% SDS-polyacrylamide gel. Proteins
were then transferred onto nitrocellulose membranes. After
transfer, membranes were blocked with 5% bovine serum albumin
(BSA)/phosphate-buffered saline (PBS) for 2 h. The membranes were
incubated with primary antibodies at 4°C overnight. The membranes
were then incubated with peroxidase-linked secondary antibody
(1:10,000; Epitomics, Burlingame, CA, USA) for 2 h at room
temperature. Proteins were then detected by enhanced
chemiluminescent reagents. GAPDH was used as an internal
control.
Primary antibodies included: rabbit anti-STAT3
(1:1000; Cell Signaling Technology, Beverly, MA, USA), rabbit
anti-pSTAT3(1:1000; Cell Signaling Technology) and rabbit
anti-GAPDH (1:2000; Epitomics).
Stable transfection of HEC-1B cells
To stably express OSM, HEC-1B cells were washed with
PBS and switched to antibiotic-free growth medium for 24 h before
transfection. All transfections used Lipofectamine 2000 reagent
(Invitrogen) as per the manufacturer’s instructions. The plasmid
that overexpressed OSM and the negative control (NC) plasmid were
generated by GeneChem (Shanghai, China), the transfection reagent
was obtained from Qiagen (Shanghai, China). The plasmid was
transfected into HEC-1B cells (1.6 µg/ml), and then the
cells were selected with G418 (1 mg/ml; Gibco, Carlsbad, CA, USA)
in the growth medium and resistant clones were chosen.
Cell migration and invasion assays
After being trypsinized, centrifuged, and
resuspended, cells were seeded in serum-free medium in the upper
compartment of individual transwell chambers (8-µm pore
size; BD Biosciences, San Jose, CA, USA) with or without Matrigel
coating (BD Biosciences) at a density of 1×105
cells/well (for the migration assay) or 2×105 cells/well
(for the invasion assay). The lower chamber was filled with 600
µl of culture medium containing 5% FBS (C5), C5 with 40
ng/ml rhOSM (C5/rhOSM), or C5 with 40 ng/ml rhOSM and 1 µM
WP1066, a STAT3 inhibitor (C5/rhOSM/WP1066). After 24 h of
incubation, cells that had failed to invade were removed from the
upper chamber. Those that were attached to the outside of the
membrane were fixed and stained with 5% crystal violet and counted
at x200 magnification. Six random fields were selected for each
membrane, and results are expressed in terms of the number of cells
per field. The migration and invasion assays were repeated at least
three times.
Evaluation of cell proliferation
Cell proliferation was evaluated with a CCK8 (Obio
Technology, Shanghai, China) assay as per the manufacturer’s
instructions. Cells (2×103 cells/well) were plated in
96-well plates and were then cultured for 24, 48, or 72 h. At each
time point, 10 µl of CCK8 stock solution was added to each
well, and the plates were further incubated for 2 h at 37°C. The
absorbance was detected at 450 nm in a microplate reader.
Tumorigenicity assays in nude mice
Female 4-week-old athymic nude BALB/c mice were
obtained from Shanghai First People’s Hospital Affiliated to
Shanghai Jiao Tong University, Shanghai, China. HEC-1B cells stably
transfected with the OSM expression plasmid or the NC plasmid were
suspended in PBS (1×107 cells in 200 µl PBS) and
injected into the subcutaneous tissue of the mice. Tumor size was
measured weekly for a 5-week period. At 35 days post-injection,
mice were euthanized, and their weights and tumor volumes were
measured prior to further histological evaluation. The tumor
volumes were calculated by using the following standard formula:
tumor volume (cm3) = (the longest diameter) × (the
shortest diameter)2 ×0.5. All experimental protocols
were approved by the Ethics Committee for Animal Experimentation of
Shanghai Jiao Tong University.
HEC-1B conditioned medium (CM) and mouse
Matrigel plug assays
HEC-1B cells were grown in C10. After 24 h, C10 was
removed and replaced with culture medium without FBS (C0), C0 with
40 ng/ml rhOSM (C0/rhOSM), or C0 with 40 ng/ml rhOSM and 1
µM WP1066 (C0/rhOSM/WP1066). Samples of conditioned medium
(CM) were obtained from each culture after an additional incubation
of 24 h.
For the plug assay, nine athymic nude BALB/c mice
were randomly divided into three groups. Growth factor-reduced
Matrigel (300 µl; BD Biosciences) was mixed with CM (100
µl) and injected subcutaneously into the flank of a mouse.
Mice were euthanized 18 days after inoculation, and plugs were
removed and photographed. Matrigel plugs were stained with
hematoxylin and eosin (H&E) for histological examination and
were analyzed by immunohistochemistry as described above using
antibodies against mouse CD34 (1:100, Abcam).
Tube formation assay
We coated a 96-well plate with 100 µl
Matrigel per well and incubated the plate at 37°C for 2 h. Then,
2×104 HUVECs were suspended in 1 ml diluted HEC-1B CM
(dilution in 1:10) and applied to the pre-coated 96-well plate at a
density of 2000 cells/well (19).
After incubation at 37°C for 16 h, morphologic changes were
observed under a microscope, and cells were photographed at x100
magnification. The total length of the capillary-like tubes that
had formed was determined and averaged across three randomly
selected microscope fields for each well.
HUVEC migration assays
To assess HUVEC migration ability, 5×104
cells suspended in 200 µl of RPMI-1640 were seeded in the
upper chamber of a Transwell. The lower chamber was filled with
HEC-1B CM from different sources as described in the plug assay.
After 24 h of incubation, cells were analyzed as described above
for the cell migration and invasion assays.
Statistical analyses
All tests were carried out with SPSS 17.0
(Microsoft, Redmond, WA, USA) or Prism (GraphPad, San Diego, CA,
USA). Each experiment was performed at least three times. Where
applicable, data are shown as the mean ± SD. Data were analyzed by
unpaired Student’s t-test or by one-way analysis of variance
(AnOVA) for multiple comparisons. The Kruskal-Wallis rank test or
Mann-Whitney U test were used for comparisons of the categorical
data. A P-value of <0.05 was considered statistically
significant.
Results
Expression of OSM in tissues and its
association with clinicopathological parameters
We assessed whether OSM is commonly upregulated in
clinical EC samples. We chose 26 samples of normal endometrium
tissue, 20 samples of atypical hyperplastic endometrium tissues, 57
cases of endometrioid endometrial cancer (EEC) tissue, and 16 cases
of papillary serous EC or clear cell EC tissue.
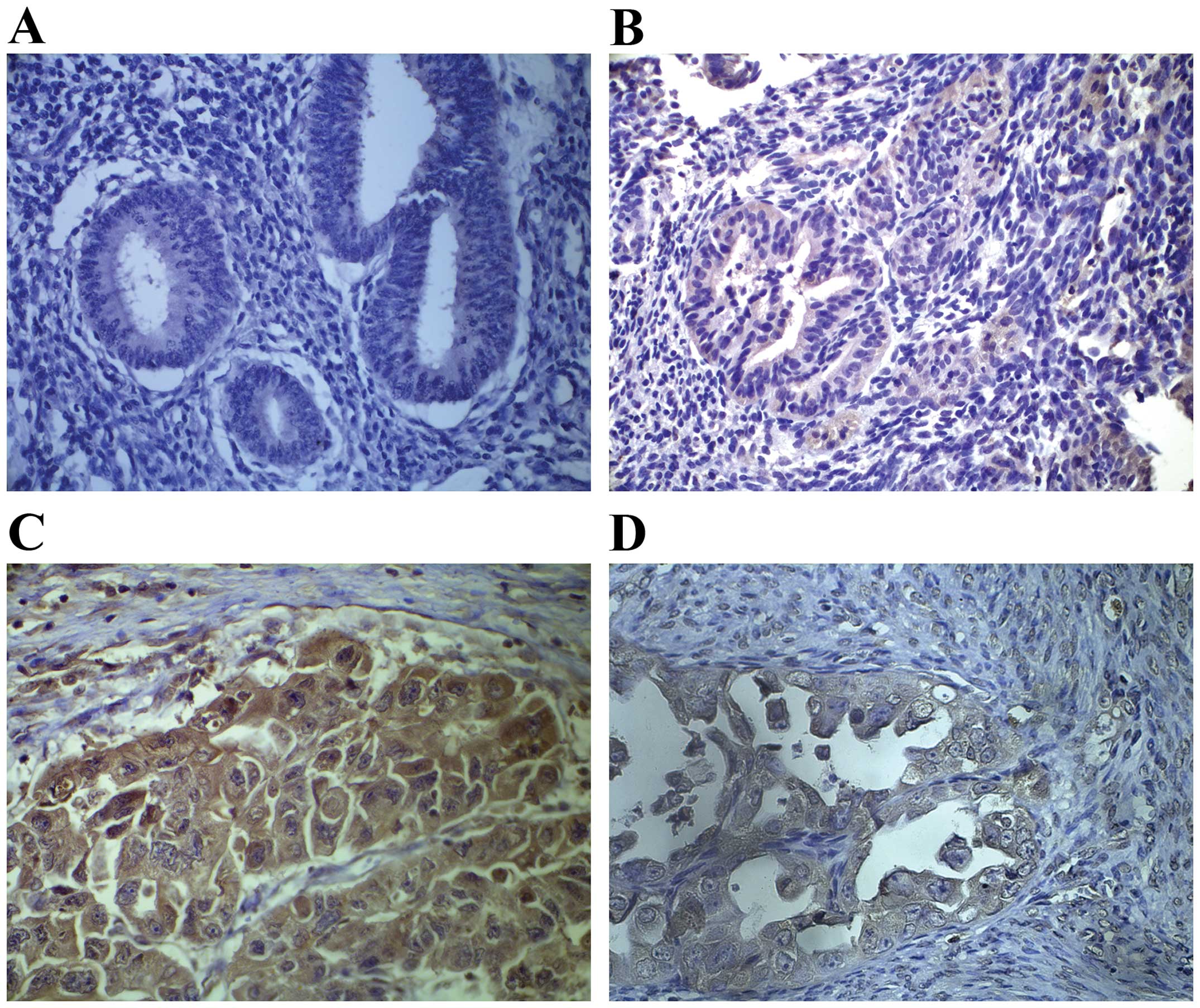
Immunohistochemistry showed that OSM expression was higher in
atypical hyperplasias and even higher in ECs when compared with
normal endometrial tissues (Fig.
1). We calculated a composite histoscore to account for both
stain intensity and uniformity to investigate whether the change in
OSM expression in ECs was associated with specific clinical
characteristics. We compared the association of OSM expression with
the clinicopathological parameters of the EC cases (Table I). Our analyses indicated a
significant association between increased OSM expression and the
depth of myometrial invasion, lymph node metastasis, advanced
disease stage (stages III or IV), and poor histological
differentiation (grade 3) (all measurements resulted in P<0.05;
Table I). However, there was no
significant difference in OSM expression when comparing
endometrioid and non-endometrioid (i.e., serous and clear cell
histological subtypes) ECs (P=0.255; Table I). These results suggested that OSM
expression is related to the development of EC and to the
risk-associated clinical features of the disease.
STAT3 is required for OSM-enhanced
migration and invasion of EC cells
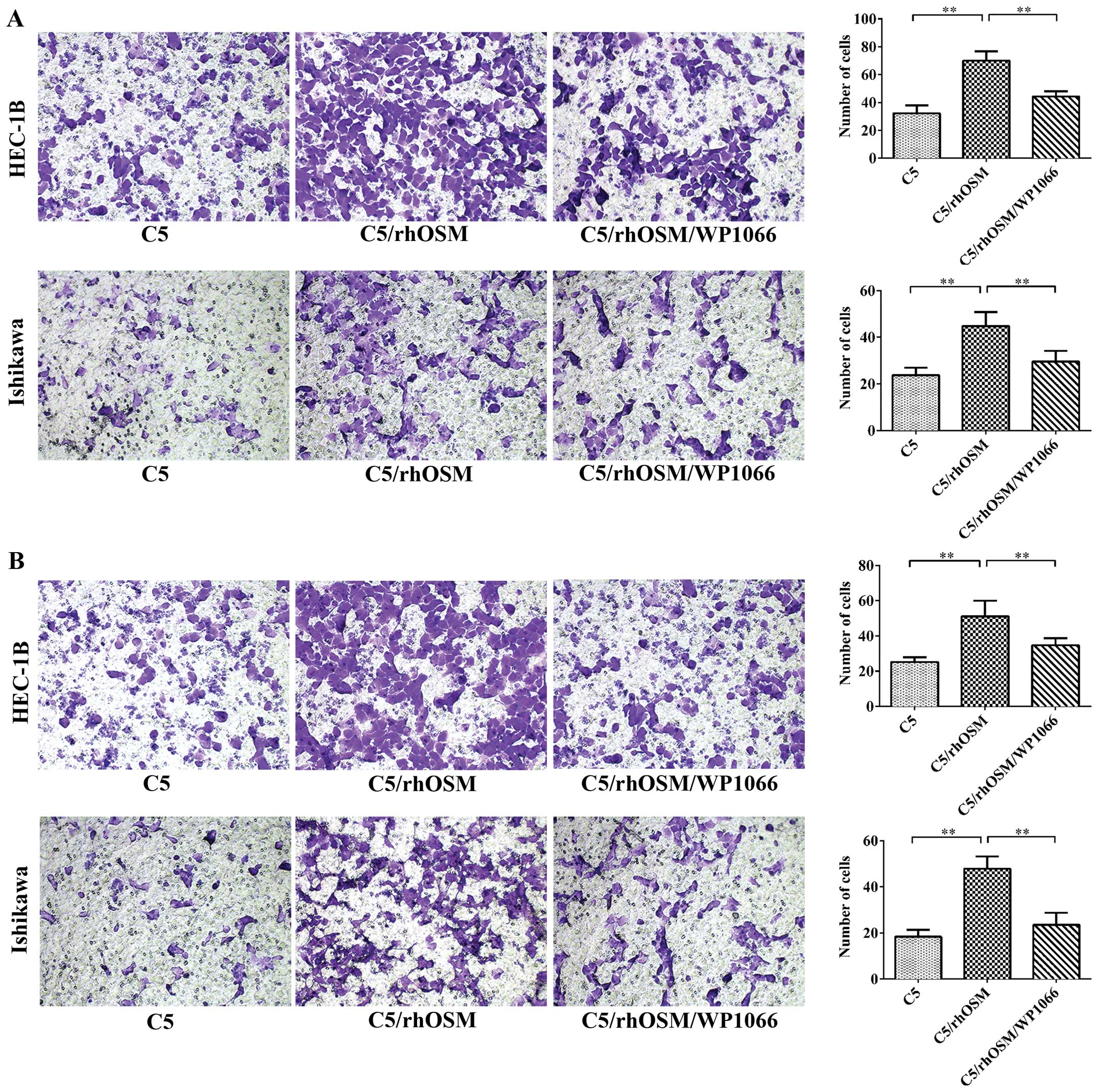
We next examined the effect of rhOSM on EC cell
migration and invasion in vitro. Transwell migration and
invasion assays were performed to study the migratory and invasive
ability of EC cells (Fig. 2). rhOSM
promoted cell migration and invasion in Ishikawa and HEC-1B cells
(P<0.01). However, when the STAT3 inhibitor WP1066 was added,
the effect was reversed. These results suggested that the effects
of rhOSM in mediating EC cell migration and invasion are mediated
by STAT3.
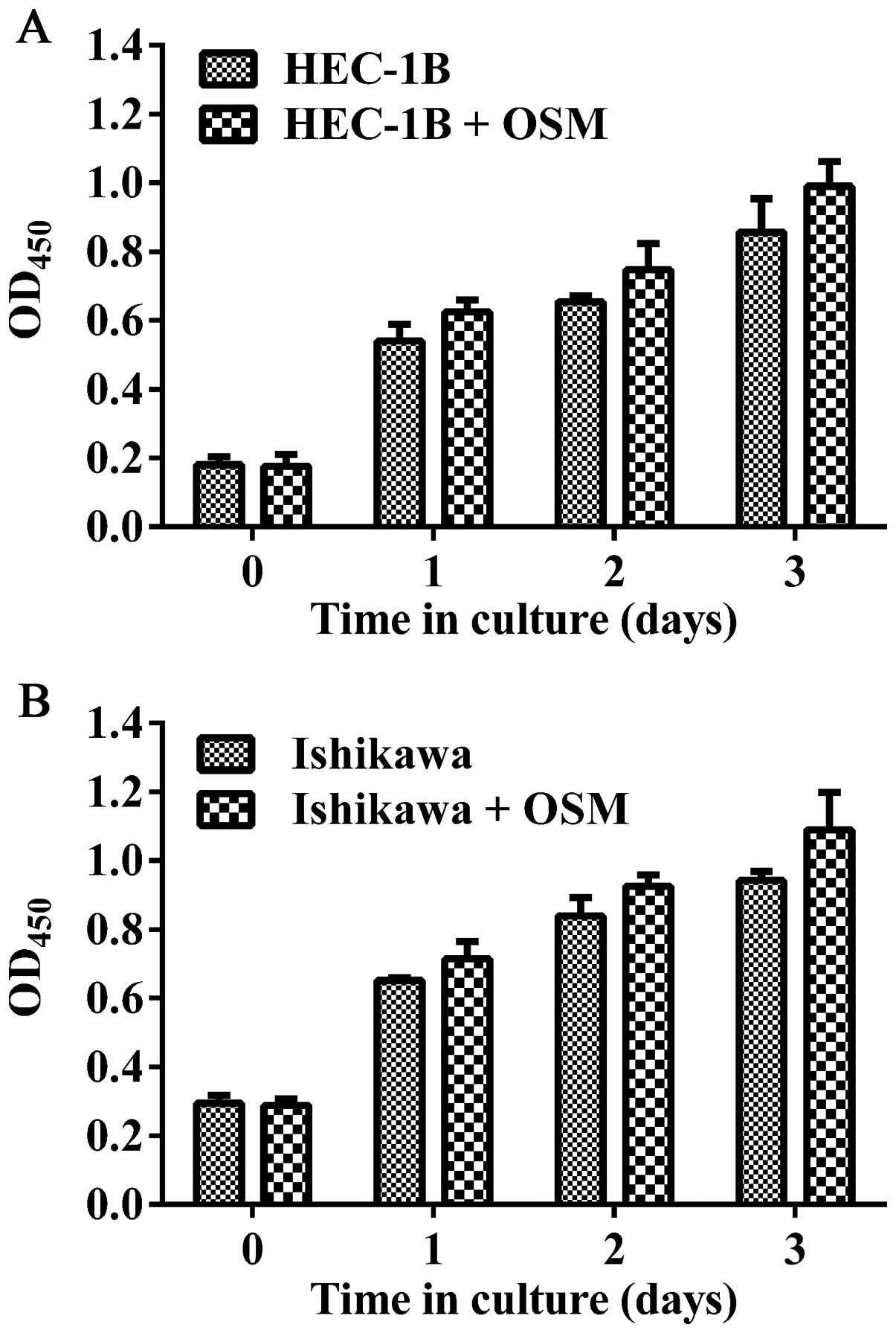
rhOSM does not directly promote cell
viability in vitro
To investigate the potential role of rhOSM in EC
cell proliferation, HEC-1B and Ishikawa cells were stimulated with
rhOSM (Fig. 3). CCK8 proliferation
assays revealed that rhOSM does not have a direct effect on HEC-1B
or Ishikawa cell growth.
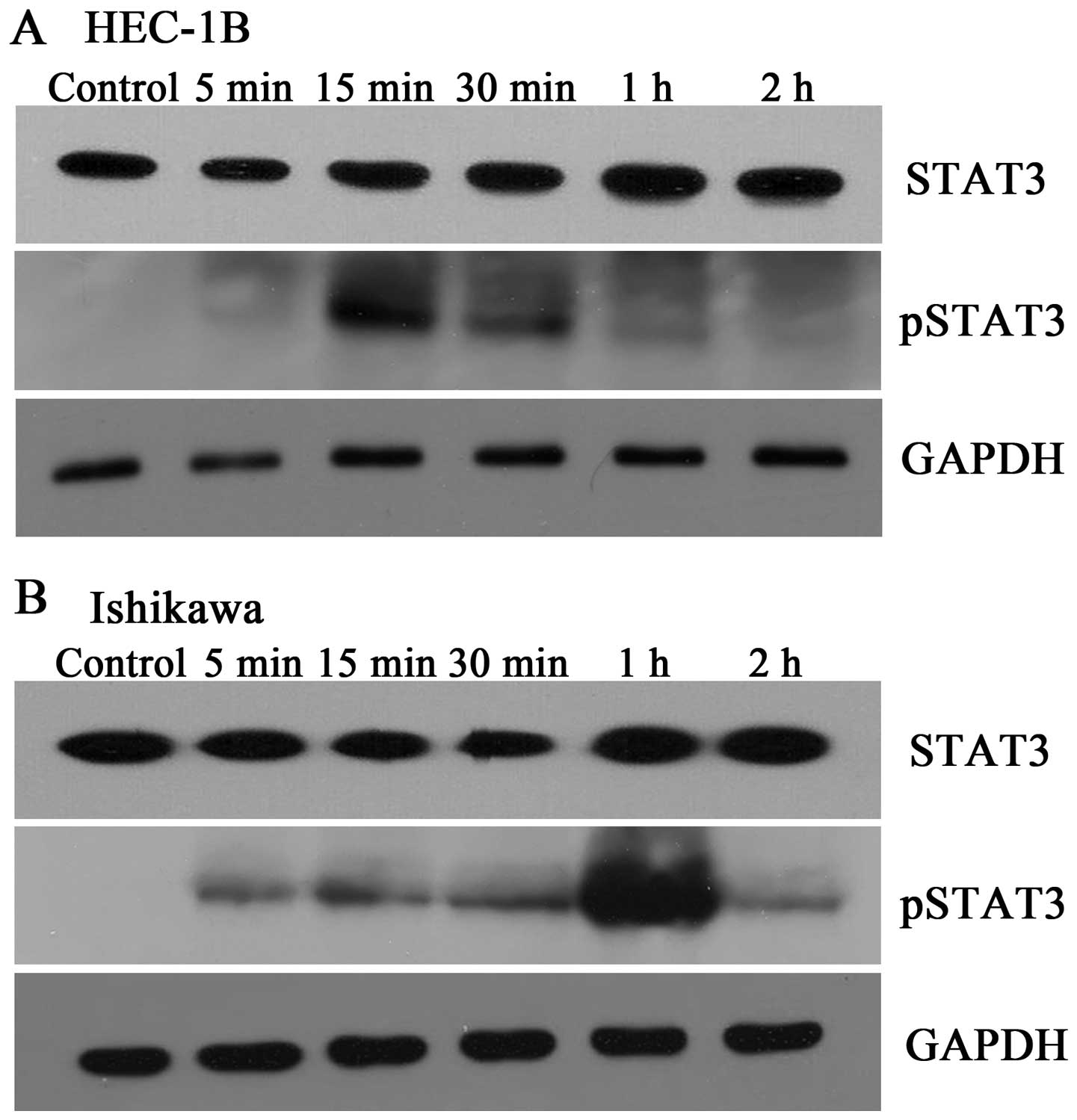
rhOSM upregulates STAT3 activity in EC
cell lines
Although activation of STAT3 by rhOSM occurs in
several cell types, it is unclear whether rhOSM can lead to STAT3
activation in EC cells. Thus, EC cells were serum starved and then
stimulated with rhOSM (40 ng/ml) for 0 min, 5 min, 15 min, 30 min,
1 h, or 2 h before cells were collected for western blotting
(Fig. 4). Basal levels of
phosphorylated STAT3 were very low in both cell lines; however,
stimulation with rhOSM led to an increase in STAT3 phosphorylation.
The peak values appeared after a 15-min (Fig. 4A) and 1-h (Fig. 4B) exposure to rhOSM in HEC-1B and
Ishikawa cells, respectively. The total STAT3 protein levels
remained fairly stable throughout the time points measured.
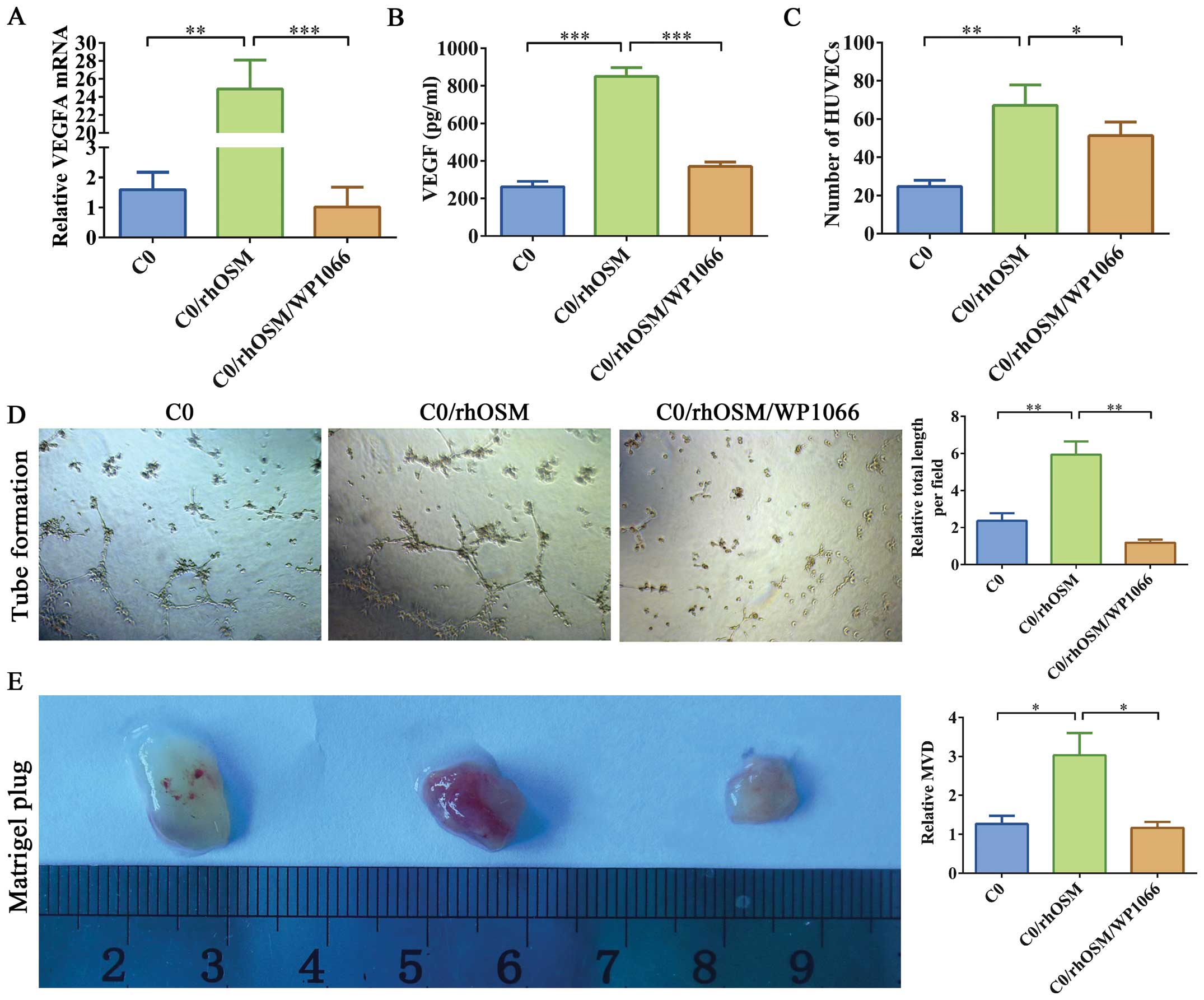
OSM increased VEGF secretion in EC
cells
VEGF is the main STAT3-regulated gene product
(20). Thus, we assessed the impact
of OSM on VEGF expression in HEC-1B cells. Based on qRT-PCR, VEGFA
mRnA expression increased in HEC-1B cells after rhOSM stimulation
(Fig. 5A). We also detected VEGF
secretion in the CM of HEC-1B cells via ELISA. rhOSM stimulation
increased VEGF secretion in HEC-1B cells (Fig. 5B). However, VEGF secretion was not
increased in cells treated with rhOSM and WP1066.
STAT3 activation mediates OSM-induced
angiogenesis
VEGF is a major regulator of tumor angiogenesis
(21). To further investigate
whether rhOSM affects VEGF-based angiogenesis in ECs, we used three
different approaches to investigate whether and how OSM could
regulate the behavior of endothelial cells. CM was collected from
HEC-1B cells cultured with C0, C0/rhOSM, or C0/rhOSM/WP1066 for 24
h. First, to investigate whether rhOSM altered the functional
behavior of endothelial cells, we used a tube formation assay.
HUVECs were placed on a basement membrane matrix in the presence of
CM. Tube formation was quantified by averaging the total length of
tubes in three randomly chosen microscope fields. CM from HEC-1B
cells stimulated by rhOSM significantly promoted tube formation
(Fig. 5D). Second, we tested
whether HUVEC migration ability was affected by differing
conditions with the use of a transwell chamber assay. The migration
ability of HUVECs increased in the C0/rhOSM group, whereas addition
of WP1066 partially inhibited these effects (Fig. 5C). Thus, OSM may act as a
pro-angiogenesis molecule in EC cells by promoting the migration of
vascular endothelial cells.
To further confirm this effect in vivo, we
performed a mouse angiogenic Matrigel plug assay. We mixed HEC-1B
CM with growth factor-reduced Matrigel and injected the resulting
plugs into mouse flanks. The plugs were removed 18 days after
injection (Fig. 5E) and were
stained with H&E (data not shown) or immunostained for CD34.
The inclusion of C0/rhOSM CM led to significantly induced blood
vessel formation in Matrigel, whereas the C0/rhOSM/WP1066 CM
inhibited these effects. These results suggested that rhOSM
stimulation leads to a large increase in angiogenic events and that
these events are STAT3 dependent.
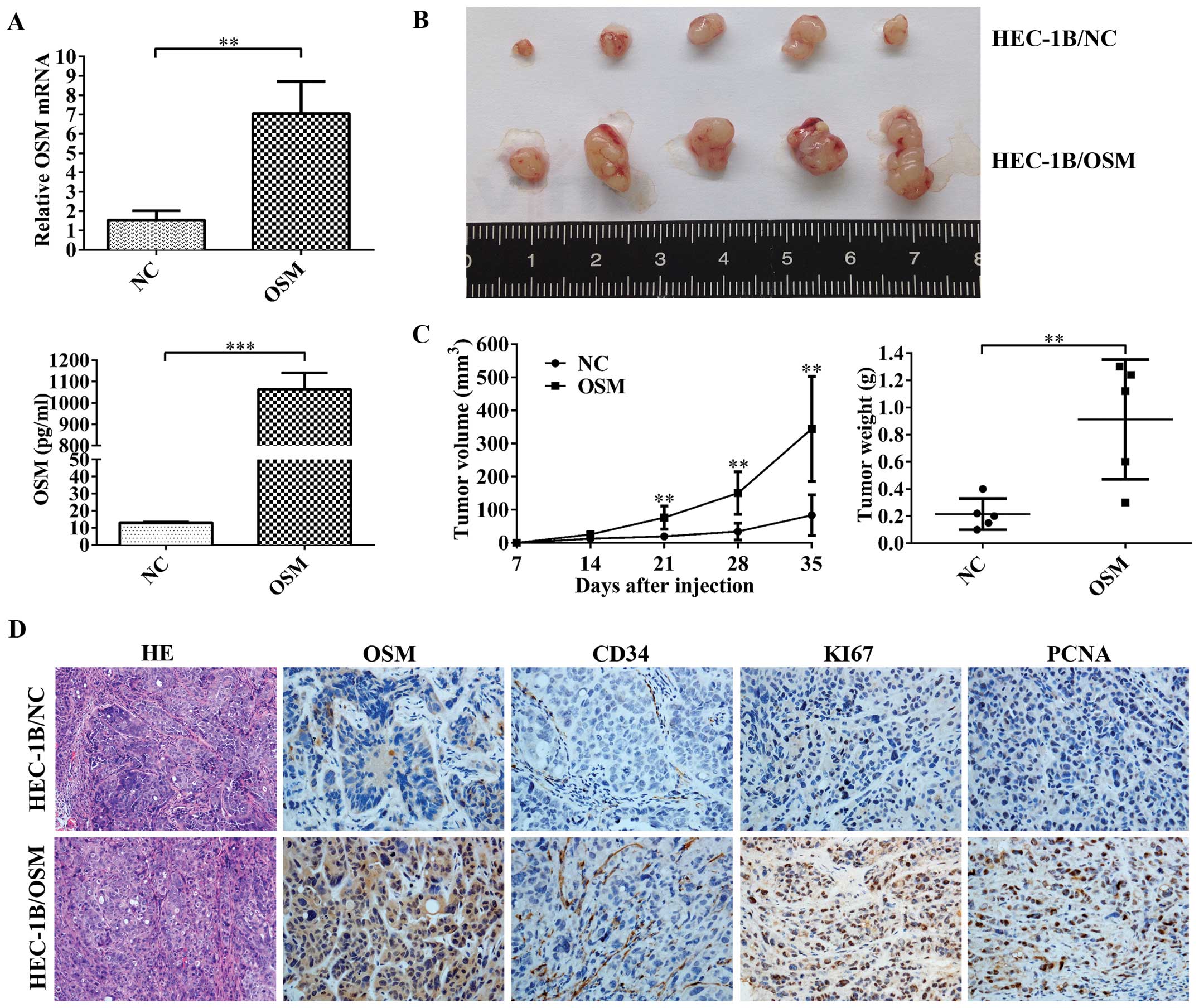
Oncogenic role of OSM in an in vivo tumor
xenograft model
To further assess the role of OSM in the progression
of EC, we performed tumorigenicity assays in nude mice. We
constructed HEC-1B cell lines that were stably transfected with
plasmid overexpressing either OSM or nC. The transfected cells were
then injected into the mice subcutaneously. Five weeks after
injection, the sizes and weights of tumors were significantly
larger in the OSM-overexpressing group as compared with the nC
group (Fig. 6B and C). Tumor
tissues were then embedded in paraffin, sectioned, and stained with
H&E and immunolabeled with antibodies against OSM, CD34, Ki67,
or PCNA (Fig. 6D). Ki67 and PCNA
expression was used to assess proliferation indices. Increased
expression of Ki67 and PCNA in the OSM-overexpressing group was
consistent with larger tumor volumes in these animals (Fig. 6B-D). Taken together, these results
suggested an important role for OSM in regulating tumor growth in
EC.
Discussion
The expression of OSM has been studied in several
human malignancies and cell types, and the results suggest it has a
dual function (22). However, the
role of OSM expression or activity in EC remains unclear. Herein,
we show that OSM expression was significantly increased in human
atypical endometrial hyperplasia and invasive cancers as compared
with normal endometrium. Moreover, increased OSM expression levels
were positively correlated with the depth of myometrial invasion,
disease stage, histological grade and lymph node metastasis of ECs,
which are important prognostic factors (23). These data suggest that
overexpression of OSM contributes to the progression of human
EC.
Solid tumor growth is largely dependent on
successful neovascularization, and several factors, the most
notable of which are VEGFs, promote tumor angiogenesis. Our
findings that VEGF secretion and VEGFA mRnA levels were correlated
with different proangiogenic abilities in the CM from the HEC-1B EC
cell line provide further support for the involvement of this
mechanism. Increased VEGF expression is found in 56–66% of EC
tumors (24). VEGFA expression is
an independent predictor of a poor prognosis in patients with EEC
(24–26). Based on the above results, we
suggest that OSM may play an important role in angiogenesis
regulation in EC.
STAT3 is a 92-kDa transcription factor, whose gene
is located on chromosome 17q21 in humans. Activated STAT3 forms
homo- and heterodimers that translocate to the nucleus, bind to the
promoter region of specific target genes, and regulate gene
transcription. Overexpression of STAT3 was found in several
cancers, such as lung (27,28), head and neck (29), stomach (30,31),
and colorectal (32,33) carcinomas. Previous studies have
shown that in certain cell lines, OSM induces the activation of
STAT3 (34).
In our study, we demonstrated that in HEC-1B and
Ishikawa cells, STAT3 was activated after exposure to rhOSM. We
further investigated the effects of rhOSM on the migration and
invasion of EC cells and found that rhOSM markedly promoted cell
migration and invasion in HEC-1B and Ishikawa cells. These findings
are consistent with our immunohistochemical analysis, which showed
that higher expression of OSM is found in tumors that displayed a
greater depth of myometrial invasion. However, the effect was
reversed when WP1066 was added. This suggests that OSM-induced
migration and invasion are both regulated by the signaling of
OSM-induced STAT3 activation. We also showed that rhOSM promoted
VEGF secretion in HEC-1B cells and that the CM taken from these
treated cells led to an increase in tube length in HUVECs in the
tube formation assay and increased MVD in plugs in the mouse
Matrigel plug assay. This phenomenon was reversed when the cells
were exposed to WP1066. Since activated STAT3 binds to the VEGF
promoter (15), rhOSM may activate
STAT3, which thereby induces VEGF expression. However, our data do
not completely rule out the possibility that other signaling
pathways, in addition to STAT3, are involved. The identification of
additional rhOSM-induced pathways and the determination of the
specific molecular mechanism responsible for the OSM-STAT3
interaction are aspects that warrant further study.
Whereas some studies have confirmed that OSM
inhibits proliferation in melanoma, osteosarcoma, and neuroblastoma
cancer cells (35,36), other studies have shown a direct
pro-proliferative effect on Kaposi’s sarcoma and Ewing sarcoma
cells (37,38). In our study, OSM did not directly
stimulate proliferation of either HEC-1B or Ishikawa cells in
vitro; however, in the mouse xenograft model, it was able to
promote proliferation and resulted in larger tumor volumes. Recent
research shows that overexpression of OSM promotes tumor growth,
and does so through alteration of the tumor environment with the
accumulation of M2 macrophages (39). Whether this kind of mechanism exists
in ECs is still unclear and requires further investigation.
In summary, our study demonstrates that OSM plays
important roles during EC progression, especially in the regulation
of angiogenesis. These findings suggest that OSM may be a valuable
prognostic biomarker for EC progression and that it could offer a
novel potential therapeutic strategy for ECs. Although our results
are promising, additional investigations are required to further
define the long-term consequences of anti-OSM treatment.
Acknowledgments
We thank Yue-qin Tang and Yan Hong (Center
Laboratory of Shanghai First People’s Hospital Affiliated to
Shanghai Jiao Tong University, Shanghai, China) and Xiao-ying He
(Department of Obstetrics and Gynecology, International Peace
Maternity and Child Health Hospital Affiliated to Shanghai Jiao
Tong University, Shanghai, China) for excellent technical
assistance. This study was supported by the national natural
Science Foundation of China (no. 81472427, no. 81272885, no.
81172476), Doctorial Innovation Fund of Shanghai Jiao Tong
University School of Medicine (no. BXJ201338).
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kandoth C, Schultz N, Cherniack AD, Akbani
R, Liu Y, Shen H, Robertson AG, Pashtan I, Shen R, Benz CC, et al
Cancer Genome Atlas Research Network: Integrated genomic
characterization of endometrial carcinoma. Nature. 497:67–73. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gadducci A, Cosio S and Genazzani AR: Old
and new perspectives in the pharmacological treatment of advanced
or recurrent endometrial cancer: Hormonal therapy, chemotherapy and
molecularly targeted therapies. Crit Rev Oncol Hematol. 58:242–256.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lahiri T, Laporte JD, Moore PE, Panettieri
RA Jr and Shore SA: Interleukin-6 family cytokines: Signaling and
effects in human airway smooth muscle cells. Am J Physiol Lung Cell
Mol Physiol. 280:L1225–L1232. 2001.PubMed/NCBI
|
|
5
|
Grenier A, Dehoux M, Boutten A,
Arce-Vicioso M, Durand G, Gougerot-Pocidalo MA and Chollet-Martin
S: Oncostatin M production and regulation by human
polymorphonuclear neutrophils. Blood. 93:1413–1421. 1999.PubMed/NCBI
|
|
6
|
Horn D, Fitzpatrick WC, Gompper PT, Ochs
V, Bolton-Hansen M, Zarling J, Malik N, Todaro GJ and Linsley PS:
Regulation of cell growth by recombinant oncostatin M. Growth
Factors. 2:157–165. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Douglas AM, Goss GA, Sutherland RL, Hilton
DJ, Berndt MC, Nicola NA and Begley CG: Expression and function of
members of the cytokine receptor superfamily on breast cancer
cells. Oncogene. 14:661–669. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Brounais B, Chipoy C, Mori K, Charrier C,
Battaglia S, Pilet P, Richards CD, Heymann D, Rédini F and
Blanchard F: Oncostatin M induces bone loss and sensitizes rat
osteosarcoma to the antitumor effect of Midostaurin in vivo. Clin
Cancer Res. 14:5400–5409. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Godoy-Tundidor S, Cavarretta IT, Fuchs D,
Fiechtl M, Steiner H, Friedbichler K, Bartsch G, Hobisch A and
Culig Z: Interleukin-6 and oncostatin M stimulation of
proliferation of prostate cancer 22Rv1 cells through the signaling
pathways of p38 mitogen-activated protein kinase and
phosphatidylinositol 3-kinase. Prostate. 64:209–216. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li Q, Zhu J, Sun F, Liu L, Liu X and Yue
Y: Oncostatin M promotes proliferation of ovarian cancer cells
through signal transducer and activator of transcription 3. Int J
Mol Med. 28:101–108. 2011.PubMed/NCBI
|
|
11
|
Mori S, Murakami-Mori K and Bonavida B:
Oncostatin M (OM) promotes the growth of DU 145 human prostate
cancer cells, but not PC-3 or LNCaP, through the signaling of the
OM specific receptor. Anticancer Res. 19:1011–1015. 1999.PubMed/NCBI
|
|
12
|
Gearing DP and Bruce AG: Oncostatin M
binds the high-affinity leukemia inhibitory factor receptor. New
Biol. 4:61–65. 1992.PubMed/NCBI
|
|
13
|
Gearing DP, Comeau MR, Friend DJ, Gimpel
SD, Thut CJ, McGourty J, Brasher KK, King JA, Gillis S, Mosley B,
et al: The IL-6 signal transducer, gp130: An oncostatin M receptor
and affinity converter for the LIF receptor. Science.
255:1434–1437. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mosley B, De Imus C, Friend D, Boiani N,
Thoma B, Park LS and Cosman D: Dual oncostatin M (OSM) receptors.
Cloning and characterization of an alternative signaling subunit
conferring OSM-specific receptor activation. J Biol Chem.
271:32635–32643. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Niu G, Wright KL, Huang M, Song L, Haura
E, Turkson J, Zhang S, Wang T, Sinibaldi D, Coppola D, et al:
Constitutive Stat3 activity up-regulates VEGF expression and tumor
angiogenesis. Oncogene. 21:2000–2008. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Folkman J: Angiogenesis in cancer,
vascular, rheumatoid and other disease. Nat Med. 1:27–31. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Risau W: Mechanisms of angiogenesis.
Nature. 386:671–674. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Creasman W: Revised FIGO staging for
carcinoma of the endometrium. Int J Gynaecol Obstet. 105:1092009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liao Y, Lu W, Che Q, Yang T, Qiu H, Zhang
H, He X, Wang J, Qiu M, Zou Y, et al: SHARP1 suppresses
angiogenesis of endometrial cancer by decreasing hypoxia-inducible
factor-1α level. PLoS One. 9:e999072014. View Article : Google Scholar
|
|
20
|
Calò V, Migliavacca M, Bazan V, Macaluso
M, Buscemi M, Gebbia N and Russo A: STAT proteins: From normal
control of cellular events to tumorigenesis. J Cell Physiol.
197:157–168. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kim KJ, Li B, Winer J, Armanini M, Gillett
N, Phillips HS and Ferrara N: Inhibition of vascular endothelial
growth factor-induced angiogenesis suppresses tumour growth in
vivo. Nature. 362:841–844. 1993. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tanaka M and Miyajima A: Oncostatin M, a
multifunctional cytokine. Rev Physiol Biochem Pharmacol. 149:39–52.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Uharcek P: Prognostic factors in
endometrial carcinoma. J Obstet Gynaecol Res. 34:776–783. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hirai M, Nakagawara A, Oosaki T, Hayashi
Y, Hirono M and Yoshihara T: Expression of vascular endothelial
growth factors (VEGF-A/VEGF-1 and VEGF-C/VEGF-2) in postmenopausal
uterine endometrial carcinoma. Gynecol Oncol. 80:181–188. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kamat AA, Merritt WM, Coffey D, Lin YG,
Patel PR, Broaddus R, Nugent E, Han LY, Landen CN Jr, Spannuth WA,
et al: Clinical and biological significance of vascular endothelial
growth factor in endometrial cancer. Clin Cancer Res. 13:7487–7495.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yokoyama Y, Sato S, Futagami M, Fukushi Y,
Sakamoto T, Umemoto M and Saito Y: Prognostic significance of
vascular endothelial growth factor and its receptors in endometrial
carcinoma. Gynecol Oncol. 77:413–418. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Haura EB, Zheng Z, Song L, Cantor A and
Bepler G: Activated epidermal growth factor receptor-Stat-3
signaling promotes tumor survival in vivo in non-small cell lung
cancer. Clin Cancer Res. 11:8288–8294. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jiang R, Jin Z, Liu Z, Sun L, Wang L and
Li K: Correlation of activated STAT3 expression with
clinicopathologic features in lung adenocarcinoma and squamous cell
carcinoma. Mol Diagn Ther. 15:347–352. 2011. View Article : Google Scholar
|
|
29
|
Seethala RR, Gooding WE, Handler PN,
Collins B, Zhang Q, Siegfried JM and Grandis JR:
Immunohistochemical analysis of phosphotyrosine signal transducer
and activator of transcription 3 and epidermal growth factor
receptor autocrine signaling pathways in head and neck cancers and
metastatic lymph nodes. Clin Cancer Res. 14:1303–1309. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gong W, Wang L, Yao JC, Ajani JA, Wei D,
Aldape KD, Xie K, Sawaya R and Huang S: Expression of activated
signal transducer and activator of transcription 3 predicts
expression of vascular endothelial growth factor in and angiogenic
phenotype of human gastric cancer. Clin Cancer Res. 11:1386–1393.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lee J, Kang WK, Park JO, Park SH, Park YS,
Lim HY, Kim J, Kong J, Choi MG, Sohn TS, et al: Expression of
activated signal transducer and activator of transcription 3
predicts poor clinical outcome in gastric adenocarcinoma. APMIS.
117:598–606. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lassmann S, Schuster I, Walch A, Göbel H,
Jütting U, Makowiec F, Hopt U and Werner M: STAT3 mRnA and protein
expression in colorectal cancer: Effects on STAT3-inducible targets
linked to cell survival and proliferation. J Clin Pathol.
60:173–179. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Morikawa T, Baba Y, Yamauchi M, Kuchiba A,
Nosho K, Shima K, Tanaka N, Huttenhower C, Frank DA, Fuchs CS, et
al: STAT3 expression, molecular features, inflammation patterns,
and prognosis in a database of 724 colorectal cancers. Clin Cancer
Res. 17:1452–1462. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fossey SL, Bear MD, Kisseberth WC, Pennell
M and London CA: Oncostatin M promotes STAT3 activation, VEGF
production, and invasion in osteosarcoma cell lines. BMC Cancer.
11:1252011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chipoy C, Berreur M, Couillaud S, Pradal
G, Vallette F, Colombeix C, Rédini F, Heymann D and Blanchard F:
Downregulation of osteoblast markers and induction of the glial
fibrillary acidic protein by oncostatin M in osteosarcoma cells
require PKCdelta and STAT3. J Bone Miner Res. 19:1850–1861. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chipoy C, Brounais B, Trichet V, Battaglia
S, Berreur M, Oliver L, Juin P, Rédini F, Heymann D and Blanchard
F: Sensitization of osteosarcoma cells to apoptosis by oncostatin M
depends on STAT5 and p53. Oncogene. 26:6653–6664. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
David E, Tirode F, Baud’huin M, Guihard P,
Laud K, Delattre O, Heymann MF, Heymann D, Redini F and Blanchard
F: Oncostatin M is a growth factor for Ewing sarcoma. Am J Pathol.
181:1782–1795. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Amaral MC, Miles S, Kumar G and Nel AE:
Oncostatin-M stimulates tyrosine protein phosphorylation in
parallel with the activation of p42MAPK/ERK-2 in Kaposi’s cells.
Evidence that this pathway is important in Kaposi cell growth. J
Clin Invest. 92:848–857. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lauber S1, Wong S, Cutz JC, Tanaka M,
Barra N, Lhoták S, Ashkar A and Richards CD: Novel function of
Oncostatin M as a potent tumour-promoting agent in lung. Int J
Cancer. 136:831–843. 2015. View Article : Google Scholar
|