Introduction
The indole chemical moiety is a backbone for several
bioactive compounds including the amino acid tryptophan, the
signaling compound melatonin, the nutritional compound
indole-3-carbinol (I3C), and several receptor or kinase agonists or
antagonists. I3C, abundant in cruciferous vegetables, is suggested
to be one of the most active ingredients responsible for the
anticancer benefits of such vegetables (1,2). Upon
exposure to acidic environments, such as that in the stomach, I3C
is converted into derivatives with variable stability and
bioactivity (3). Diindolylmethane
(DIM), an acid condensate of I3C, is thought to be one of the
bioactive derivatives (4). I3C has
antimicrobial activity against Staphylococcus,
Enterococcus, Escherichia, and Pseudomonas
microorganisms (5).
Indole compounds have potential as chemopreventive
and chemotherapeutic agents. Cellular targets identified for
natural or derivatized indole compounds include PPARγ and Nur77
(6), transcription factors
(7–10), cyclin-dependent kinase (CDK)
complexes (11,12), PKB/Akt (13,14),
and hormone receptors (15,16). Indole compounds have activity
against colon cancer cells, suggesting their potential use in
chemoprevention or therapy (17–20). A
full description of cellular targets and potential mechanisms of
actions of indole compounds is available (21,22).
Despite the biological relevance of indole
compounds, the bioactivities of many indole derivatives, especially
those related to I3C, remain unknown. To evaluate the activities of
indoles with a structural relationship to I3C, 14 compounds were
selected from an indole library and their effects were tested on
cells derived from human colon cancers. After an initial screening
of these at 50 µm, BEI-9 was identified as a potent inhibitor of
cell proliferation. We also identified BEI-9 as an inhibitor of the
NF-κB signaling pathway at submicromolar concentrations. A
preliminary test to determine a safe dose to mice showed that BEI-9
could be administered at doses below 10 mg/kg without obvious
pathological changes or toxicological signs. These results suggest
that BEI-9 and its derivatives or analogues could be developed into
bioactive drug entities.
Materials and methods
Cell culture
SW480 and HCT116 cells were purchased from the
American Type Culture Collection (ATCC) and maintained in McCoy’s
5A medium containing antibiotics and fetal bovine serum (FBS).
Luciferase reporter cells were generated and used for experiments
as described previously (23).
HepG2 human liver carcinoma cells were obtained from the ATCC and
grown in dulbecco’s modified Eagle’s medium (DMEM) supplemented
with 10% FBS, 100 U/ml penicillin, 100 µg/ml streptomycin, 2
mM L-glutamine, and 1 mM sodium pyruvate. The assay media included
phenol red-free DMEM supplemented with 5% charcoal/dextran-treated
FBS and the other additives. All cells were cultured in an
incubator with a humidified atmosphere under 5% Co2 and
95% air at 37°C.
Chemicals and plasmids
Dimethyl sulfoxide (DMSO), rifampicin, and SR12813
were purchased from Sigma-Aldrich (St. Louis, MO, USA).
pcdna3-human pregnane X receptor (hPXR) and pGL3-CYP3A4-luc
plasmids were as previously described (24,25).
hPXR transactivation assays
HepG2 cells were transfected with pGL3-CYP3A4-luc
reporter and pcDNA3-hPXR plasmids using FuGENE 6 (Promega, Madison,
WI, USA). After 24 h of transfection in growth media,
104 cells in the assay media were plated into 96-well
culture plates (PerkinElmer) and exposed to DMSO (vehicle) or a PXR
agonist, rifampicin or SR 12813, for an additional 20 h. At 10 min
before the luciferase activity assay using the Neolite Reporter
Gene Assay system (PerkinElmer), DMSO or BEI-9 (10 µM) was
added to the cells, which were incubated at 37°C and room
temperature for 5 min each. Luminescence was measured with a
FLUOstar Optima microplate reader (BMG Labtech).
MTS and CellTiter-Glo assays
[3-(4,5-Dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl]-2H-tetrazolium
(MTS) cell proliferation assay and CellTiter-Glo®
Luminescent Cell Viability Assay kit (Promega) were used, according
to the manufacturer’s instructions, to evaluate the viability of
cancer cells. For both assays, 104 cells/well of 96-well
plates were exposed to the test compounds for 24 or 48 h, after
which the assays were performed. Readings from vehicle-treated
cells were used to normalize the data. The results were expressed
as viability indices representing relative percentages compared to
the controls. For experiments with HepG2 cells, cells in the assay
media were plated into 96-well culture plates (PerkinElmer) at a
density of 104 cells/well and exposed to DMSO or a PXR
agonist (rifampicin or SR12813) for 20 h. BEI-9 (10 µM) was
added to the cells 10 min prior to measuring the luminescence with
the CellTiter-Glo luminescent assay system and a FLUOstar Optima
microplate reader.
Microscopy
Phase-contrast images of cells were captured at x20
magnification (and a 10x eyepiece) using an Olympus IX71 inverted
microscope fitted with a digital camera equipped with
CellSens® Image Capture software (Olympus America, Inc.,
Center Valley, PA, USA). Images were stored in TIFF format and
subsequently cropped and resized using Microsoft PowerPoint.
Cell cycle analysis
Cells were prepared for flow cytometry as described
previously (26). Cells were
harvested by trypsinization with 0.25% trypsin-EDTA (Invitrogen
Corp., Carlsbad, CA, USA) and then centrifuged. Pellets were
suspended in 300 µl of phosphate-buffered saline (PBS;
Invitrogen Corp.) and fixed by addition of 700 µl of 100%
ethanol while vortexing. Next, the cells were stored at −20°C for a
minimum of 12 h. Fixed cells were centrifuged and stained in FACS
staining solution (320 mg/ml RNase A and 0.4 mg/ml propidium
iodide) in PBS without calcium and magnesium. Stained cells were
filtered through 70-µm filters and analyzed by flow
cytometry on a C6 Accuri® flow cytometer (Accuri
Cytometers, Ann Arbor, MI, USA). Data were analyzed, and histograms
were prepared using CFlow™ software (Accuri Cytometers).
Scratch wound assays
Cells were grown to confluency in 6-well plates, and
the monolayers were wounded by scratching the layer of cells with
the tip of a 200-µl pipette. Scratch positions were
microphotographed every 24 h for 96 h. At 48-h intervals, depleted
culture medium was replaced with fresh medium containing the same
concentration of BEI-9. Widths of the wound gaps were measured by
an electronic micrometer scale, and the results were plotted on a
graph.
NF-κB reporter luciferase assays
For luciferase assays, cells were seeded and treated
in 96-well plates. Before reading the plates, the culture medium
was removed by aspiration, and 50 µl of 1X luciferin-PBS
substrate solution was added to each well. With a luminometer set
at 37°C, plates were read immediately after addition of substrate
solution and subsequently after 5 and 10 min. The time-points at
which peak readings for the wells were obtained were selected for
calculation of relative luciferase units (RLU). Luciferase
expression was quantified as RLU, normalized to readings of control
wells, and expressed as relative NF-κB reporter activity.
Immunoblotting
Cell lysates were prepared in RIPA cell lysis buffer
(Sigma-Aldrich) containing a protease inhibitor cocktail. Protein
concentrations were determined using a detergent-compatible protein
assay (Bio-Rad Laboratories Inc., Hercules, CA, USA). Samples
containing equivalent protein concentrations were mixed with
Laemmli’s buffer and boiled for 5 min. Proteins were resolved by
SDS-PAGE, transferred to PVDF membranes (GE Healthcare Life
Sciences, Piscataway, NJ, USA), and blocked in 5% non-fat dry milk.
Primary antibodies for cyclin A (Upstate Biotechnology, Lake
Placid, NY, USA) and cyclin D (Millipore, Darmstadt, Germany) were
used at 1:1,000 dilutions. Mouse anti-tubulin antibody was
purchased from Sigma-Aldrich. Peroxidase-conjugated anti-rabbit and
anti-mouse IgG secondary antibodies were purchased from GE
Healthcare Life Sciences and used at 1:5,000 dilutions.
Chemiluminescent detections were performed with Classico or
Crescendo Premixed Chemiluminescent HRP substrates (Millipore,
Billerica, MA, USA).
Experiments with mice
All animal procedures were approved by the Tuskegee
University Animal Care and Use Committee. As a prelude to
performing anticancer studies on BEI-9, a safe-to-administer dose
for nude mice was determined by intraperitoneal injection of BEI-9
to nude mice at 6 weeks of age. To prepare the injections, volumes
of BEI-9 stock solution were mixed with sterile PBS to a final
volume of 1 ml. Solutions (100 µl) containing the desired
BEI-9 concentrations were injected. As controls, DMSO injections
were prepared similarly. Portions of these solutions prepared for
injections were saved to determine if the amounts used for
injection of mice were effective in vitro. Initially, small
numbers of mice were injected once with a dose of 1, 50, or 100
mg/kg of BEI-9. After elimination of single injections of 50 and
100 mg/kg doses due to toxic outcomes, additional mice were
administered the vehicle or 1, 5, or 10 mg/kg of BEI-9 once a week
for 6 weeks. Body weights and general conditions of health were
followed to assess any unusual effects of the treatment.
Results
BEI-9 inhibits cell proliferation
A subset of 14 compounds (Table I) from a commercial indole library
(Sigma-Aldrich) was screened by use of cell viability assays. To
measure their bioactivities, a starting concentration of 50
µM was tested. Two independent assays for cell viability,
MTS assays and CellTiter-Glo assays, were performed. The MTS assay
measures a reduction in tetrazolium dye by NAD(P) H-dependent
oxidoreductases, whereas the CellTiter-Glo assay is dependent on
the relative amount of ATP available in the cells, which is
required for the activity of the enzyme luciferase to catalyze a
luminescent reaction. BEI-9 was the most potent, with activity
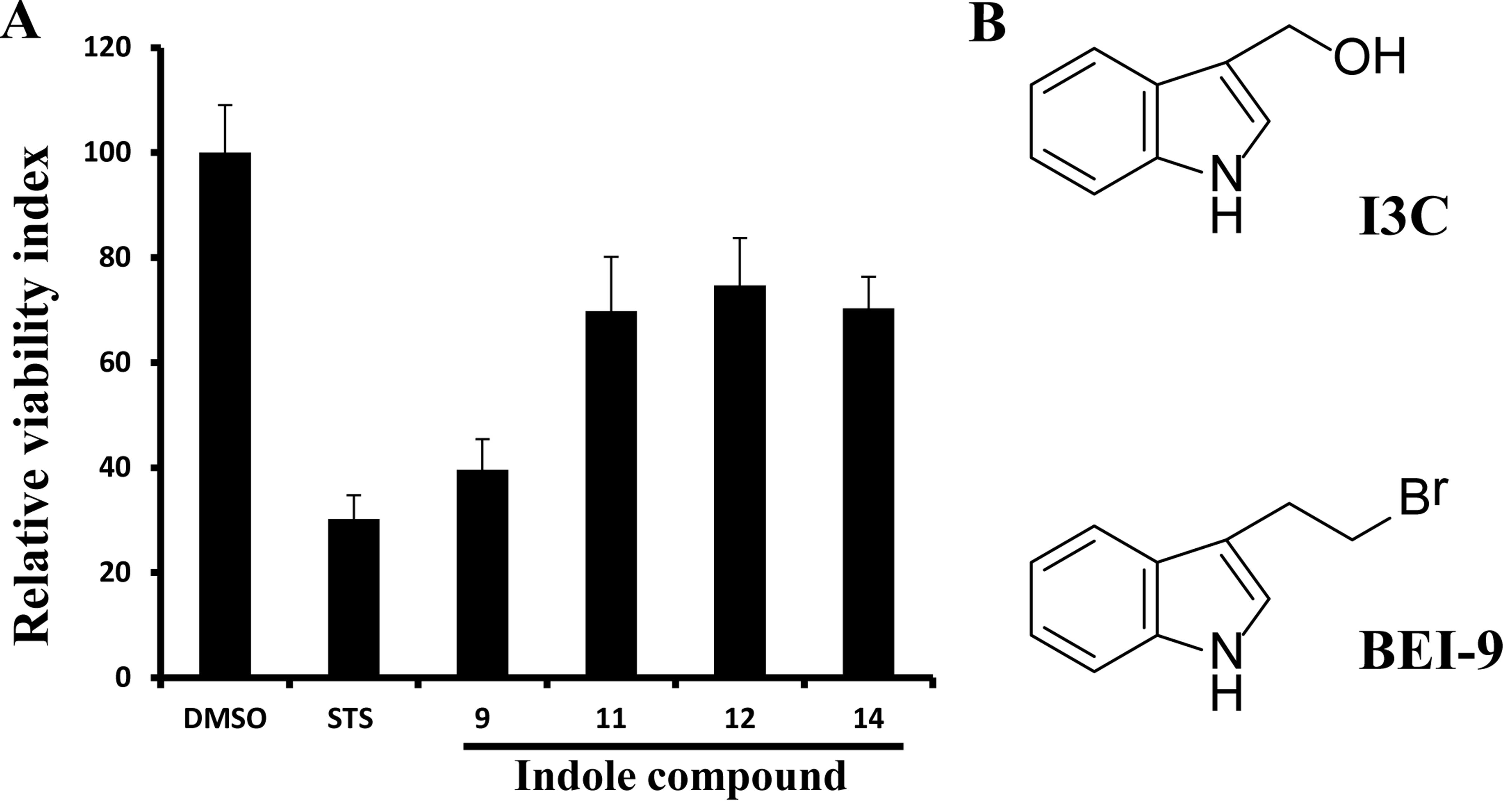
greater than I3C. In the 24-h assay (Fig. 1A), BEI-9 reduced the proliferation
index of SW480 cells by ~60%, relative to 30% for I3C and was
chosen for further testing. The structures of I3C and BEI-9 are
shown in Fig. 1B.
 | Table IFourteen indole compounds tested for
bioactivity on cancer cells at a concentration of 50 µM. |
Table I
Fourteen indole compounds tested for
bioactivity on cancer cells at a concentration of 50 µM.
| Chemical name | Designation |
|---|
|
5-Methoxyindole | Indole-1 |
| 5-Chloroindole-3
carboxaldehyde | Indole-2 |
|
Indole-5-carboxaldehyde | Indole-3 |
|
1-Methylindole-2-carboxaldehyde | Indole-4 |
| Indole-4-carboxylic
acid | Indole-5 |
|
Ethyl-5-hydroxy-2-methylindole-3-carboxylate | Indole-6 |
|
5-Bromoindole-3-acetic acid | Indole-7 |
|
Methyindole-3-carboxylate | Indole-8 |
| 3-(2-Bromoethyl)
indole | BEI-9 |
| Indole-3-carboxylic
acid | Indole-10 |
|
Indole-3-carboxyladehyde | Indole-11 |
|
6-Methoxyindole | Indole-12 |
|
5-Hydroxy-indole-3-acetic acid | Indole-13 |
|
Indole-3-carbinol | Indole-14 |
To determine whether the reduction in the
proliferation index by BEI-9 was due to cell death or to decreased
proliferation, cells were seeded in 24-well dishes at
104 cells/well and exposed to either the vehicle (DMSO)
or BEI-9 (50 µM). Phase-contrast images of the cells were
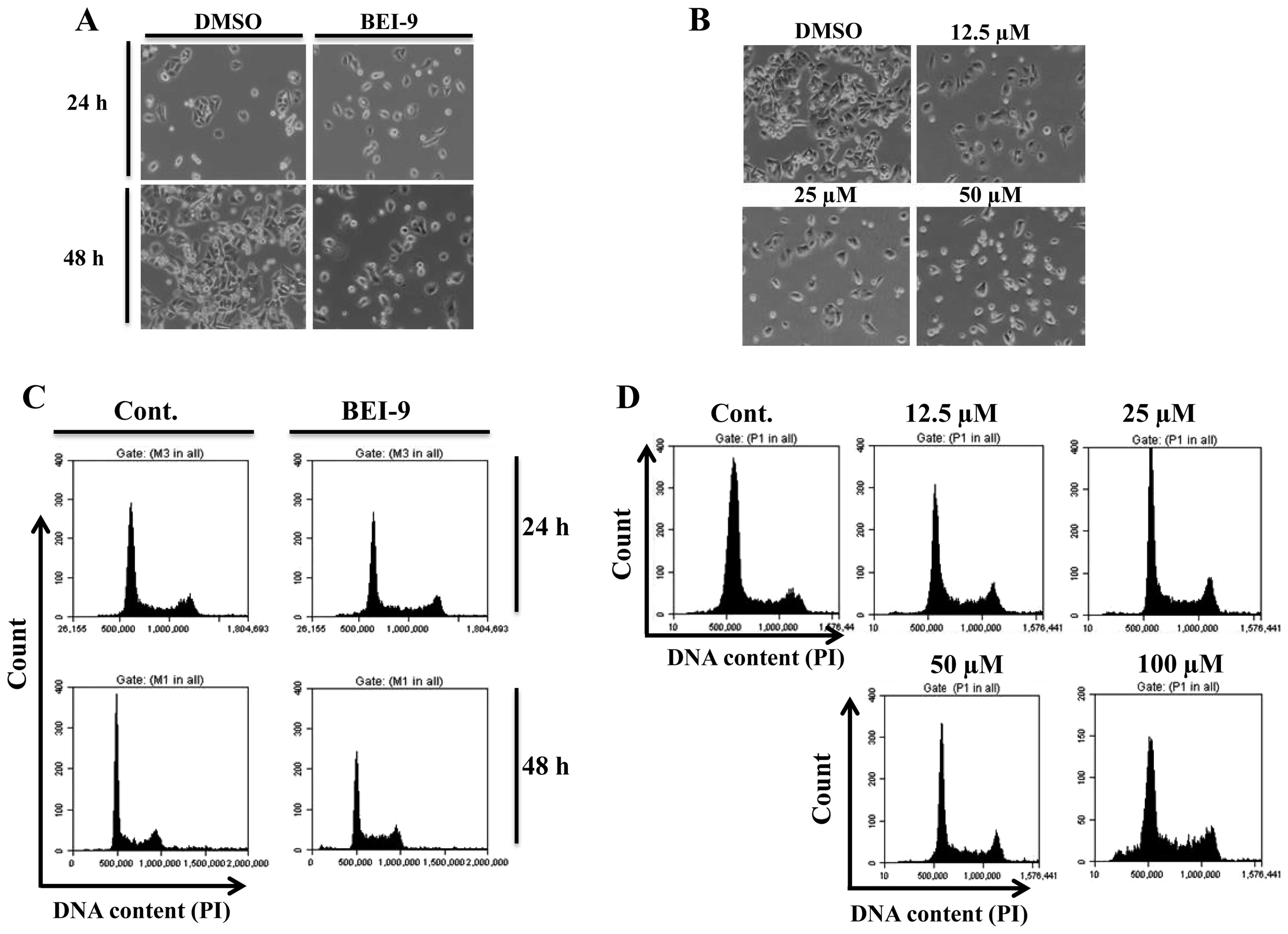
recorded at 24 and 48 h after treatment. The monolayers were not
washed, in order to preserve any dead cells. BEI-9-treated SW480
cells failed to proliferate, but vehicle-treated cells continued to
proliferate, an effect most evident at 48 h (Fig. 2A). Since there was no increase in
the number of floating (detached or dead) cells, BEI-9 apparently
blocked SW480 cell proliferation without causing cell death. A
dose-response experiment involving concentrations ranging from 12.5
to 50 µM indicated that amounts <50 µM inhibited
cell proliferation (Fig. 2B).
Furthermore, the possibility of inhibited cell proliferation
without death of SW480 cells was assessed by cell cycle analysis of
cells exposed to BEI-9 (50 µM) for 24 or 48 h. In agreement
with the phenotypic observation, the cell cycle profiles of
BEI-9-treated cells were indistinguishable from the control cells
at 24 and 48 h after treatment (Fig.
2C). SW480 cells were also exposed for 24 h to BEI-9
concentrations ranging from 12.5 to 100 µM, and their cell
cycle profiles were analyzed by flow cytometry. Evidence for
increased cell death was evident only at 100 µM (Fig. 2D). Since such a high concentration
is unlikely to be achieved in animals, the primary mechanism of
action of BEI-9 apparently does not involve induction of cell
death.
BEI-9 inhibits cell motility in wound
scratch assays
Since BEI-9 is an inhibitor of cell proliferation,
its effects on cell motility, as determined by a scratch wound
assay, were assessed. SW480 cells were grown to 100% confluency and
wounded by scratching the monolayers with a pipette tip. Cells were
exposed to BEI-9 (25 µM) or DMSO, the vehicle. Healing of
the wound by migration of cells from the wound edge was followed by
imaging at 24-h intervals. The DMSO-treated cells migrated, closing
the wound gap within 96 h (Fig. 3A and
B). In contrast, BEI-9-treated cells failed to close the gap.
Thus, BEI-9 inhibited the migratory capacity of these cells.
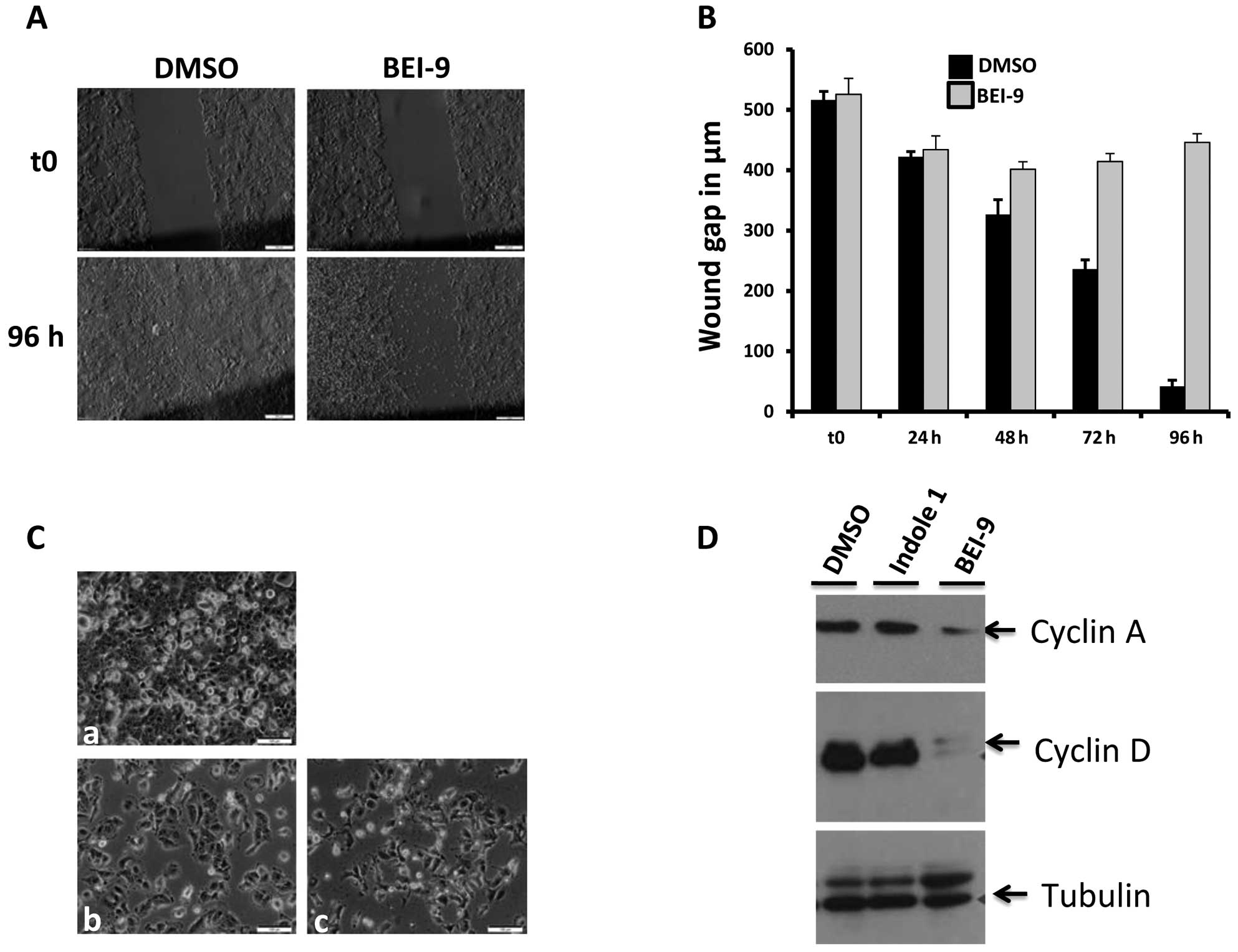 | Figure 3Effects of BEI-9 on motility,
survival, recovery from treatment, and cyclin levels in treated
cells. (A and B) The scratch wound assay was performed as described
in Materials and methods. Phase contrast images of the wounded and
then dimethyl sulfoxide (DMSO) or BEI-9 treated SW480 cell
monolayers were taken at 24-h intervals. Panel a shows images of
the same spots taken immediately after scratching (upper panels)
and then 96 h after the first treatment (lower panels). Panel B
shows widths of scratch wound gaps measured at 24-h intervals for
96 h. (C) Sparsely seeded SW480 cell monolayers were exposed to
either DMSO or BEI-9 (25 µM) for 48 h. Then the monolayers
were washed with culture medium and further incubated for up to 4
days. DMSO-treated cells became 100% confluent within 24 h of
washing (a), whereas BEI-9-treated cells did not recover from the
treatment, as shown on micrographs at the wash time (b) or 4 days
after washing (c). (D) SW480 cells were exposed to DMSO, indole 1
(25 µM, inactive), or BEI-9 (25 µM) for 24 h. Protein
levels of cyclin A or D were detected by immunoblotting. Tubulin
served as a loading control. (E and F) Effects of BEI-9 on HCT116
colon carcinoma cells. The viability of cells exposed for 24 h to
the indicated concentrations of BEI-9 was measured with the MTS
assay. Absorbance values at 490 nm wavelength are shown on the
y-axis (E). Photomicrographs of cells exposed for 48 h to DMSO or
BEI-9 (0.2, 1, 5, or 10 µM) are shown (F). |
SW480 cells fail to recover from the
effect of BEI-9
Since the phenotypic outcome of treating SW480 cells
with BEI-9 was to ‘freeze’ the cells at the status quo at the time
of treatment, it was possible that the cells, after a period of
exposure, could recover if the agent was removed from the culture
medium. SW480 cells were exposed to BEI-9 (25 µM) and left
in the medium for 48 h. After the treatment, cell monolayers were
washed 5 times with regular growth medium, and then left in the
same medium for up to 4 days. Vehicle-treated cells grew to
confluency in 48 h, but BEI-9-treated cells did not recover from
the treatment even at 4 days after withdrawal of the drug (Fig. 3C). Although the cells did not
recover and were more flattened, they maintained their adhesion to
the surface of the culture dish (Fig.
3C).
BEI-9 downregulates cyclin D1
Progression through the cell cycle is regulated by
cyclins and their CDKs. Regulation of cyclin-CDK activity is
controlled by synthesis and degradation of the cyclins. Cyclin D1,
a regulator involved in the G1-S transition of cells, is an
oncogene (27). Since it regulates
cell cycle progression, its expression could be affected by
treatment with BEI-9, partly accounting for lack of cell cycle
progression in treated cells. To this end, the levels of cyclin D1
protein were assessed in the control and treated cells by
immunoblotting. Also assessed were the levels of cyclin A, which
regulates both G1-S and G2-M transitions. BEI-9 treatment decreased
the expression of cyclin D1, as well as that of cyclin a (Fig. 3D), indicating that the ‘freeze’
effect of BEI-9 is mediated by inhibition of cell cycle progression
caused by inhibition of the expression of cyclins that drive the
transitions.
Similar to the effect for SW480, BEI-9, at 5 and 10
µM, decreased the viability of HCT116 colon cancer cells, as
determined by MTS assays (Fig. 3E),
and stopped their proliferation (Fig.
3F).
BEI-9 inhibits NF-κB signaling
pathway
Since the multifunctional transcription factor NF-κB
is a regulator of cyclin D1 (28),
its effects on NF-κB signaling in SW480 cells exposed to BEI-9 were
determined. To this end, NF-κB reporter SW480 cells (SW-NFL) stably
transduced with a construct containing NF-κB-response elements
linked to the luciferase gene as a reporter were used. As
demonstrated previously, these cells activate NF-κB in response to
TNFα and to some cancer chemotherapeutic drugs (23). As a response to NF-κB activation,
these cells express increased amounts of luciferase, which can be
measured by luminescence assays.
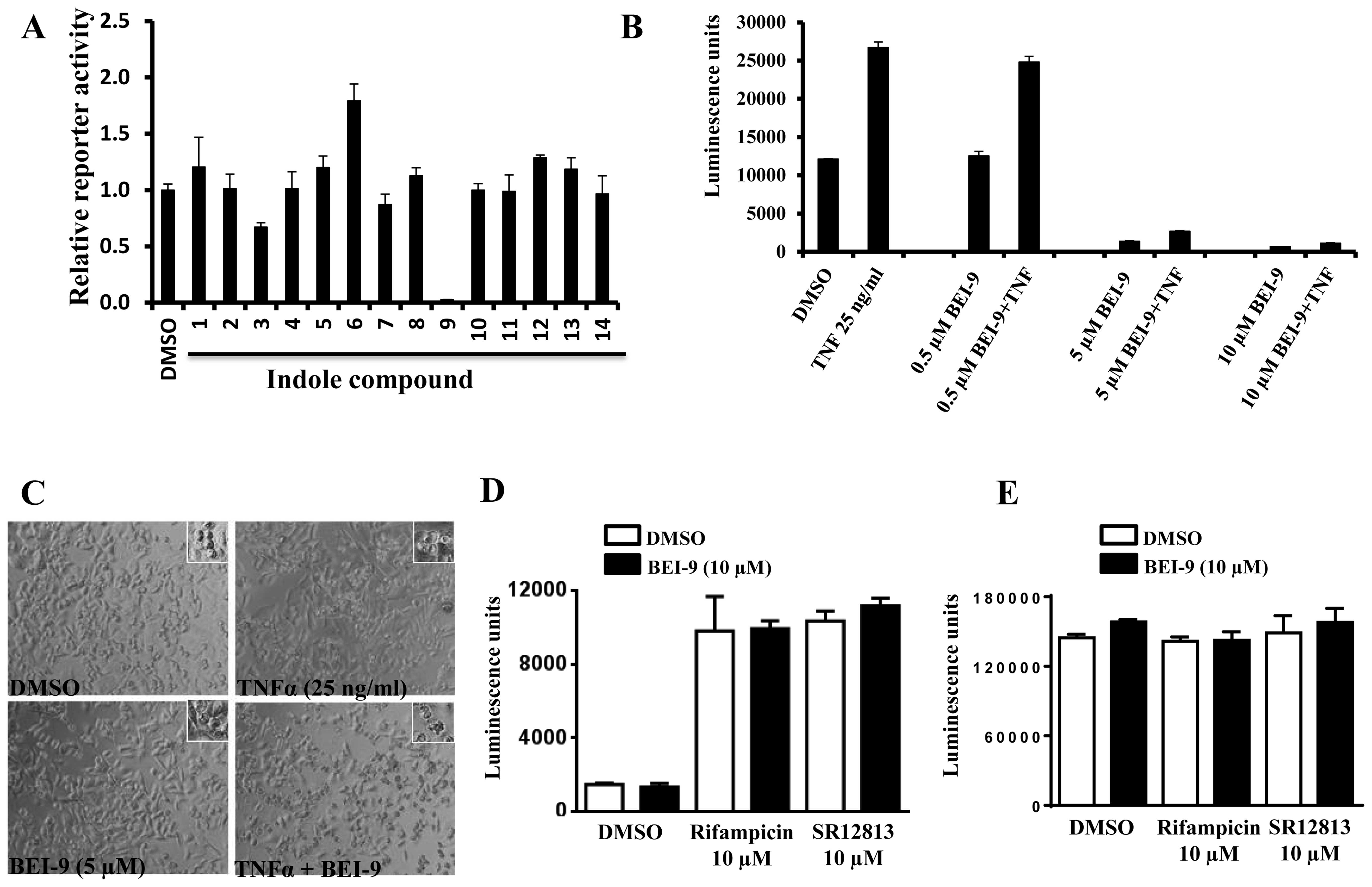
BEI-9 and the other 13 compounds were assessed for
their effects on the basal levels of luciferase activity. Equal
numbers of SW480-NFL cells were seeded in 96-well plates and
exposed to DMSO or to the test compounds. Luciferase activity was
measured at 24 h after the treatment. Among the 14 compounds, only
BEI-9 reduced the reporter activity (Fig. 4A). Then the possibility that
cytokine-induced NF-κB activation is blocked by BEI-9 was
determined. Since we previously established that these cells are
responsive to TNFα, an inducer of the NF-κB pathway, and to various
chemotherapeutic drugs (23), TNFα
was used to activate NF-κB in these reporter cells. Cells were
exposed to TNFα (25 ng/ml) and to 0.5, 5 or 10 µM BEI-9. At
5 or 10 µM, BEI-9 abolished the activation of NF-κB by TNFα
(Fig. 4B). The difference between
the effects of 0.5 and 5 µM BEI-9 was <10-fold, showing
that, for SW480 cells, effective concentrations of BEI-9 are in a
low micromolar range. Although, as single agents, neither of the
compounds caused cell death, the combination of TNFα (25 ng/ml) and
BEI-9 (5 µM) resulted in the appearance of cells with
membrane blebs, which are a characteristic of apoptosis (Fig. 4C). Thus, in addition to inhibiting
NF-κB activation, BEI-9 added to TNFα-treated cells may divert TNF
receptor-initiated signaling toward apoptosis.
Luciferase reporter assays are dependent on the
activity of the luciferase enzyme to catalyze the conversion of
luciferin to oxyluciferin in the presence of ATP and oxygen,
generating light in the process. Therefore, compounds that directly
interfere with the enzyme activity should be distinguished from
those that inhibit the signaling activity inside the cells. To test
this, the PXR-luciferase reporter system expressed in hepg2 cells
was used, and BEI-9 (10 µM) was added to the cells 5 min
before measuring luciferase. ATP-dependent cell viability was
measured with CellTiter-Glo kits, to determine if BEI-9 competes
with cellular ATP, which is required for luciferase activity. The
results from both assays (Fig. 4D and
E) suggest that, at bioactive concentrations, BEI-9 does not
inhibit luciferase directly or indirectly by competing with
ATP.
To rule out the possibility that BEI-9 directly
inhibits luciferase enzyme activity, the possibility that BEI-9
affects the luciferase activity in HepG2 cell-based luciferase
reporter gene assays was assessed. hPXR is a ligand-dependent
nuclear receptor that regulates the expression of drug-metabolizing
enzymes, including cytochrome p450 (CYP3A4) (29). HPXR transactivation assays were
accomplished with HepG2 cells transiently transfected with hPXR and
CYP3A4-luc, in which the expression of luciferase was controlled by
the hPXR-regulated CYP3A4 promoter. The human PXR agonists,
rifampicin and SR12813, induced PXR transactivation of CYP3A4
promoter activity (Fig. 4D). BEI-9
(10 µM) did not affect the luminescence either under basal
(DMSO) or stimulated (rifampicin or SR12813) conditions (Fig. 4D), showing that it does not directly
inhibit luciferase activity.
We previously demonstrated that camptothecin (CPT),
a drug used to treat various types of cancers, activates NF-κB in
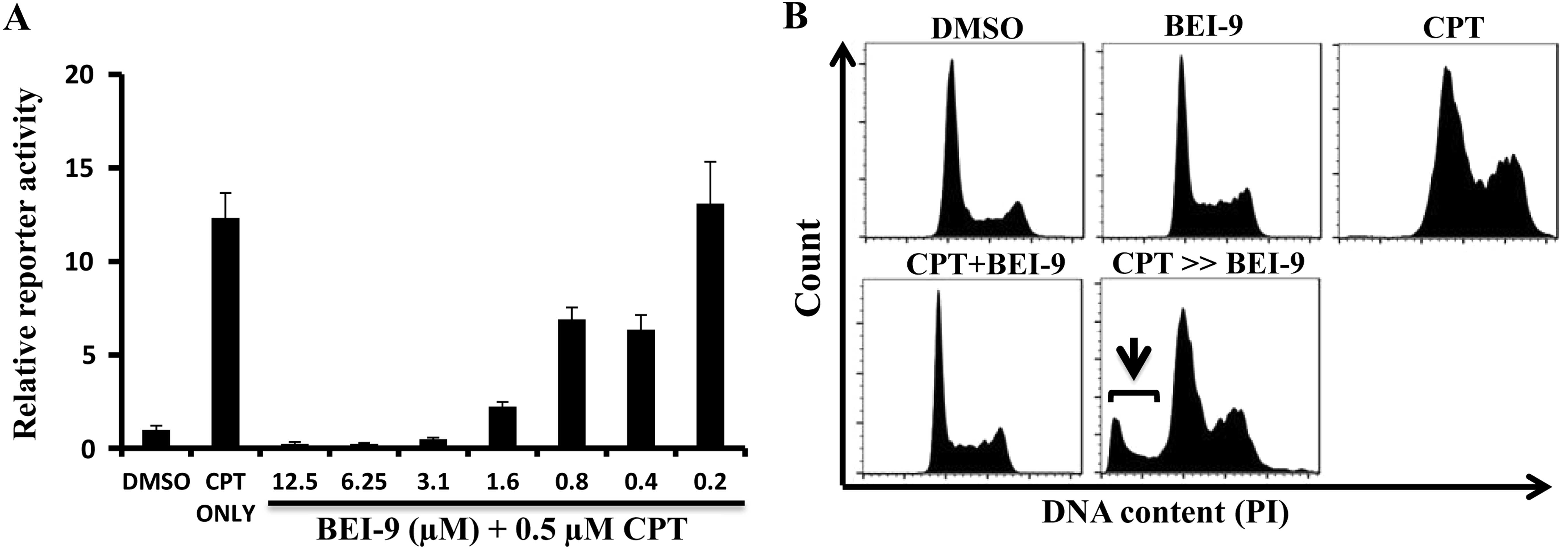
SW480 cells at peak concentrations of 0.5–1 µM (23). To examine the possibility that BEI-9
suppresses the drug-induced NF-κB response in these cells, SW480
reporter cells were exposed to CPT (0.5 µM) and to varying
concentrations (0.2–12.5 µM) of BEI-9. BEI-9 inhibited the
NF-κB response >50% at concentrations >0.8 µM
(Fig. 5A). Although CPT and BEI-9,
separately or together, did not induce cell death in these cells,
as determined by flow cytometry, sequential treatment of CPT for 24
h followed by BEI-9 for 24 h resulted in the appearance of a
distinct sub-G1 population, an indication of cell death (Fig. 5B).
BEI-9 was well tolerated by nude mice at
intraperitoneal doses up to 10 mg/kg, but 100 mg/kg was toxic,
causing death within 24 h (Table
II). A dose of 50 mg/kg, although not as lethal, resulted in
loss of body weight and noticeable discomfort to the mice,
requiring termination. Doses of 1 and 5 mg/kg did not result in
body weight loss, nor did they induce any noticeable change in
physical state or behavior. The most obvious gross pathology after
repeated intraperitoneal injections was irritation of the
peritoneum in the 10 mg/kg dose group, resulting in localized
peritoneal adhesions and moderate weight loss. Histopathological
analyses of the tissues collected from kidneys, liver, heart,
spleen, pancreas, lungs, and intestines did not show pathological
abnormalities for doses <10 mg/kg.
| Table IISummary of the experiments to
determine a safe dose range for BEI-9 in nude mice. |
Table II
Summary of the experiments to
determine a safe dose range for BEI-9 in nude mice.
| Dosage group | No. of animals | No. of once-a-week
injections | Gross
pathology | Histopathology
(major findings) | Remarks |
|---|
| Control | 5 | 6 | None | | |
| Vehicle (DMSO) | 5 | 6 | None | | |
| 1 mg/kg | 10 | 6 | None | | |
| 5 mg/kg | 10 | 6 | None | | |
| 10 mg/kg | 5 | 6 | Peritoneal
localized adhesions | | Adhesions noted at
necropsy |
| 50 mg/kg | 2 | 2 | Weight loss,
discomfort | Hepatocyte
vacuolation, swelling and some degenerative changes | Terminated |
| 100 mg/kg | 1 | 1 | Acute toxicity
within 24 h | Not performed | Lethal |
Discussion
In the present study, BEI-9 was characterized as an
inhibitor of cell proliferation. It was more potent than I3C, which
has anticancer activities (1,2). The
mechanisms involved in its anti-proliferative and anti-NF-κB
signaling activities remain to be determined. Nevertheless, since
NF-κB activation is implicated in carcinogenesis and in reducing
sensitivity to anticancer drugs, BEI-9 should be investigated in
combination with drugs, such as CPT and docetaxel, which activate
NF-κB (23,30–32).
BEI-9 was found to be an inhibitor of cell proliferation but did
not cause cell death, as evaluated by microscopy and cell cycle
analysis. The bioactivity of BEI-9 appears to depend on inhibiting
the progression of cells through the cell cycle. Part of this
effect may be through enhanced degradation or reduced production of
cyclins, which are cell cycle regulators. The possibility of BEI-9
acting as a metabolic inhibitor of cell growth, resulting in cells
‘frozen’ at the phase in which they existed at the time of
treatment, needs further examination, particularly in view of the
involvement of tryptophan, an indole, in cellular metabolism.
The effects of combinations of CPT and BEI-9 appear
to be dependent on the sequence of treatments. This is in
accordance with the different mechanisms and dynamics of actions of
TNFα and CPT; the slow CPT-induced cellular effects could be
overcome by the ‘freeze’ effects of BEI-9, and the rapid receptor
effects of TNFα could be modulated by BEI-9 as a second step.
However, when CPT was given time to act on the cells, subsequent
addition of BEI-9 led to cell death. Further examination of the
dynamics of such interactions is needed to identify the mechanisms
of BEI-9 interaction with other anticancer agents.
Of note, necrostatins, similar compounds with the
indole backbone, are regulators of necroptosis, a form of cell
death (33). The cellular targets
for necrostatins are the RIPK1 and IDO proteins (34–36),
which have potential applications in inflammatory diseases and
neuroprotection (37,38). Further research is needed to
determine whether common signaling proteins are targeted by
necrostatins and BEI-9. Given the similar interference with
inflammatory signaling by necrostatins and BEI-9, the latter should
be tested for neurovascular and cardiovascular protection.
Since BEI-9 did not have noticeable physical or
pathological effects in nude mice at doses below 10 mg/kg,
subsequent studies with mice can be designed with 10 mg/kg as the
upper limit of dosage. Calculated for a 2-ml volume of mouse blood,
the highest tolerable dose we administered to mice (10 mg/kg)
corresponds to ~10 µM, well above the effective
concentrations that had biological effects such as NF-κB
inhibition. Furthermore, since nude mice were used in these
experiments, the potential effects of BEI-9 on the immune system or
cells thereof need to be evaluated.
Acknowledgments
This study was supported through NIH grant nos.
SC2CA13787, U54CA118948, and SC3GM109314. Support for the TU
RCMI/CBR Imaging Core Facility was obtained through grant no.
G12MD007585. We thank Dr Donald L. Hill for editorial assistance
with the manuscript and Jason White for technical assistance at the
Core Facility. We would like to thank Patrick Flannery (Auburn
University) for technical assistance and Dr Tao at the Auburn
University for sharing the FLUOstar Optima plate reader.
References
|
1
|
Talalay P and Fahey JW: Phytochemicals
from cruciferous plants protect against cancer by modulating
carcinogen metabolism. J Nutr. 131(Suppl 11): S3027–S3033.
2001.
|
|
2
|
Murillo G and Mehta RG: Cruciferous
vegetables and cancer prevention. Nutr Cancer. 41:17–28. 2001.
View Article : Google Scholar
|
|
3
|
De Kruif CA, Marsman JW, Venekamp JC,
Falke HE, Noordhoek J, Blaauboer BJ and Wortelboer HM: Structure
elucidation of acid reaction products of indole-3-carbinol:
detection in vivo and enzyme induction in vitro. Chem Biol
Interact. 80:303–315. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bradlow HL: Review. Indole-3-carbinol as a
chemoprotective agent in breast and prostate cancer. In Vivo.
22:441–445. 2008.PubMed/NCBI
|
|
5
|
Sung WS and Lee DG: Mechanism of decreased
susceptibility for Gram-negative bacteria and synergistic effect
with ampicillin of indole-3-carbinol. Biol Pharm Bull.
31:1798–1801. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Safe S, Papineni S and Chintharlapalli S:
Cancer chemotherapy with indole-3-carbinol, bis(3′-indolyl)methane
and synthetic analogs. Cancer Lett. 269:326–338. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Takada Y, Andreeff M and Aggarwal BB:
Indole-3-carbinol suppresses NF-kappaB and IkappaBalpha kinase
activation, causing inhibition of expression of NF-kappaB-regulated
anti-apoptotic and metastatic gene products and enhancement of
apoptosis in myeloid and leukemia cells. Blood. 106:641–649. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cram EJ, Liu BD, Bjeldanes LF and
Firestone GL: Indole-3-carbinol inhibits CDK6 expression in human
MCF-7 breast cancer cells by disrupting Sp1 transcription factor
interactions with a composite element in the CDK6 gene promoter. J
Biol Chem. 276:22332–22340. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rahman KM, Ali S, Aboukameel A, Sarkar SH,
Wang Z, Philip PA, Sakr WA and Raz A: Inactivation of NF-kappaB by
3,3′-diindolylmethane contributes to increased apoptosis induced by
chemotherapeutic agent in breast cancer cells. Mol Cancer Ther.
6:2757–2765. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Song JM, Qian X, Teferi F, Pan J, Wang Y
and Kassie F: dietary diindolylmethane suppresses
inflammation-driven lung squamous cell carcinoma in mice. Cancer
Prev Res (Phila). 8:77–85. 2015. View Article : Google Scholar
|
|
11
|
Matsuzaki Y, Koyama M, Hitomi T, Kawanaka
M and Sakai T: Indole-3-carbinol activates the cyclin-dependent
kinase inhibitor p15(INK4b) gene. FEBS Lett. 576:137–140. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Garcia HH, Brar GA, Nguyen DH, Bjeldanes
LF and Firestone GL: Indole-3-carbinol (I3C) inhibits
cyclin-dependent kinase-2 function in human breast cancer cells by
regulating the size distribution, associated cyclin E forms, and
subcellular localization of the CDK2 protein complex. J Biol Chem.
280:8756–8764. 2005. View Article : Google Scholar
|
|
13
|
Chinni SR and Sarkar FH: Akt inactivation
is a key event in indole-3-carbinol-induced apoptosis in PC-3
cells. Clin Cancer Res. 8:1228–1236. 2002.PubMed/NCBI
|
|
14
|
Howells LM, Gallacher-Horley B, Houghton
CE, Manson MM and Hudson EA: Indole-3-carbinol inhibits protein
kinase B/Akt and induces apoptosis in the human breast tumor cell
line MDA MB468 but not in the nontumorigenic HBL100 line. Mol
Cancer Ther. 1:1161–1172. 2002.PubMed/NCBI
|
|
15
|
Sundar SN, Kerekatte V, Equinozio CN, Doan
VB, Bjeldanes LF and Firestone GL: Indole-3-carbinol selectively
uncouples expression and activity of estrogen receptor subtypes in
human breast cancer cells. Mol Endocrinol. 20:3070–3082. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hsu JC, Zhang J, Dev A, Wing A, Bjeldanes
LF and Firestone GL: Indole-3-carbinol inhibition of androgen
receptor expression and downregulation of androgen responsiveness
in human prostate cancer cells. Carcinogenesis. 26:1896–1904. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li Y, Li X and Guo B: Chemopreventive
agent 3,3′-diindolylmethane selectively induces proteasomal
degradation of class I histone deacetylases. Cancer Res.
70:646–654. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bonnesen C, Eggleston IM and Hayes JD:
Dietary indoles and isothiocyanates that are generated from
cruciferous vegetables can both stimulate apoptosis and confer
protection against DNA damage in human colon cell lines. Cancer
Res. 61:6120–6130. 2001.PubMed/NCBI
|
|
19
|
Chintharlapalli S, Smith R III, Samudio I,
Zhang W and Safe S:
1,1-Bis(3′-indolyl)-1-(p-substitutedphenyl)methanes induce
peroxisome proliferator-activated receptor gamma-mediated growth
inhibition, transactivation, and differentiation markers in colon
cancer cells. Cancer Res. 64:5994–6001. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Frydoonfar HR, McGrath DR and Spigelman
AD: Inhibition of proliferation of a colon cancer cell line by
indole-3-carbinol. Colorectal Dis. 4:205–207. 2002. View Article : Google Scholar
|
|
21
|
Weng JR, Tsai CH, Kulp SK and Chen CS:
Indole-3-carbinol as a chemopreventive and anti-cancer agent.
Cancer Lett. 262:153–163. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ahmad A, Sakr WA and Rahman KW: Mechanisms
and therapeutic implications of cell death induction by indole
compounds. Cancers (Basel). 3:2955–2974. 2011. View Article : Google Scholar
|
|
23
|
Samuel T, Fadlalla K, Gales DN, Putcha BD
and Manne U: Variable NF-κB pathway responses in colon cancer cells
treated with chemotherapeutic drugs. BMC Cancer. 14:5992014.
View Article : Google Scholar
|
|
24
|
Pondugula SR, Flannery PC, Apte U, Babu
JR, Geetha T, Rege SD, Chen T and Abbott KL:
Mg2+/Mn2+-dependent phosphatase 1A is
involved in regulating pregnane X receptor-mediated cytochrome p450
3A4 gene expression. Drug Metab Dispos. 43:385–391. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pondugula SR, Flannery PC, Abbott KL,
Coleman ES, Mani S, Samuel T and Xie W: Diindolylmethane, a
naturally occurring compound, induces CYP3A4 and MDR1 gene
expression by activating human PXR. Toxicol Lett. 232:580–589.
2015. View Article : Google Scholar
|
|
26
|
Samuel T, Fadlalla K, Mosley L, Katkoori
V, Turner T and Manne U: Dual-mode interaction between quercetin
and DNA-damaging drugs in cancer cells. Anticancer Res. 32:61–71.
2012.PubMed/NCBI
|
|
27
|
Casimiro MC, Velasco-Velázquez M,
Aguirre-Alvarado C and Pestell RG: Overview of cyclins D1 function
in cancer and the CDK inhibitor landscape: past and present. Expert
Opin Investig Drugs. 23:295–304. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Guttridge DC, Albanese C, Reuther JY,
Pestell RG and Baldwin AS Jr: NF-kappaB controls cell growth and
differentiation through transcriptional regulation of cyclin D1.
Mol Cell Biol. 19:5785–5799. 1999.PubMed/NCBI
|
|
29
|
Pondugula SR and Mani S: Pregnane
xenobiotic receptor in cancer pathogenesis and therapeutic
response. Cancer Lett. 328:1–9. 2013. View Article : Google Scholar
|
|
30
|
Codony-Servat J, Marín-Aguilera M, Visa L,
García-Albéniz X, Pineda E, Fernández PL, Filella X, Gascón P and
Mellado B: Nuclear factor-kappa B and interleukin-6 related
docetaxel resistance in castration-resistant prostate cancer.
Prostate. 73:512–521. 2013. View Article : Google Scholar
|
|
31
|
Tsang PS, Cheuk AT, Chen QR, Song YK,
Badgett TC, Wei JS and Khan J: Synthetic lethal screen identifies
NF-κB as a target for combination therapy with topotecan for
patients with neuroblastoma. BMC Cancer. 12:1012012. View Article : Google Scholar
|
|
32
|
Huang TT, Wuerzberger-Davis SM, Seufzer
BJ, Shumway SD, Kurama T, Boothman DA and Miyamoto S: NF-kappaB
activation by camptothecin. A linkage between nuclear DNA damage
and cytoplasmic signaling events. J Biol Chem. 275:9501–9509. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Degterev A, Huang Z, Boyce M, Li Y, Jagtap
P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA and Yuan J:
Chemical inhibitor of nonapoptotic cell death with therapeutic
potential for ischemic brain injury. Nat Chem Biol. 1:112–119.
2005. View Article : Google Scholar
|
|
34
|
Takahashi N, Duprez L, Grootjans S,
Cauwels A, Nerinckx W, DuHadaway JB, Goossens V, Roelandt R, Van
Hauwermeiren F, Libert C, et al: Necrostatin-1 analogues: critical
issues on the specificity, activity and in vivo use in experimental
disease models. Cell Death Dis. 3:e4372012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Degterev A, Maki JL and Yuan J: activity
and specificity of necrostatin-1, small-molecule inhibitor of RIP1
kinase. Cell Death Differ. 20:3662013. View Article : Google Scholar :
|
|
36
|
Degterev A, Hitomi J, Germscheid M, Ch’en
IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, et al:
Identification of RIP1 kinase as a specific cellular target of
necrostatins. Nat Chem Biol. 4:313–321. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
King MD, Whitaker-Lea WA, Campbell JM,
Alleyne CH Jr and Dhandapani KM: Necrostatin-1 reduces
neurovascular injury after intracerebral hemorrhage. Int J Cell
Biol. 2014:4958172014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Vandenabeele P, Grootjans S, Callewaert N
and Takahashi N: Necrostatin-1 blocks both RIPK1 and IDO:
consequences for the study of cell death in experimental disease
models. Cell Death Differ. 20:185–187. 2013. View Article : Google Scholar :
|