Introduction
One of the many mechanisms by which cancer cells
acquire drug resistance is by apoptosis evasion (1,2).
Antineoplastic agents activate the apoptotic intrinsic pathway
which results in the permeabilization of the outer mitochondrial
membrane. The subsequent release of cytochrome c and other
pro-apoptotic molecules leads to the formation of a large protein
complex, the apoptosome, containing cytochrome c, apoptotic
protease activating factor 1 (Apaf-1) and caspase-9. Regulation of
these processes occurs through the activity of members of the BCL-2
family of proteins (3).
Upregulation of anti-apoptotic proteins, such as BCL-2, BCL-XL, and
members of the inhibitor of apoptosis (IAP) family, and
inactivating mutations in pro-apoptotic proteins Bax and Bak, or
p53 have been described in both the development of cancer and drug
resistance (2,4). Among several therapeutic strategies,
the restoration of p53-mediated apoptosis in cancer cells has been
sought for some time, so far without a clinical application
(5,6). Therefore, targeting inhibitors of
apoptosis offer another opportunity for drug development (7).
TP53-regulated inhibitor of apoptosis 1 (TRIAP1;
alias p53CSV, p53-inducible cell-survival factor) is a small,
evolutionarily conserved protein that is 76 amino acids long and
contains a twin Cx9C motif. Its expression is induced by
p53 following low levels of genotoxic stress (8). In yeast it is located in the
mitochondrial intermembrane space where it can interact with both
Ups1 and Ups2, protecting them against proteolysis and regulating
cardiolipin and phosphatidylethanolamine levels within mitochondria
(9). In its extramitochondrial
role, TRIAP1 modulates apoptotic pathways through interaction with
HSP70, inhibition of the interaction of cytochrome c with
Apaf-1 and activation of caspase-9, to inhibit apoptosis and permit
DNA damage repair (8,10). Despite its name, TRIAP1 does not
share any similarity with XIAP, c-IAP, survivin or other members of
the IAP family (11). TRIAP1
is upregulated in myelomas with unfavorable prognosis (12) and TRIAP1 mRNA has been found
in all breast cancer cell lines tested (13). However, the role of TRIAP1 in breast
or other solid tumors has been poorly addressed.
Breast cancer is one of the most common tumors
affecting women, and chemotherapy, endocrine therapy and
radiotherapy regimens are part of the present mainstream treatment.
Even those cancers that initially respond well to treatment may
become resistant and give rise to secondary tumors, normally with
fatal consequences (14). Thus, it
is crucial to be able to predict which patients will respond to
therapy in order to design the best clinical strategy available and
to develop drugs for the treatment of resistance (15).
Here we describe the upregulation of TRIAP1 in
drug-resistant breast cancer cells. Experimental modulation of
TRIAP1 in breast cancer cells, either by overexpression or
downregulation by RNA interference, changed cellular sensitivity to
doxorubicin, thus confirming TRIAP1 as a novel effector of drug
resistance.
Materials and methods
Cell culture, transduction and
transfection
The breast cancer cell lines CAL51 (German Resource
Centre for Biological Material, DSMz, Braunschweig, Germany) and
MCF7 (European Collection of Cell Cultures, Health Protection
Agency, UK) were grown in low glucose (1 g/l) Dulbecco’s modified
Eagle’s medium (DMEM)-GlutaMax (Invitrogen, Carlsbad, CA, USA)
supplemented with 10% foetal calf serum (FCS). Drug-resistant
derivatives CALDOX, MCFDOX and MLET5 have been previously described
(16–18). RNA from MDA-MB-231, T47D, ZR75 and
226-L-U19 cells (19) was a
generous gift from Y. Zhou. Human primary mammary epithelial cells
(HMECs) were purchased from Invitrogen and immortalized by
expression of the catalytic subunit of human telomerase as
previously described (20) to
generate HMEC-TERT cells. HMECs were grown on Mammary Epithelial
Cell Growth Medium (Invitrogen). GP2-293 pantropic retroviral
packaging (Clontech Laboratories, Mountain View, CA, USA) and
HEK293T cells (American Type Culture Collection, LGC Standards,
Teddington, UK) were maintained in DMEM supplemented with 4.5 g/l
glucose, 10% FCS and 4 mM L-glutamine (Invitrogen). For culturing
in estrogen-free conditions, MCF-7 cells were cultured in DMEM
lacking phenol red (DMEM-PR; Gibco-BRL, UK), supplemented with 10%
dextran-coated charcoal-stripped FCS (DSS; First Link Ltd., UK).
MLET5 cells were routinely cultured in DMEM-PR, containing 10% DSS.
All cells were routinely tested for the presence of mycoplasma
using the Lonza MycoAlert kit (Basel, Switzerland).
DNA procedures
A human TRIAP1 cDNA clone with a C-terminal GFP tag
cloned into pCMV6-AC-GFP was purchased from Origene and used for
transient transfections. For stable transfections, the genomic
region encompassing TRIAP1 (first and second exons including
both5′- and 3′-UTRs as well as the single intervening sequence) was
amplified from genomic DNA isolated from IMR-90 fibroblasts by PCR
using Platinum® Taq DNA Polymerase High Fidelity
(Invitrogen) with primers OLEY335 and OLEY336 (Table I). The amplified 2.6-kbp fragment
was initially cloned into pCRII-TOPO (Invitrogen) and the
TRIAP1 open reading frame verified by sequencing. Then, the
genomic TRIAP1 fragment was liberated by digestion with
BamHI and EcoRI and cloned into the BamHI- and
EcoRI-digested lentiviral vector FUW. Retroviral plasmids
pGFP-V-RS expressing short hairpins targeting TRIAP1
(5′-AGAGATTCCTATTGAAGGACTGG AGTTCA-3′) and non-targeting scrambled
(TR30013) were from Origene.
 | Table IOligonucleotides used in this
study, |
Table I
Oligonucleotides used in this
study,
| Primer | Sequence
(5′-3′) | Use |
|---|
| OLEY335 |
GGGGGATCCCGACGCGCCTGAGAGTGATGACATCA | TRIAP1
genomic amplification |
| OLEY336 |
GGGGTCGACTGTGAGGTTTCTGATTGCCATACTG | TRIAP1
genomic amplification |
| OLEY287 |
AGGATTTCGCAAGTCCAGAA | TRIAP1
QPCR |
| OLEY288 |
GCTGATTCCACCCAAGTAT | TRIAP1
QPCR |
| OLEY373 |
TCACCGCCCTACACATCAAACT | RPS14
QPCR |
| OLEY374 |
CTGCGAGTGCTGTCAGAGG | RPS14
QPCR |
| OLEY375 |
AGGGTTATGTGGTCCGAATCA | RPS6
QPCR |
| OLEY376 |
TGCCCCTTACTCAGTAGCAGG | RPS6
QPCR |
Viral transduction
Viral transductions were essentially as previously
described (16). Briefly,
retroviral transfections were performed using 20 µg
retroviral plasmid, 2 µg pVSV-G (Clontech Laboratories) and
55 µg Polyethylenimine (MW 25,000) (Polysciences,
Warrington, PA, USA) prior to co-transfection into the GP2-293
pantropic retroviral packaging cells. Lentiviral transfections were
performed using 20 µg lentiviral plasmid, 2 µg
pVSV-G, 8 µg pPAX2 (Addgene, Cambridge, MA, USA) and 75
µg Polyethylenimine (MW 25,000) (Sigma-Aldrich, St. Louis,
MO, USA) prior to co-transfection into the HEK293T cells. Viral
supernatants were collected 24–48 h after transfections, filtered
through 0.45-µm cellulose acetate filters and supplemented
with 8 µg/ml Polybrene (Sigma-Aldrich) prior to adding to
recipient cells. Puromycin (1 µg/ml; Sigma-Aldrich) was
added 96 h after the first infection to recipient cells for
selection of stable transgene expression.
Transient transfections
Cells (1×105 in 6-well plates) were
transfected with a final concentration of 40 nM ON-TARGETplus
SMARTpool TRIAP1 siRNA oligonucleotides (Thermo Scientific)
using HiPerFect (Qiagen, UK) following the manufacturer’s
instructions. EGFP siRNA (Ambion, UK) was used as a negative
control (21). A sulphorhodamine B
(Sigma-Aldrich) assay (22) was
used to screen for drug cytotoxicity as previously described
(19). For transient
overexpression, cells (1×104) were seeded in 96-well
plates and transfected with FuGENE HD (Promega, Madison, WI, USA)
using 2 µg DNA as recommended by the manufacturer. Then
cells were treated for 24 h with 0.4 µM doxorubicin, and
caspase-9 activity was determined using a Caspase-Glo 9 assay
(Promega) following the manufacturer’s instructions and normalized
to cell density obtained after sulphorhodamine B staining.
Gene expression analysis
Total RNA (isolated using RNAzol B; Biogenesis,
Poole, UK) was reverse transcribed with RNase H+ MMLV
Reverse Transcriptase (iScript cDNA Synthesis kit; Bio-Rad) and
real-time quantitative PCR was performed using SYBR-Green (Bioline
Reagents, London, UK) and gene specific primers (Table I) on an ABI Prism 7700 Detection
system (PerkinElmer Life Sciences, Waltham, MA, USA). PCR
conditions included an initial step at 95°C for 10 min followed by
40 cycles of 95°C for 30 sec, 60°C for 30 sec and 72°C for 30 sec.
A comparative threshold cycle was used to determine the relative
gene expression as previously described (23). TFF1 expression was performed
using TaqMan Gene Expression assay Hs00907239_m1 (Invitrogen)
following the manufacturer’s instructions.
Yeast procedures
Wild-type Saccharomyces cerevisiae BY4743
(MATa/α his3Δ1/his3Δ1 leu2Δ0/leu2Δ0 met15Δ0/MET15 lys2Δ0/LYS2
ura3Δ0/ura3Δ0) and Mdm35 knockout strain were purchased
from Open Biosystems. Screening for drug toxicity was performed in
YPD plates with doxorubicin after 3 days of growth.
Drug resistance clonogenic assay
Cells were seeded, at least in duplicate, at a
density of 1–5×105 cells in 25-cm2 culture
flasks and exposed to a single dose of doxorubicin for 24 h. Cells
were kept in culture for 2–4 weeks with drug-free medium changes
every week. Drug-resistant clones were fixed with 4%
paraformaldehyde and stained with 0.2% crystal violet. Crystal
violet retained in the cells was quantified by solubilization with
33% acetic acid and measured at OD592 nm.
Statistical analysis
Statistical evaluations were performed using the
Student’s t-test for paired data, and data were considered
significant at a P-value <0.05.
Results
TRIAP1 is ubiquitously expressed in
normal human tissues and upregulated in breast cancer cells
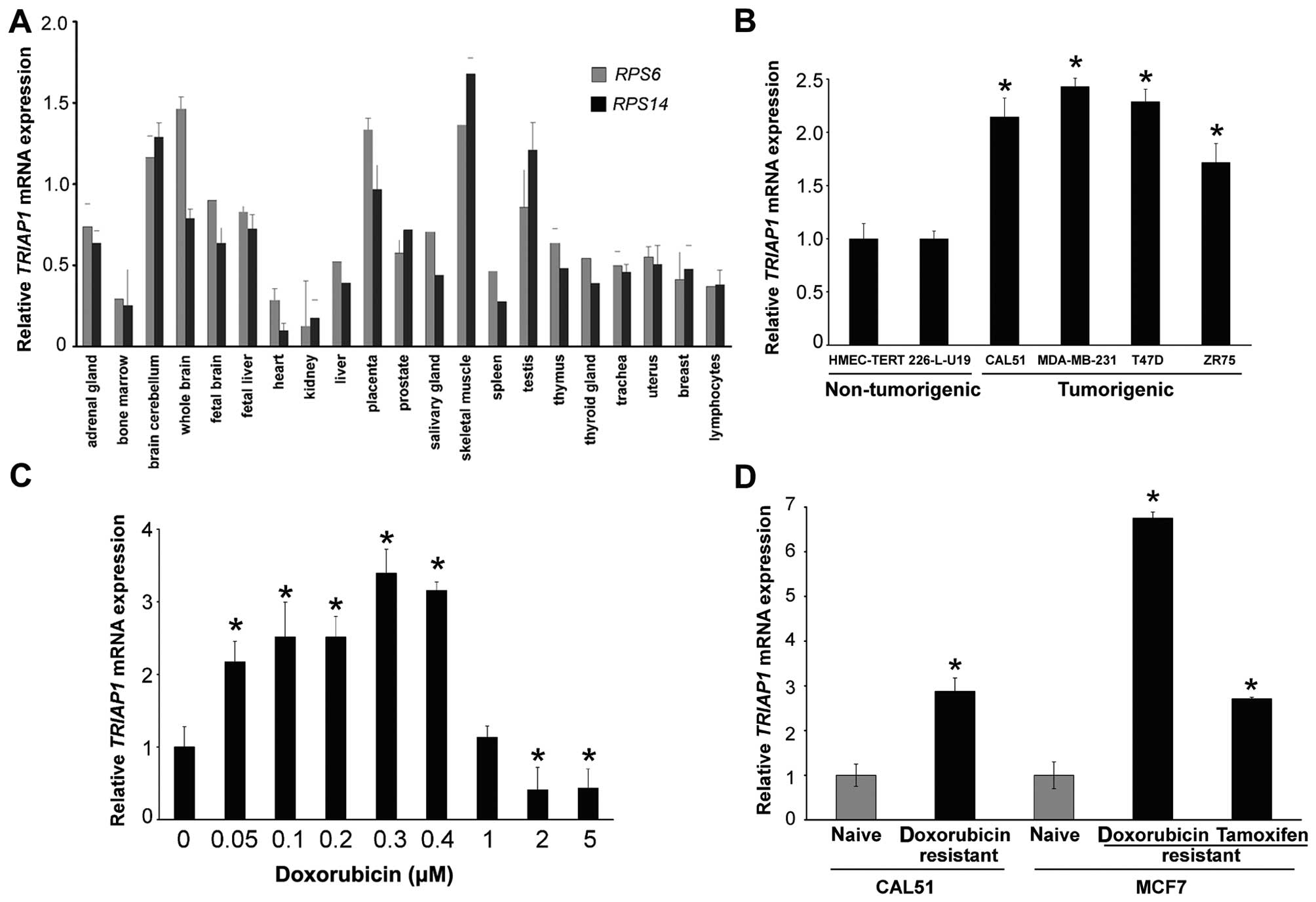
We initially tested TRIAP1 expression in a panel of
RNAs from normal human tissues (24) and found it ubiquitously expressed
(Fig. 1A). Although there was a
lack of tissue specificity, bone marrow, heart and kidney showed
very low expression, whereas brain, placenta and testis showed high
expression, with that from skeletal muscle being the highest.
Normal breast tissue expressed TRIAP1 mRNA at intermediate
levels (Fig. 1A). Next we tested
TRIAP1 mRNA expression in four breast cancer cell lines
(CAL51, MDA-MB-231, T47D and ZR75) and in two immortal, but
non-tumorigenic, breast cell lines (HMEC-TERT and 226-L-U19). An
~2-fold upregulation was found in the cancerous cell lines,
indicating that malignant transformation of breast epithelial cells
is, at least in vitro, associated with TRIAP1 upregulation
(Fig. 1B).
A polyclonal antibody raised to the synthetic
peptide CVQKAIKEKEIPIEGLEF (amino acids 47–64) as well as three
monoclonal antibodies raised to the epitopes HGKEKPENSS (amino
acids 67–76), IKEKEIPIEG (amino acids 52–61) and FAEKFLKGDS (amino
acids 23–32), all failed to robustly detect TRIAP1 as a band at ~10
kDa (data not shown). Equally, a few commercial antibodies were
tested and failed in the same way (data not shown). Thus, TRIAP1
expression analyses throughout the present study were performed
exclusively at the mRNA level.
TRIAP1 is upregulated in drug-resistant
breast cancer cell lines
TRIAP1 upregulation following genotoxic stress has
been exclusively demonstrated in a colon cancer cell line (8). We confirmed that CAL51 breast cancer
cells exhibited upregulation of TRIAP1 mRNA expression after
24 h of doxorubicin treatment at low to moderate doses (up to 0.4
µM). Higher doses (2–5 µM) repressed TRIAP1
expression (Fig. 1C). Then we
examined whether TRIAP1 was associated with a drug-resistance
phenotype. For this, we tested TRIAP1 mRNA expression in two
doxorubicin-resistant breast cancer cell lines derived from CAL51
(16) and MCF7 (17) cells, and in a third cell line
derived from MCF7 cells after estrogen deprivation, which are
resistant to tamoxifen and etoposide (18). Doxorubicin and etoposide are two
topoisomerase II inhibitors producing genotoxic stress, ultimately
leading to apoptosis, which form part of many chemotherapeutic
cancer regimes. TRIAP1 mRNA was upregulated in these three
drug-resistant cell types between 3- and 8-fold (Fig. 1D).
TRIAP1 is an effector of drug
resistance
In order to determine whether TRIAP1 is an effector
of drug resistance, we tested cell susceptibility to doxorubicin
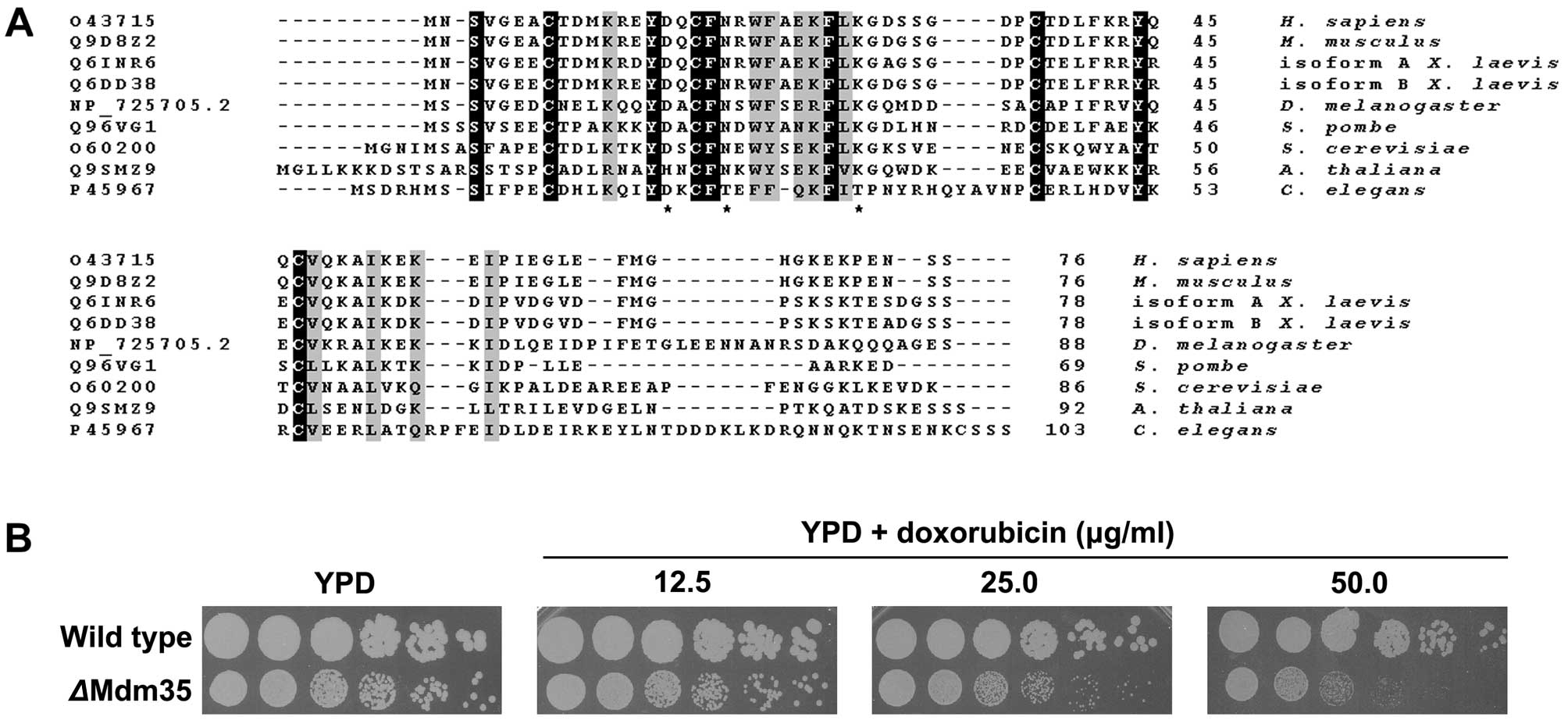
after experimental modulation of TRIAP1 expression. Comparison of
TRIAP1 protein sequences revealed a striking conservation across
kingdoms (Fig. 2A), albeit not at
the C-terminus, indicating that this protein must perform a
fundamental role in cellular physiology. The TRIAP1 homologue in
S. cerevisiae, Mdm35, shares 33% identity with human TRIAP1.
Since yeast cells harbour an ancestral programmed cell death
machinery that shares many key molecules that are part of the
apoptotic intrinsic pathway in mammalian cells (25), and because genetic manipulation of
yeast allows the knockout of genes, we initially tested our
hypothesis in Mdm35 knockout yeast cells. Indeed,
ΔMdm35 cell growth was greatly inhibited by the presence of
doxorubicin in the yeast growth medium compared to wild-type cell
growth (Fig. 2B).
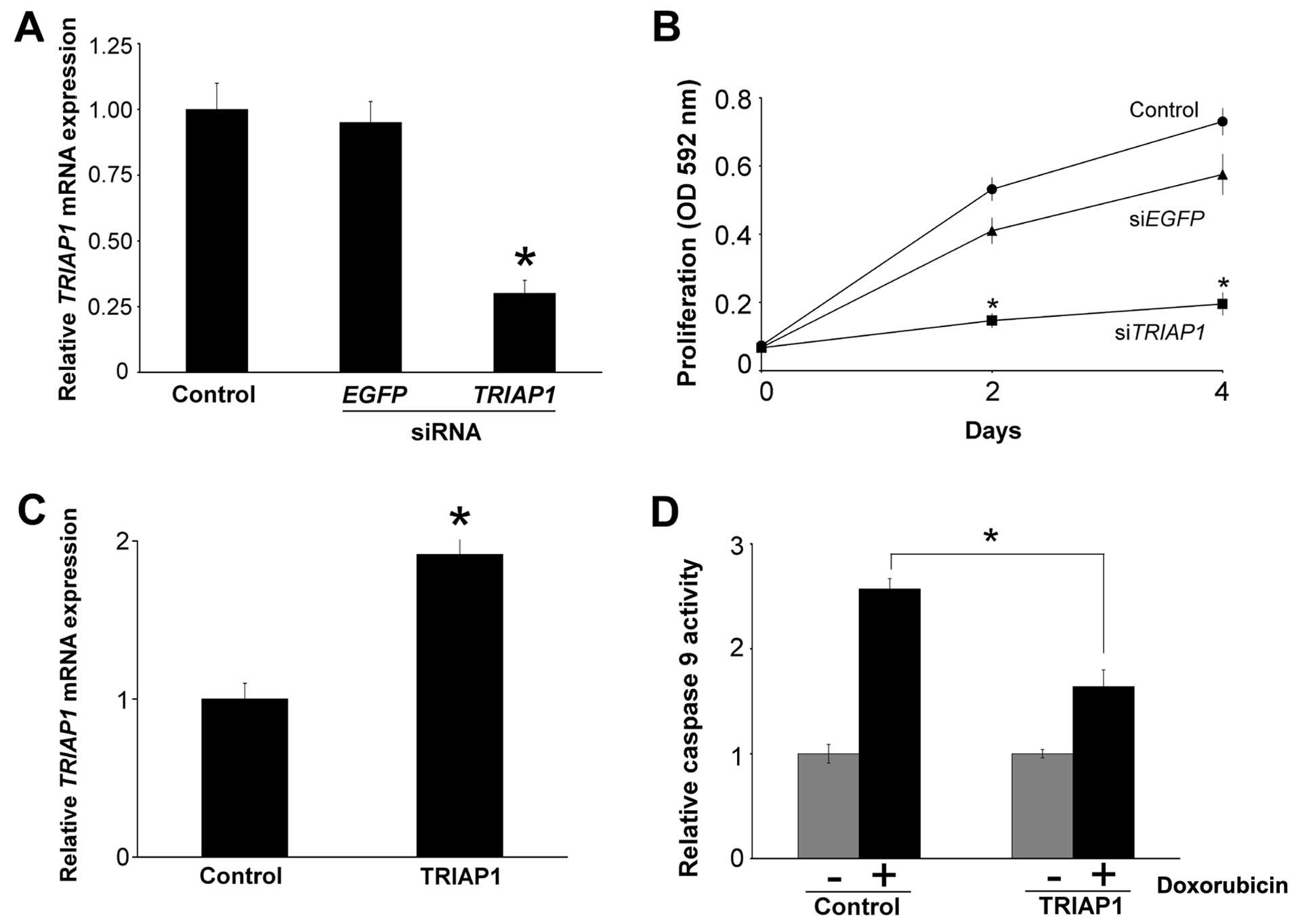
Next, we determined whether experimental
downregulation of TRIAP1 would have a similar effect in breast
cancer cells. For this, we transiently transfected siRNAs targeting
TRIAP1 mRNA in doxorubicin-resistant CALDOX cells and
obtained ~60% downregulation in TRIAP1 mRNA levels (Fig. 3A). Transfection of siRNAs targeting
EGFP mRNA, which is not expressed in these cells, was used
as a negative control. This transient decrease in TRIAP1
mRNA levels was sufficient to inhibit the proliferation of cells in
the presence of doxorubicin compared to the slight decrease in
proliferation obtained with the negative control (Fig. 3B).
As TRIAP1 blocks the formation of the apoptosome and
activation of caspase-9 (8), we
next ascertained whether caspase-9 activity would be reduced by
TRIAP1 in breast cancer cells. To do this, we transiently
overexpressed TRIAP1 in CAL51 cells (Fig. 3C) and treated them with 0.4
µM doxorubicin for 24 h. As expected, the control cells
transfected with an empty vector exhibited increased caspase-9
activity, and this increase was reduced in the cells overexpressing
TRIAP1 (Fig. 3D).
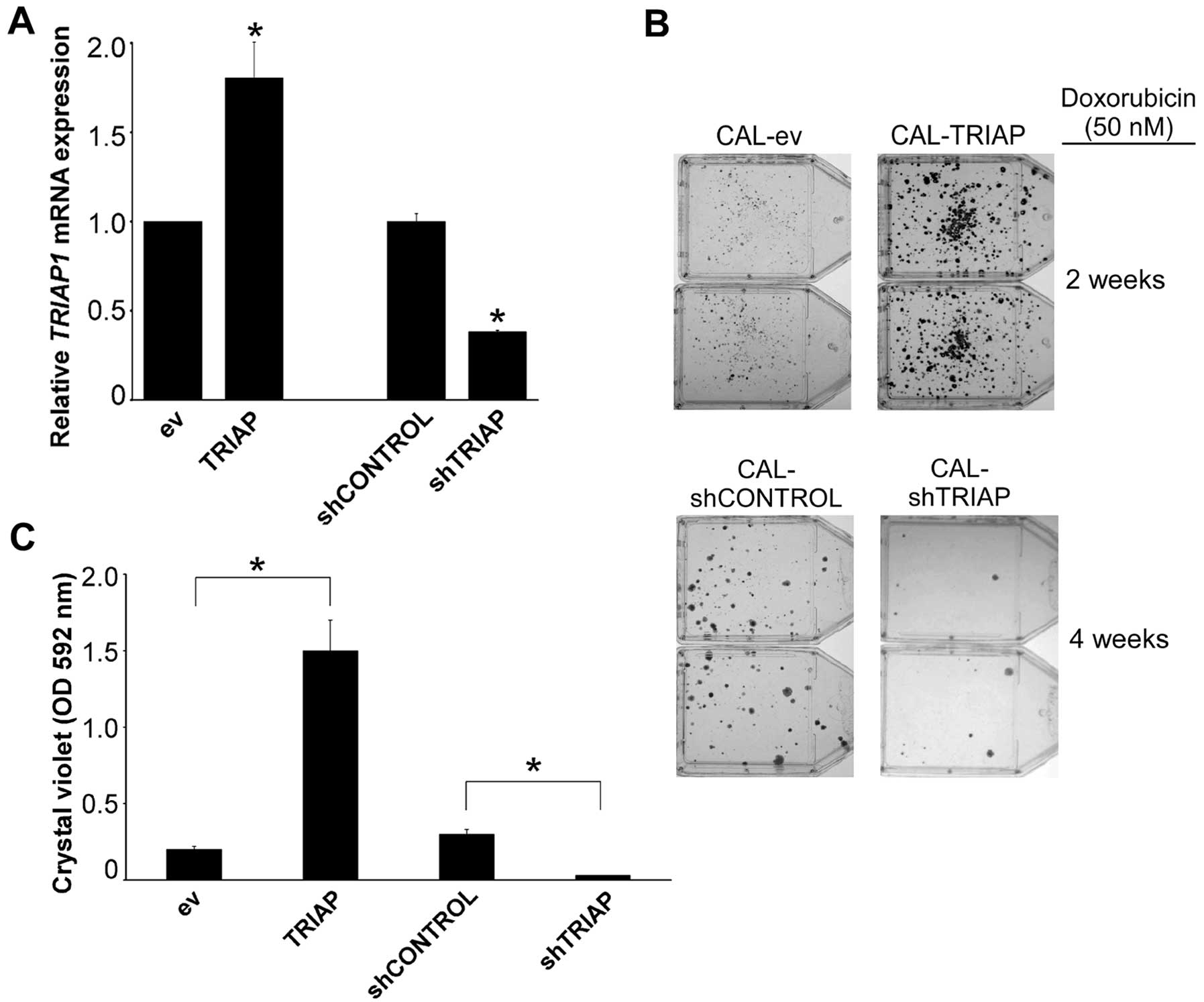
Although experiments performed with transient
transfections are informative to assess the short-term
susceptibility to cytotoxic drugs, the generation of drug
resistance is a slower process, often taking several weeks in
vitro, and required a different approach. We generated CAL51
derivative cells that had either permanently downregulated TRIAP1
by stable expression of hairpins targeting TRIAP1 mRNA
(shTRIAP) or permanently upregulated TRIAP1. CAL-shTRIAP cells
showed decreased levels of TRIAP1 mRNA by ~60% (Fig. 4A). In addition, we managed to
reproduce the slight increase in TRIAP1 mRNA expression
found in drug-resistant CALDOX cells by using low multiplicity of
infection of the lentivirus used for TRIAP1 overexpression
(Fig. 4A). Thus, although these
experimental changes in TRIAP1 expression after stable gene
expression were modest, they reflect very accurately the difference
found between naïve and drug-resistant cells (Fig. 1D). Next we performed clonogenic
assays after doxorubicin treatment. For this, doxorubicin was added
to cell cultures and the emergence of doxorubicin-resistant clones
was monitored over a period of 2–4 weeks. Stable experimental
overexpression of TRIAP1 led to an increase in
doxorubicin-resistant clones, and, conversely, downregulation of
TRIAP1 led to fewer drug-resistant clones than the control cells
(Fig. 4B and C).
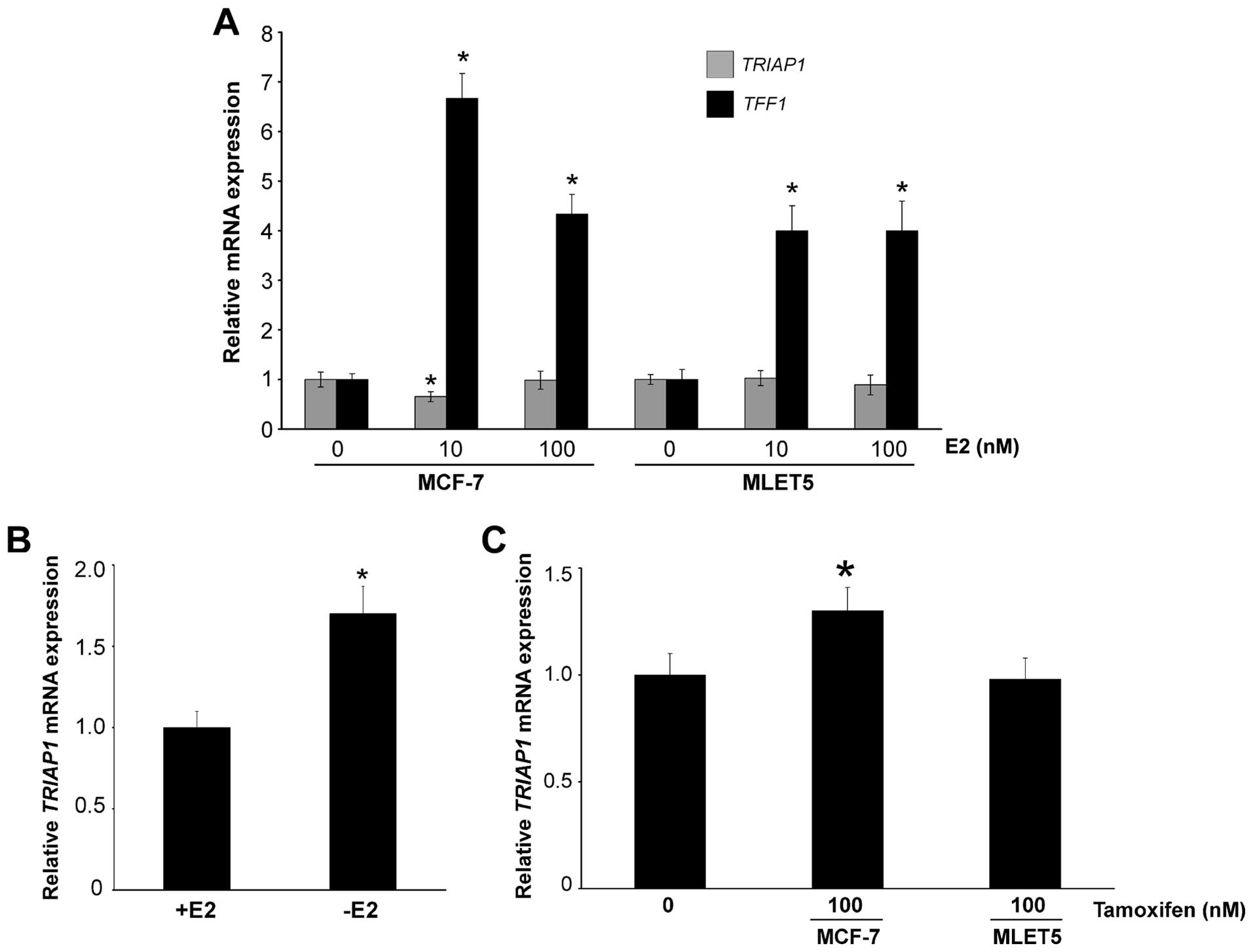
TRIAP1 is upregulated by lack of
estrogen
As tamoxifen-resistant cells have elevated levels of
TRIAP1 mRNA (Fig. 1D), we
investigated whether a link exists between estrogen and TRIAP1
expression. We cultured MCF-7 and MLET5 cells for 3 days in
estrogen-deprived medium, re-introduced estrogen for 16 h, and then
determined TRIAP1 mRNA levels. There was no upregulation of
TRIAP1 in any cell type; in fact, MCF-7 cells treated with 10 nM
estrogen showed a slight decrease in TRIAP1 expression (Fig. 5A). As expected, the
estrogen-responsive gene TFF1 (26) was upregulated in both cell lines
(Fig. 5A). This indicates that
TRIAP1 is not an estrogen-responsive gene. Importantly, when
MCF-7 cells were cultured in the absence of estrogen, TRIAP1
expression was upregulated (Fig.
5B). Moreover, the addition of tamoxifen, which blocks estrogen
receptor activation, led to a small but significant upregulation of
TRIAP1 mRNA in the MCF-7 cells, but not, as expected, in the
tamoxifen resistant MLET5 cells (Fig.
5C).
Overall, these results establish TRIAP1 as a
novel effector of drug resistance in breast cancer cells.
Discussion
One of the many mechanisms by which cancer cells
develop resistance to therapeutic intervention is by apoptosis
evasion. This is not surprising since the apoptotic machine is
complex, with multiple layers of regulation (1,2). The
IAP gene family encodes a group of structurally-related proteins
sharing a BIR domain, such as XIAP or survivin, which can be
upregulated in breast cancer (27,28).
However, other inhibitors of apoptosis do not share structural
similarities with members of the IAP family. One such protein,
TRIAP1, is a small, 76-amino acid protein in humans that binds to
HSP70 and inhibits the formation of the apoptosome. Apoptosis
evasion is one of the hallmarks of cancer (29), and is thus not surprising to find
TRIAP1 mRNA upregulated in a variety of malignancies
(30–32) and in breast cancer cell lines, but
not in TERT-immortalized primary mammary epithelial cells or in
cells transformed but not yet fully tumorigenic (Fig. 1B). Although the mechanisms
upregulating TRIAP1 during tumorigenesis are not known, a low to
moderate amount of several genotoxic agents triggers its expression
via p53 activation (8). Importantly
we also found that estrogen deprivation upregulated TRIAP1.
Estrogen deprivation reduces p53 activity (33,34),
and in normal mammary epithelial cells tamoxifen activates p53
(35), although not in MCF-7 cells
(36). Thus, the upregulation of
TRIAP1 in these cases is very likely to be p53-independent. Our
data clearly demonstrated that TRIAP1 is a novel effector of drug
resistance. Its experimental overexpression confers cells the
capacity to develop doxorubicin-resistant derivatives, whereas its
downregulation by RNA interference generates cells more susceptible
to the cytotoxic effects of the drug.
Due to its ubiquitous tissue expression in humans
and extremely high conservation throughout evolution it is likely
that TRIAP1 has an important function in eukaryotic cells. Research
on yeast indicates that TRIAP1 (Mdm35) normally resides in the
mitochondrial intermembrane space where it interacts with Usp1 and
Usp2 (PRELI1 and PRELI2 in humans) to regulate the synthesis of
cardiolipin and phosphatidylethanolamine, respectively (9). Although TRIAP1/PRELI complexes
(Mdm35/Usp homologues in yeast) have been shown to prevent
apoptosis by mediating intramitochondrial transport of phosphatidic
acid in HeLa cells (37), it is
tempting to speculate that the upregulation of TRIAP1 expression
following moderate genotoxic stress (Fig. 1C), or that found in drug-resistant
cells (Fig. 1D), helps to maintain
the balance towards apoptosis inhibition by blocking apoptosome
formation and caspase-9 inactivation (8). However, after stronger genotoxic
stress, which probably leads to irreparable DNA damage, there is no
activation of TRIAP1 expression. A large escape of cytochrome
c from the mitochondria would be enough to change the
balance towards apoptosome formation and the background levels of
TRIAP1 protein would not be enough to block the interaction
cytochrome c-Apaf1-caspase-9 (38). In addition, it is not clear whether,
in addition to cytochrome c, TRIAP1 or TRIAP1/PRELID
complexes can also escape from the intermembrane space following
stress to interact with HSP70. Nonetheless, yeast data as well as
those presented here indicate a dual role for TRIAP1 in
mitochondrial lipid biosynthesis and drug resistance. This may
offer a potential drug development strategy for the synthesis of
compounds blocking TRIAP1 activity.
Acknowledgments
We thank Yuan Zhou for a generous gift of
MDA-MB-231, T47D, ZR75 and 226-L-U19 RNA, Hetal Dattani for advice
on TFF1 expression, Andres Clemente Blanco for advice on
yeast work, and Eric Lam and Laki Buluwela for the MCFDOX and MLET5
cells, respectively. G.C. was the recipient of an Erasmus
postgraduate scholarship. Y.H. was partially supported by the China
Scholarship Council. We thank the Breast Cancer Research Trust
(C.A.), Breast Cancer Campaign (2011MaySP01 to E.Y.), Cancer
Research UK (R.C.C.) and the Experimental Cancer Medicine Centre
Network (R.C.C.) for their financial support.
Abbreviations:
|
Apaf-1
|
apoptotic protease activating factor
1
|
|
DMEM
|
Dulbecco’s modified Eagle’s medium
|
|
DMEM-PR
|
DMEM lacking phenol red
|
|
DSS
|
dextran-coated charcoal-stripped
FCS
|
|
FCS
|
foetal calf serum
|
|
HMECs
|
human primary mammary epithelial
cells
|
|
IAP
|
inhibitor of apoptosis
|
|
TRIAP1
|
TP53-regulated inhibitor of apoptosis
1
|
References
|
1
|
Mashima T and Tsuruo T: Defects of the
apoptotic pathway as therapeutic target against cancer. Drug Resist
Updat. 8:339–343. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Indran IR, Tufo G, Pervaiz S and Brenner
C: Recent advances in apoptosis, mitochondria and drug resistance
in cancer cells. Biochim Biophys Acta. 1807:735–745. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Elmore S: Apoptosis: a review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cotter TG: Apoptosis and cancer: the
genesis of a research field. Nat Rev Cancer. 9:501–507. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yu Q: Restoring p53-mediated apoptosis in
cancer cells: new opportunities for cancer therapy. Drug Resist
Updat. 9:19–25. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kastan MB: Wild-type p53: tumors can’t
stand it. Cell. 128:837–840. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
de Graaf AO, de Witte T and Jansen JH:
Inhibitor of apoptosis proteins: new therapeutic targets in
hematological cancer? Leukemia. 18:1751–1759. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Park WR and Nakamura Y: p53CSV, a novel
p53-inducible gene involved in the p53-dependent cell-survival
pathway. Cancer Res. 65:1197–1206. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tamura Y, Iijima M and Sesaki H: Mdm35p
imports Ups proteins into the mitochondrial intermembrane space by
functional complex formation. EMBO J. 29:2875–2887. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Niu P, Liu L, Gong Z, Tan H, Wang F, Yuan
J, Feng Y, Wei Q, Tanguay RM and Wu T: Overexpressed heat shock
protein 70 protects cells against DNA damage caused by ultraviolet
C in a dose-dependent manner. Cell Stress Chaperones. 11:162–169.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Salvesen GS and Duckett CS: IAP proteins:
blocking the road to death’s door. Nat Rev Mol Cell Biol.
3:401–410. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Felix RS, Colleoni GW, Caballero OL,
Yamamoto M, Almeida MS, Andrade VC, Chauffaille MdeL, Silva WA Jr,
Begnami MD, Soares FA, et al: SAGE analysis highlights the
importance of p53csv, ddx5, mapkapk2 and ranbp2 to multiple myeloma
tumorigenesis. Cancer Lett. 278:41–48. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Neve RM, Chin K, Fridlyand J, Yeh J,
Baehner FL, Fevr T, Clark L, Bayani N, Coppe JP, Tong F, et al: A
collection of breast cancer cell lines for the study of
functionally distinct cancer subtypes. Cancer Cell. 10:515–527.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ali S and Coombes RC: Endocrine-responsive
breast cancer and strategies for combating resistance. Nat Rev
Cancer. 2:101–112. 2002. View
Article : Google Scholar
|
|
15
|
Eccles SA, Aboagye EO, Ali S, Anderson AS,
Armes J, Berditchevski F, Blaydes JP, Brennan K, Brown NJ, Bryant
HE, et al: Critical research gaps and translational priorities for
the successful prevention and treatment of breast cancer. Breast
Cancer Res. 15:R922013. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Raguz S, Adams C, Masrour N, Rasul S,
Papoutsoglou P, Hu Y, Cazzanelli G, zhou Y, Patel N, Coombes C, et
al: Loss of O’-methylguanine-DNA methyltransferase confers
collateral sensitivity to carmustine in topoisomerase II-mediated
doxorubicin resistant triple negative breast cancer cells. Biochem
Pharmacol. 85:186–196. 2013. View Article : Google Scholar
|
|
17
|
Chen J, Gomes AR, Monteiro LJ, Wong SY, Wu
LH, Ng TT, Karadedou CT, Millour J, Ip YC, Cheung YN, et al:
Constitutively nuclear FOXO3a localization predicts poor survival
and promotes Akt phosphorylation in breast cancer. PLoS One.
5:e122932010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tolhurst RS, Thomas RS, Kyle FJ, Patel H,
Periyasamy M, Photiou A, Thiruchelvam PT, Lai CF, Al-Sabbagh M,
Fisher RA, et al: Transient over-expression of estrogen receptor-α
in breast cancer cells promotes cell survival and
estrogen-independent growth. Breast Cancer Res Treat. 128:357–368.
2011. View Article : Google Scholar
|
|
19
|
Rasul S, Balasubramanian R, Filipović A,
Slade MJ, Yagüe E and Coombes RC: Inhibition of gamma-secretase
induces G2/M arrest and triggers apoptosis in breast cancer cells.
Br J Cancer. 100:1879–1888. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yagüe E, Arance A, Kubitza L, O’Hare M,
Jat P, Ogilvie CM, Hart IR, Higgins CF and Raguz S: Ability to
acquire drug resistance arises early during the tumorigenesis
process. Cancer Res. 67:1130–1137. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Filipović A, Gronau JH, Green AR, Wang J,
Vallath S, Shao D, Rasul S, Ellis IO, Yagüe E, Sturge J, et al:
Biological and clinical implications of nicastrin expression in
invasive breast cancer. Breast Cancer Res Treat. 125:43–53. 2011.
View Article : Google Scholar
|
|
22
|
Vichai V and Kirtikara K: Sulforhodamine B
colorimetric assay for cytotoxicity screening. Nat Protoc.
1:1112–1116. 2006. View Article : Google Scholar
|
|
23
|
Yague E, Armesilla AL, Harrison G, Elliott
J, Sardini A, Higgins CF and Raguz S: P-glycoprotein (MDR1)
expression in leukemic cells is regulated at two distinct steps,
mRNA stabilization and translational initiation. J Biol Chem.
278:10344–10352. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Raguz S, De Bella MT, Slade MJ, Higgins
CF, Coombes RC and Yagüe E: Expression of RPIP9 (Rap2 interacting
protein 9) is activated in breast carcinoma and correlates with a
poor prognosis. Int J Cancer. 117:934–941. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Almeida B, Silva A, Mesquita A,
Sampaio-Marques B, Rodrigues F and Ludovico P: Drug-induced
apoptosis in yeast. Biochim Biophys Acta. 1783:1436–1448. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sun JM, Spencer VA, Li L, Yu Chen H, Yu J
and Davie JR: Estrogen regulation of trefoil factor 1 expression by
estrogen receptor alpha and Sp proteins. Exp Cell Res. 302:96–107.
2005. View Article : Google Scholar
|
|
27
|
Nachmias B, Ashhab Y and Ben-Yehuda D: The
inhibitor of apoptosis protein family (IAPs): an emerging
therapeutic target in cancer. Semin Cancer Biol. 14:231–243. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang S, Bai L, Lu J, Liu L, Yang CY and
Sun H: Targeting inhibitors of apoptosis proteins (IAPs) for new
breast cancer therapeutics. J Mammary Gland Biol Neoplasia.
17:217–228. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: the next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Garber ME, Troyanskaya OG, Schluens K,
Petersen S, Thaesler Z, Pacyna-Gengelbach M, van de Rijn M, Rosen
GD, Perou CM, Whyte RI, et al: Diversity of gene expression in
adenocarcinoma of the lung. Proc Natl Acad Sci USA. 98:13784–13789.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lu KH, Patterson AP, Wang L, Marquez RT,
Atkinson EN, Baggerly KA, Ramoth LR, Rosen DG, Liu J, Hellstrom I,
et al: Selection of potential markers for epithelial ovarian cancer
with gene expression arrays and recursive descent partition
analysis. Clin Cancer Res. 10:3291–3300. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Skrzypczak M, Goryca K, Rubel T, Paziewska
A, Mikula M, Jarosz D, Pachlewski J, Oledzki J and Ostrowski J:
Modeling oncogenic signaling in colon tumors by multidirectional
analyses of microarray data directed for maximization of analytical
reliability. PLoS One. 5:e130912010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Molinari AM, Bontempo P, Schiavone EM,
Tortora V, Verdicchio MA, Napolitano M, Nola E, Moncharmont B,
Medici N, Nigro V, et al: Estradiol induces functional inactivation
of p53 by intracellular redistribution. Cancer Res. 60:2594–2597.
2000.PubMed/NCBI
|
|
34
|
Fernández-Cuesta L, Anaganti S, Hainaut P
and Olivier M: Estrogen levels act as a rheostat on p53 levels and
modulate p53-dependent responses in breast cancer cell lines.
Breast Cancer Res Treat. 125:35–42. 2011. View Article : Google Scholar
|
|
35
|
Somaï S, Chaouat M, Jacob D, Perrot JY,
Rostène W, Forgez P and Gompel A: Antiestrogens are pro-apoptotic
in normal human breast epithelial cells. Int J Cancer. 105:607–612.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang GJ, Kimijima I, Onda M, Kanno M,
Sato H, Watanabe T, Tsuchiya A, Abe R and Takenoshita S:
Tamoxifen-induced apoptosis in breast cancer cells relates to
down-regulation of bcl-2, but not bax and bcl-X(L), without
alteration of p53 protein levels. Clin Cancer Res. 5:2971–2977.
1999.PubMed/NCBI
|
|
37
|
Potting C, Tatsuta T, König T, Haag M, Wai
T, Aaltonen MJ and Langer T: TRIAP1/PRELI complexes prevent
apoptosis by mediating intramitochondrial transport of phosphatidic
acid. Cell Metab. 18:287–295. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cain K: Chemical-induced apoptosis:
formation of the Apaf-1 apoptosome. Drug Metab Rev. 35:337–363.
2003. View Article : Google Scholar
|