Introduction
A total of 989,600 new gastric cancer (GC) cases and
738,000 mortalities are estimated to have occurred in 2008,
accounting for 8% of the total cases and 10% of total deaths
worldwide. Over 70% of new cases and deaths occur in developing
countries and GC is a leading cause of cancer-associated mortality
in China (1,2). Dysregulation of normal signaling
pathways is a critical process in the pathogenesis of GC (3,4).
Mitogen-activated protein kinase (MAPK) signaling activities play
important roles in many of the processes involved in the initiation
and genesis of cancer, and MAPK signaling pathway abnormalities
have been shown to be involved in various human malignancies,
including GC (5–7).
MAPK pathways constitute a highly conserved family
of kinase modules (8,9) that it serve to relay information from
extracellular signals to the effectors which control various cell
processes such as proliferation, differentiation, migration and
apoptosis (10–13). MAPKs are activated by
phosphorylation on the threonine and tyrosine residues of a
conserved signature T-X-Y motif within the activation loop of the
kinase (5). There are MAPK
phosphatases (MKPs) that act as negative regulators of MAPK
activity in mammalian (14,15). The MKPs constitute a distinct
subgroup of 10 catalytically active enzymes within the larger
family of cysteine-dependent dual-specificity protein phosphatases
(DUSPs) (16,17). The dual-specificity phosphatase 9
(DUSP9) gene, also known as mitogen-activated protein kinase
phosphatase 4 (MKP4), was first described in 1997 by Muda et
al (18). This gene is a member
of the large family of protein tyrosine phosphatases (PTPs)
(18). It consists of 4 exons and
has a size of 8,884 bp and is localized on chromosome Xq28, which
codes for a functional protein of 41.9 kDa.
DUSP9 has been shown to be downregulated in
colorectal cancer, renal and hepatocellular carcinomas, and
squamous cell carcinoma (19–22).
In a mouse model, the tumor-suppressor capacity of DUSP9 was
confirmed on squamous cell carcinoma and NSCLC tissues (17). However, to the best of our
knowledge, no studies have been performed regarding DUSP9
expression in human GC tissues. Additionally, no studies have been
performed with the regard to the mechanism of DUSP9 inactivation in
GC.
The present study was performed using 30 matched GC
and normal gastric mucosa tissues. The analysis was conducted using
bisulfite sequencing PCR (BSP) after bisulfite treatment of DNA.
The lentiviral transfection of these cells with an inducible DUSP9
transfect led to a re-expression of DUSP9, was resulting in a
repression of proliferation in GC cell lines.
Materials and methods
Ethics statement
For use of these clinical materials for the present
study, written informed consent from all the patients and approval
from the Ethics Committees of the Nanfang Hospital of Guangdong
Province were obtained. The subjects involved in this study
provided signed written informed consent. The study was approved by
the Ethics Committee of Southern Medical University, Guangzhou,
Guangdong, China. Animal experimentation was approved by the Ethics
Committee of Animal Research at Southern Medical University. The
experimental protocol was established according to the associated
national guidelines from the Ministry of Science and Technology of
China.
Cell culture and tissue collection
GC cell lines MKN-1, SCP26, SCP17, SCP51, M5, SW480,
SW620, and LOVO were maintained in RPMI-1640 supplemented with 10%
FCS (MP Biomedicals) at 37°C in a 5% CO2 incubator. The
GC and normal mucosa samples were collected from the Nanfang
Hospital, the Affiliated Hospital of Southern Medical University,
Guangzhou, China. All the specimens had confirmed pathological
diagnosis and were staged according to the 2009 UICC-TNM
classification of malignant tumors.
RNA isolation, reverse transcription, and
RT-qPCR
Total RNA was extracted from frozen tumor samples
using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). cDNA was
synthesized from 500 ng total RNA using the PrimeScript RT Master
Mix (Perfect Real Time, DRR036A; Takara, Tokyo, Japan). mRNA
expression levels were quantified using quantitative PCR in a
96-well format by a SYBR-Green-based approach using a 7500 Fast
Real-Time PCR system (Applied Biosystems) and SYBR®
Premix Ex Taq™ II (Takara Bio, Inc., Ohtsu, Japan) in a final
volume of 20 µl including 100 ng cDNA and 0.4 pmol/µl
of each primer. The thermal cycling conditions included an initial
denaturation for 30 sec at 95°C and 40 cycles consisting of an
annealing step at 95°C for 5 sec and an extension step at 60°C for
20 sec. Each sample was analyzed in triplicate. The sequences
primer used for PCR are shown in Table
I. The relative expression of genes were calculated by the
2−ΔΔCt method. Data are presented as the relative
quantity of target mRNA normalized to the expression of GAPDH mRNA
and relative to a calibrator sample. Each assay was performed three
times.
 | Table IPCR primer sequences for RT-qPCR and
bisulfite sequencing PCR assay. |
Table I
PCR primer sequences for RT-qPCR and
bisulfite sequencing PCR assay.
| Primer | Sequence | Sequence product size
(bp) | Annealing temperature
(°C) |
|---|
| RT-qPCR | | | |
| DUSP9 | F:
5′-CAGCCGTTCTGTCACCGTC-3′ | 208 | 60 |
| R:
5′-CAAGCTGCGCTCAAAGTCC-3′ | | |
| GAPDH | F:
5′-GGAGCGAGATCCCTCCAAAAT-3′ | 197 | 60 |
| R:
5′-GGCTGTTGTCATACTTCTCATGG-3′ | | |
| CCND1 | F:
5′-GCTGCGAAGTGGAAACCATC-3′ | 135 | 60 |
| R:
5′-CCTCCTTCTGCACACATTTGAA-3′ | | |
| CDK6 | F:
5′-TCTTCATTCACACCGAGTAGTGC-3′ | 130 | 61 |
| R:
5′-TGAGGTTAGAGCCATCTGGAAA-3′ | | |
| p21 | F:
5′-TGTCCGTCAGAACCCATGC-3′ | 139 | 60 |
| R:
5′-AAAGTCGAAGTTCCATCGCTC-3′ | | |
| Bisulfite
sequencing PCR assay | | | |
| BSP-DUSP9 | F:
5′-AATAGAGGTTTGTAGGTGGGAG-3′ | 138 | 58 |
| R:
5′-CCCTACCCAAAAAAAACACT-3′ | | |
Bisulfite sequencing
Clinical samples were homogenized and digested
overnight with proteinase K. Clinical sample DNA was modified by
the bisulfite reaction using an EpiTect Bisulfite kit (Qiagen,
Germany) and quantified using a NanoDrop instrument (Thermo
Scientific, Rockford, IL, USA). Modified DNA (40 ng/reaction) was
amplified by PCR, using 0.2 µM of each primer, 2 units of
HotStart Taq DNA polymerase, and 0.2 mM of each dNTP per reaction.
Cycling programs (Applied Biosystems, Life technologies, USA) were
95°C for 5 min, then 40 cycles of 95°C for 10 sec, 60°C for 30 sec,
and 72°C for 20 sec, followed by a 5-min incubation at 72°C. PCR
products were examined after gel electrophoresis in 1.5% agarose to
confirm that a single band was obtained. Forward and reverse primer
sequences of DUSP9 for bisulfite sequencing (Invitrogen) are shown
in Table I, and together they
amplified a 138-bp product containing 11 CpG sites in the promoter.
Sequence homologies were identified using the BLAST program of the
National Center for Biotechnology Information available at
http://www.ncbi.nlm.nih.gov/BLAST/.
Western blot analysis
Cell lysate was prepared using RIPA buffer (50 mM
Tris pH 7.4, 0.15 M NaCl, 1% Triton X-100, 1% sodium deoxycholate
and 0.1% SDS with 1 mM PMSF) with protease inhibitors and
quantified using the BCA protein assay (BioTek, China). Protein (20
µg) was loaded onto a 10% SDS-PAGE gel (Bio-Rad, Hercules,
CA, USA) that was then transferred onto a PVDF membrane (Amersham
Biosciences, Buckinghamshire, UK) and incubated with rabbit
anti-DUSP9 (1:2,000 diluted, cat no. ab54941-100; Abcam, Cambridge,
UK), rabbit anti-p21 (1:1,000 diluted; Cell Signaling Technology,
Danvers, MA, USA), rabbit anti-CDK4 (1:2,000 diluted), rabbit
anti-CDK6 (1:3,000 diluted), rabbit anti-CCND1 (1:2,000 diluted)
and rabbit anti-c-Jun (1:1,000 diluted) (all from Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA) at 4°C overnight in
blocking buffer (3% non-fat dry milk/BSA in TBS) followed by
incubation with HRP-conjugated secondary anti-mouse anti-body
(1:5,000 diluted; ZSGB-Bio, China). The bands were visualized by an
ECL Western Blot kit (CWBio Technology, Beijing, China). The images
were captured using a ChemiDoc™ XRS+ Molecular Imager
(Bio-Rad).
Tumor growth assay
Female BALB/c nude mice aged 4–5 weeks were
purchased from the Central Animal Facility of Southern Medical
University. Animal handling and experimental procedures were
approved by the Animal Experimental Ethics Committee of CUHK.
Following fluorescence-activated cell sorting, the green
fluorescence protein-positive cells were isolated and a total of
5×105 infected cells were injected subcutaneously into
the dorsal flank of nude mice. Each group contained 7 mice and the
experiment was repeated three times.
Plasmid construction and
transfection
Eukaryotic expression wild-type vectors
(pEGFP-DUSP9) were constructed by Guangzhou Sagene (Sagene
Incorporation, Guangzhou, China). MKN-1 cells were transfected with
empty vector (pEGFP-C1), and the wild-type vector (pEGFP-DUSP9)
using Lipofectamine 2000 (Invitrogen). Culture medium containing
G418 (Sigma-Aldrich, St. Louis, MO, USA) was used to select stable
transfectants.
Establishment of lentivirus-delivered
LV-sh-DUSP9 and LV-DUSP9 in GC cells
The preparation of lentivirus-expressing human DUSP9
short hairpin RNA (shRNA-1015, Table
II) was performed using the pLVTHM-GFP lentiviral RNAi
expression system. The lentiviral particles were used to infect
MKN-1 GC cell lines. Lentivirus (GV208, Ubi-MCS-EGFP) particles
carrying DUSP9 and its control were purchased from GeneChem
(Shanghai, China). The lentiviral transduction of MKN-1 cells was
carried out according to the manufacturer’s instructions. The
resulting cells were seeded in 96-well plates and cultured for 3
weeks to produce a stable DUSP9-overexpressing MKN-1 cells and
MKN-1 control cells. The high expression of DUSP9 was validated by
RT-qPCR.
| Table IIshRNA sequences for DUSP9. |
Table II
shRNA sequences for DUSP9.
| DUSP9 | Sequence |
|---|
| 1015 | Sense |
5′-AGGCCATTGAGTTCATTGATTCAAGAGATCAATGAACTCAATGGCCTTTTTTTACGCGT-3′ |
| Antisense |
5′-ACGCGTAAAAAAAGGCCATTGAGTTCATTGATCTCTTGAATCAATGAACTCAATGGCCT-3′ |
Transient transfection with siRNAs for
DUSP9
siRNAs were transfected at a working concentration
of 100 nmol/l using Lipofectamine 2000 reagent (Invitrogen). The
siRNA for DUSP9, a non-specific control were all purchased from
Guangzhou RiboBio Co., Ltd. (Guangzhou, China). The sequence of
each gene and their controls are shown in Table III. Twenty-four hours before
transfection, MKN-1 cells were plated onto a 96-well plate or a
6-well plate at a 30–50% confluence. GC cells were then transfected
into MKN-1 cells using TurboFect siRNA transfection reagent
(Fermentas, Vilnius, Lithuania) according to the manufacturer’s
instructions. The cells were collected after 48–72 h for subsequent
experiments.
| Table IIIsiRNA sequences of DUSP9. |
Table III
siRNA sequences of DUSP9.
| DUSP9 | Sequence |
|---|
| 921 | Sense |
5′-CUCCCAAACUUCUUCGAGAdTdT-3′ |
| Antisense |
3′-dTdTTCUCGAAGAAGUUUGGGAG-5′ |
Colony formation assay
After 72 h of infection, the cells were plated in
6-well plates at a concentration of 200 cells/well and grown for 2
weeks. After 2 weeks, the cells were washed twice with PBS, fixed
with methanol/acetic acid (3:1, v/v), and stained with 0.5% crystal
violet. The number of colonies was counted under the microscope
(Olympus FV1000; Olympus, Tokyo, Japan).
Cell proliferation and cell-cycle
analyses
Cell proliferation was analyzed using an MTT assay
(Sigma-Aldrich). Briefly, MKN-1 cells (5×103) were
plated onto 96-well plates, respectively, in 100 µl of
growth medium and allowed to adhere overnight. The cells were then
transfected with DUSP or Si-DUSP9 and control, respectively. At
different time-points (24, 48, and 72 h), the culture medium was
removed and replaced with culture medium containing 10 µl of
sterile MTT dye (5 mg/ml). After incubation at 37°C for 4 h, the
MTT solution was removed, and 150 µl dimethyl sulfoxide
(DMSO) was added to dissolve the formazan crystals. Spectrometric
absorbance at 490 nm was measured by a BioTek ELx800 microplate
photometer (SN211805; BioTek Instruments, Winooski, VT, USA).
For the cell-cycle analysis, LV-DUSP9-infected MKN-1
cells, LV-sh-DUSP9-infected MKN-1 cells and negative control cells
were fixed in 70% ice-cold ethanol for 48 h at 4°C, stained by
incubation with PBS containing 10 µg/ml propidium iodide and
0.5 mg/ml RNase A for 15 min at 37°C, and analyzed for the DNA
content of labeled cells by a FACSCalibur cytometer. Each
experiment was performed in triplicate.
Statistical analysis
Data are presented as mean ± SD. SPSS 13.0 software
(SPSS Inc., Chicago, IL, USA) and GraphPad software (GraphPad
Software, Inc., La Jolla, CA, USA) were used to analyze the data
for statistical significance. A two-tailed Student’s t-test was
used for comparisons of two independent groups. One-way ANOVA was
used to determine the differences between groups for all in
vitro analyses and the log-rank Mantel-Cox test for in
vitro and in vivo analyses. P<0.05 was considered to
be statistically significant.
Results
DUSP9 expression is reduced in GC cell
lines
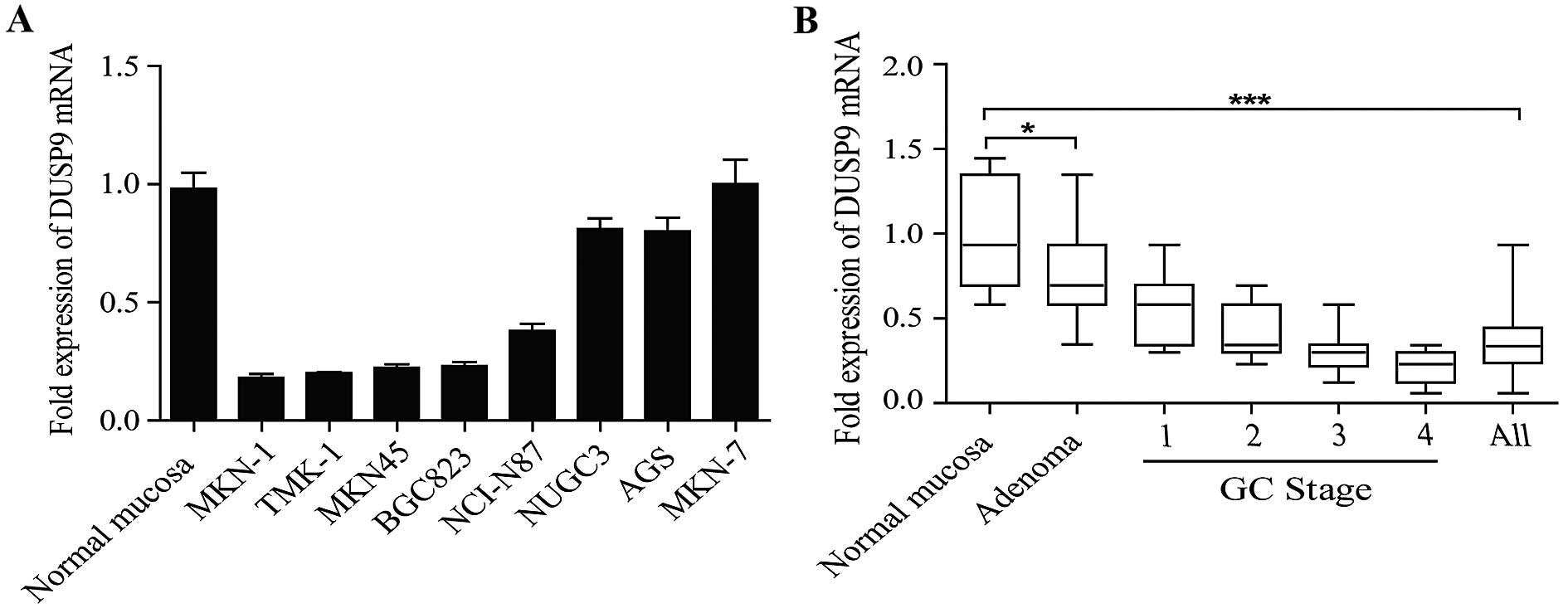
A panel of human GC cell lines was first analyzed to
quantify the expression level of DUSP9. The results showed that the
expression level of DUSP9 was decreased in the seven GC cell lines
examined, compared with the normal gastric mucosa tissues (Fig. 1A). DUSP9 transcript levels were
significantly decreased in all the stages of GC compared with the
normal gastric mucosa tissues (Fig.
1B). DUSP9 transcript levels were also reduced in premalignant
lesions (adenomas) compared with those in normal gastric mucosa
tissues, indicating that DUSP9 loss occurs early in the progression
to tumorigenesis. These data supported that the DUSP9 transcript
level was downregulated in GC.
DUSP9 is silenced via hypermethylation in
malignancy
The DUSP9 promoter contains a large CpG island from
−1297 to +1104 from the transcription start site (TSS, Fig. 2A) based on the criteria and
algorithm described by Li and Dahiya (23). We performed BSP in 30 matched GC and
normal gastric mucosa tissues. BSP of the DUSP9 promoter included
11 CpG sites (Fig. 2B, left panel).
In normal gastric mucosa tissues, the DUSP9 promoter demonstrated
low methylation levels (average methylation level was 20.3%). By
contrast, DUSP9 promoter methylation was significantly increased at
each individual CpG site examined and reached an average of 74.5%
in the cancerous tissues (Fig. 2B,
right panel, P<0.001).
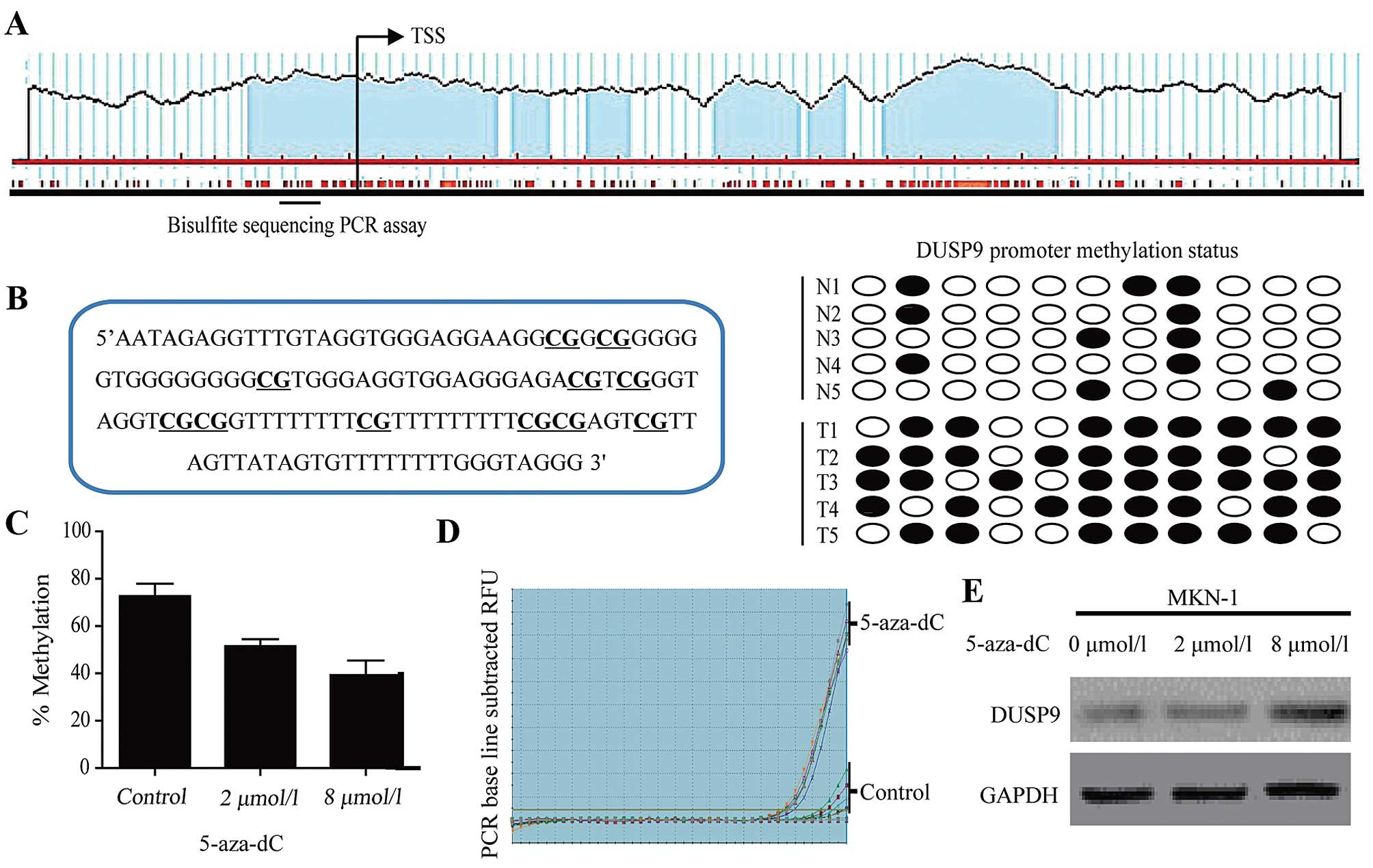 | Figure 2DUSP9 expression is silenced via
promoter hypermethylation in GC. (A) Schematic representation of
the human DUSP9 promoter. The CpG island of DUSP9 extends from
−1297 to +1104 from TSS. Each red tick mark represents is one CpG
site. The arrows indicate the TSS. To determine the methylation
level, BSP was carried out on 11 CpG sites extending from −215 to
−78 from TSS (underlined). (B) Bisulfite sequencing evaluation of
CpG island methylation of the 11 CpG sites of DUSP9 promoter in GC
(T=30) and normal mucosa tissues (N=30). Left panel, location of 11
CpG sites in the DUSP9 promoter. Right panel, open circles,
unmethylated CpG; closed circles, methylated CpG. (C) BSP in MKN-1
cells showed detection of DUSP9 methylation status following
treatment with 5-aza-dC for 72 h. To determine the methylation
level (%), BSP was carried out on 11 CpG sites. (D) RT-qPCR in
MKN-1 cells showed detection of DUSP9 mRNA following treatment with
vehicle or 5-aza-dC for 72 h. (E) Treatment with 5-aza-dC restoring
DUSP9 expression in MKN-1. 5-aza-dC treatment can lead to DNA
demethylation via inhibition of DNA methyltransferase activity.
Data are presented as the average of triplicate measurements from
duplicate experiments. TSS, transcriptional start site; 5-aza-dC,
5-aza-2′-deoxycytidine; DUSP9, dual-specificity phosphatase 9; GC,
gastric cancer; BSP, bisulfite sequencing PCR. |
To confirm the role of DNA methylation in the
transcriptional regulation of DUSP9, we treated MKN-1 cells with 2
or 8 µM 5-aza-2′-deoxycytidine for 72 h and examined DUSP9
promoter methylation and mRNA expression changes. DUSP9 gene
expression was restored following 5-aza-2′-deox-ycytidine treatment
(Fig. 2D and E). This re-expression
was accompanied by a decrease in promoter DNA methylation from 94
to 72% (Fig. 2C). These results
indicated that promoter hypermethylation is one mechanism mediating
transcriptional silencing of DUSP9 in GC.
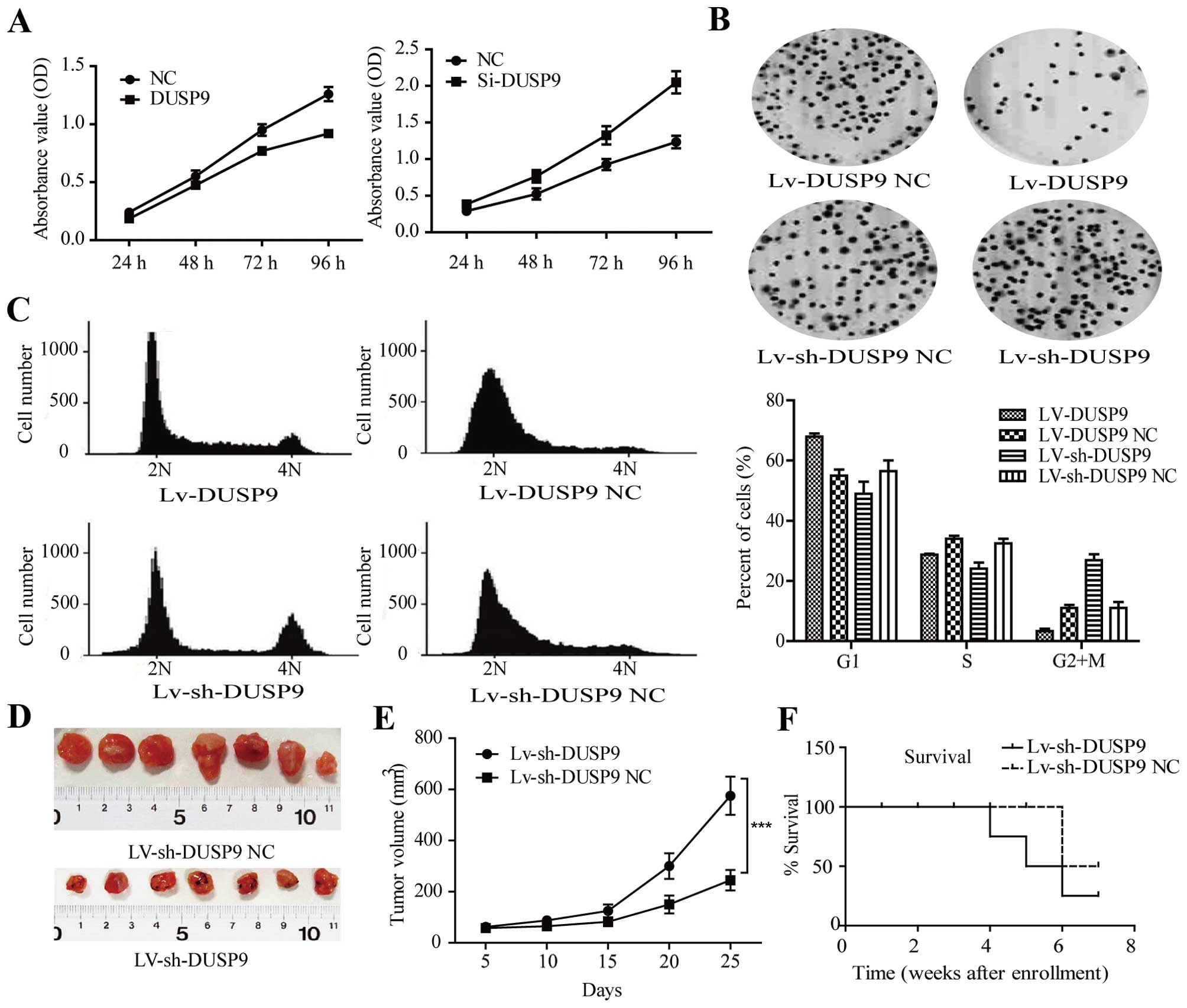
DUSP9 induces growth inhibition in GC
cells in vitro and in vivo
To examine the effect of DUSP9 on cell growth, MKN-1
cells were transiently transfected with DUSP9 vector or negative
control vector, respectively. As shown in Fig. 3A, the results of MTT assay showed
that DUSP9 inhibited cell growth in MKN-1 cells by 52% (P<0.01),
whereas Si-DUSP9 promoted cell growth in MKN-1 cells by 74%
(P<0.01). By contrast, the DUSP9-negative control or Si-DUSP9
control had no effect on cell growth, indicating that the effect
caused by DUSP9 was highly specific. As demonstrated by the colony
formation assay, DUSP9-infected MKN-1 cells exhibited much fewer
and smaller colonies compared with LV-con-infected cells (Fig. 3B, P<0.01).
We used lentiviral vectors to stably restore the
expression of DUSP9 in MKN-1 cells and examined cell-cycle
distribution. Compared with the negative control, LV-DUSP9-infected
MKN-1 cells showed an increased percentage of cells in the G1 phase
and fewer cells in the S phase (Fig.
3C, P<0.01), while the cell-cycle distribution had a
significant difference between LV-sh-DUSP9 control and
LV-sh-DUSP9-transfected cells (Fig.
3C, P<0.0001). These results suggested that the
growth-suppressive effect of DUSP9 was partly due to a G1 phase
arrest.
MKN-1 cells were infected with LV-sh-DUSP9 and then
injected subcutaneously into the dorsal flank of nude mice. As
early as 2 weeks post-implantation, the growth of transplanted
tumors between two groups became statistically significant. At 25
days after implantation, those mice injected with LV-sh-DUSP9
carried larger burdens. As compared with the LV-sh-DUSP9-treated
group, the average tumor volume of the control group was markedly
reduced by >60% (Fig. 3D and E,
P<0.001). The loss of animals in both arms of the study is
visible in Fig. 3F, where the
survival curve for the control arm decreased considerably after
week 4, with the difference between the two groups approaching
significance, as determined by the log-rank Mantel-Cox test
(P<0.01).
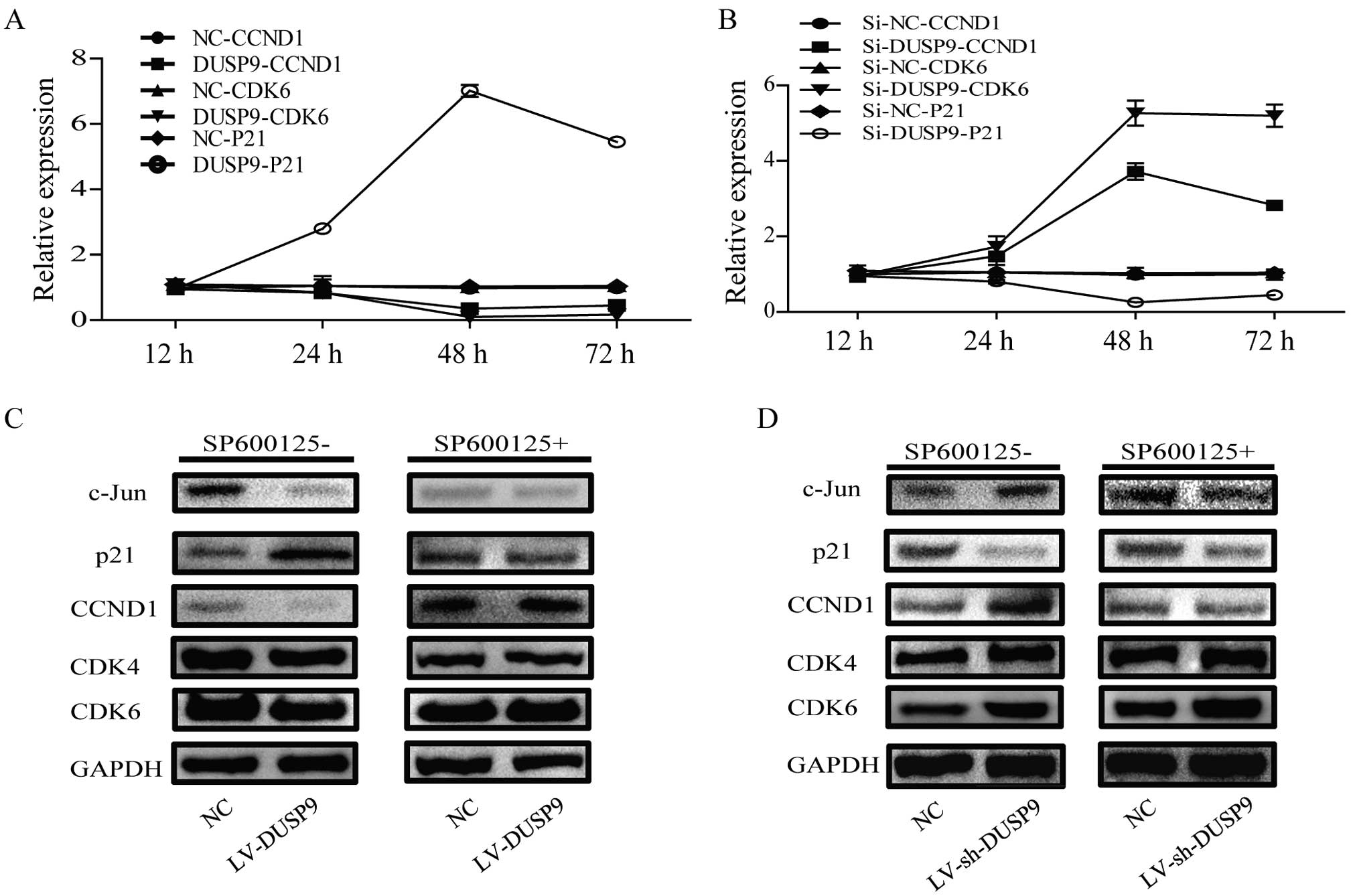
DUSP9 inhibits progression of the cell
cycle at S-G2/M phase by regulating cell cycle-related
molecules
To investigate the mechanisms leading to loss of
cell proliferation by DUSP9, we assessed whether the observed
inhibitory effects of DUSP9 on cell proliferation were due to
induction of cell-cycle arrest. As previously described, infection
with LV-sh-DUSP9 increased the percentage of cells in the S and
G2/M phase. DUSP-mediated cycle-related molecules downregulation
was observed in the transient expression system in MKN-1 cells
(Fig. 4A). RT-qPCR showed that, the
mRNA expression level of the CCND1 gene was shown to be decreased
0.81-fold at 24 h and 0.35-fold at 48 h, and this level was
sustained up to 72 h after the transfection of DUSP9 into MKN-1
cells. The mRNA expression levels of CDK4 and CDK6 in DUSP9
gene-transfected MKN-1 cells were at the same level as that of
CCND1 mRNA expression throughout the time course. By contrast,
Si-DUSP9-mediated cycle-related molecules upregulation was observed
in the transient expression system in MKN-1 cells (Fig. 4B).
We also examined the effect of DUSP9 on cell
cycle-regulatory molecules, including c-Jun, CCND1, CDK4, CDK6, and
p21 (Fig. 4C, left). The infection
of MKN-1 cells with LV-DUSP9 resulted in downregulation in the
levels of c-Jun protein as well as the levels of CCND1.
Downregulation of CDK4 and CDK6 was also observed in MKN-1 cells.
The level of p21 protein increased markedly following the infection
of MKN-1 cells with LV-DUSP9. By contrast, the infection of MKN-1
cells with LV-sh-DUSP9 resulted in upregulation in the levels of
c-Jun protein as well as the levels of CCND1. Upregulation of CDK4
and CDK6 was also observed in MKN-1 cells. In addition, the
expression of p21 increased in LV-sh-DUSP9-infected cells in
comparison with the controls. However, the infection of MKN-1 cells
with LV-sh-DUSP9/LV-DUSP9 resulting in regulation of the levels of
cycle-related molecules was inhibited when the JNK inhibitor
SP600125 was added (Fig. 4C, right
and 4D, right), suggesting that the drug inhibits the
DUSP9-mediated JNK activation of c-Jun, CCND1, CDK4, CDK6 and
inactivation of p21. Thus, the results of the present study confirm
that DUSP9 induces regulation in the levels of cycle-related
molecules via JNK signaling.
Discussion
DNA methylation of promoter-associated CpG islands
may function as an alternate mechanism of silencing
tumor-suppressor genes in numerous neoplasias, including GC. De
novo methylation of genes appears to be an early and frequent
event in most neoplasias (24–28).
Hypermethylation occurs at different stages in the development of
cancer and in different cell networks (29). In the genesis of many types of
cancer, hypermethylation of the CpG islands in the promoter regions
of tumor-suppressor genes is a major event, and is able to affect
genes involved in the cell cycle, DNA repair, the metabolism of
carcinogens, cell-to-cell interaction, apoptosis, and angiogenesis
(30,31). In the present study, the expression
of DUSP9 has been reported to be reduced in different types of
cancer (19–22). The results show that, DUSP9 was
frequently methylated in human GC and the expression of DUSP9 was
suppressed by promoter region methylation.
The aim of this study was to examine the CpG island
methylation status of DUSP9, on 30 clinical GC samples and selected
corresponding tumor-free tissues, using BSP for the determination
of the promoter methylation status. The methylation status of the
tumor samples was compared to that of the corresponding tumor-free
samples. To the best of our knowledge, this is the first study
using BSP for a DUSP9 methylation analysis on clinical GC samples.
The determination of the methylation status for each sample and CpG
island was successful.
DUSP9 was silenced by promoter region
hypermethylation and G2/M phase arrest was induced by DUSP9 in
MKN-1 cells. DUSP9 inhibits GC growth, suggesting that DUSP9 is a
tumor suppressor in human GC. The expression of CCND1, c-Jun, CDK4,
CDK6 was upregulated whereas p21 was down-regulated by DUSP9 in
MKN-1 cells. However, the infection of MKN-1 cells with
LV-sh-DUSP9/LV-DUSP9 resulted in the regulation in the levels of
cycle-related molecules being suppressed when the JNK inhibitor
SP600125 was added. Our results suggest that cell proliferation was
suppressed by DUSP9 by inhibiting c-Jun, which was induced by JNK
signaling. Loss of DUSP9 with progression of GC allows the
growth-promoting activities of JNK to persist, while the invasive
and growth inhibitory effects are lost. The increased levels of
DUSP9 expression observed in human GC cell lines were inversely
correlated with JNK activity and markers of apoptosis, indicating
that DUSP9 may be anti-proliferative in GC via its activity towards
JNK.
In conclusion, DUSP9 was frequently methylated in
human GC and the expression of DUSP9 was silenced by the promoter
region hypermethylation. DUSP9 suppresses GC proliferation by
inhibiting JNK signaling pathways. As was the case in the present
study, overexpression of DUSP9 correlated with reduced JNK
activity. However, DUSP9-induced JNK kinase inactivation can be
specifically blocked by the inhibitor SP600125. This results
suggest that therapeutic intervention to increase the expression or
activity of DUSP9 may enable the activation of the
anti-proliferative signals in malignant cells.
Acknowledgments
This study was supported by the Medicine and Health
grant from Wenzhou Bureau of Science and Technology (grant no.
Y20140281). The authors thank Ming Li (Sun Yat-Sen University
Cancer Center, Guangzhou, People’s Republic of China) and Liang
Wang (Zhejiang University, Hangzhou, Zhejiang, People’s Republic of
China) for technical support.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hartgrink HH, Jansen EP, van Grieken NC
and van de Velde CJ: Gastric cancer. Lancet. 374:477–490. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wu WK, Cho CH, Lee CW, Fan D, Wu K, Yu J
and Sung JJ: Dysregulation of cellular signaling in gastric cancer.
Cancer Lett. 295:144–153. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Venerito M, Nardone G, Selgrad M, Rokkas T
and Malfertheiner P: Gastric cancer - epidemiologic and clinical
aspects. Helicobacter. 19(Suppl 1): 32–37. 2014. View Article : Google Scholar
|
|
5
|
Wagner EF and Nebreda AR: Signal
integration by JNK and p38 MAPK pathways in cancer development. Nat
Rev Cancer. 9:537–549. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dong HW, Zhang S, Sun WG, Liu Q, Ibla JC,
Soriano SG, Han XH, Liu LX, Li MS and Liu JR: β-ionone arrests cell
cycle of gastric carcinoma cancer cells by a MAPK pathway. Arch
Toxicol. 87:1797–1808. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen P, Zhao D, Sun Y, Huang L, Zhang S
and Yuan Y: Protein inhibitor of activated STAT-1 is downregulated
in gastric cancer tissue and involved in cell metastasis. Oncol
Rep. 28:2149–2155. 2012.PubMed/NCBI
|
|
8
|
Chen L, Hu W, Tan S, Wang M, Ma Z, Zhou S,
Deng X, Zhang Y, Huang C, Yang G, et al: Genome-wide identification
and analysis of MAPK and MAPKK gene families in Brachypodium
distachyon. PLoS One. 7:e467442012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Berlier JL, Rigutto S, Dalla Valle A,
Lechanteur J, Soyfoo MS, Gangji V and Rasschaert J: Adenosine
triphosphate prevents serum deprivation-induced apoptosis in human
mesenchymal stem cells via activation of the MAPK signaling
pathways. Stem Cells. 33:211–218. 2015. View Article : Google Scholar
|
|
10
|
Dhillon AS, Hagan S, Rath O and Kolch W:
MAP kinase signalling pathways in cancer. Oncogene. 26:3279–3290.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen Z, Cai Y, Zhang W, Liu X and Liu S:
Astragaloside IV inhibits platelet-derived growth
factor-BB-stimulated proliferation and migration of vascular smooth
muscle cells via the inhibition of p38 MAPK signaling. Exp Ther
Med. 8:1253–1258. 2014.PubMed/NCBI
|
|
12
|
Bernardo-Faura M, Massen S, Falk CS, Brady
NR and Eils R: Data-derived modeling characterizes plasticity of
MAPK signaling in melanoma. PLoS Comput Biol. 10:e10037952014.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Park GB, Choi Y, Kim YS, Lee HK, Kim D and
Hur DY: ROS-mediated JNK/p38-MAPK activation regulates Bax
translocation in sorafenib-induced apoptosis of EBV-transformed B
cells. Int J Oncol. 44:977–985. 2014.PubMed/NCBI
|
|
14
|
Favata MF, Horiuchi KY, Manos EJ, Daulerio
AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F,
et al: Identification of a novel inhibitor of mitogen-activated
protein kinase kinase. J Biol Chem. 273:18623–18632. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Caunt CJ and Keyse SM: Dual-specificity
MAP kinase phosphatases (MKPs): shaping the outcome of MAP kinase
signalling. FEBS J. 280:489–504. 2013. View Article : Google Scholar :
|
|
16
|
Wu GS: Role of mitogen-activated protein
kinase phosphatases (MKPs) in cancer. Cancer Metastasis Rev.
26:579–585. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Keyse SM: Dual-specificity MAP kinase
phosphatases (MKPs) and cancer. Cancer Metastasis Rev. 27:253–261.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Muda M, Boschert U, Smith A, Antonsson B,
Gillieron C, Chabert C, Camps M, Martinou I, Ashworth A and
Arkinstall S: Molecular cloning and functional characterization of
a novel mitogen-activated protein kinase phosphatase, MKP-4. J Biol
Chem. 272:5141–5151. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu Y, Lagowski J, Sundholm A, Sundberg A
and Kulesz-Martin M: Microtubule disruption and tumor suppression
by mitogen-activated protein kinase phosphatase 4. Cancer Res.
67:10711–10719. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wu S, Wang Y, Sun L, Zhang Z, Jiang Z, Qin
Z, Han H, Liu Z, Li X, Tang A, et al: Decreased expression of
dual-specificity phosphatase 9 is associated with poor prognosis in
clear cell renal cell carcinoma. BMC Cancer. 11:4132011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu J, Ni W, Xiao M, Jiang F and Ni R:
Decreased expression and prognostic role of mitogen-activated
protein kinase phosphatase 4 in hepatocellular carcinoma. J
Gastrointest Surg. 17:756–765. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jenner S, Wiedorn KH and Techel D:
Development of a DUSP9 methylation screening assay. Pathol Oncol
Res. 21:123–130. 2015. View Article : Google Scholar
|
|
23
|
Li LC and Dahiya R: MethPrimer: designing
primers for methylation PCRs. Bioinformatics. 18:1427–1431. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Baylin SB, Herman JG, Graff JR, Vertino PM
and Issa JP: Alterations in DNA methylation: a fundamental aspect
of neoplasia. Adv Cancer Res. 72:141–196. 1998. View Article : Google Scholar
|
|
25
|
Baylin SB and Herman JG: DNA
hypermethylation in tumorigenesis: epigenetics joins genetics.
Trends Genet. 16:168–174. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Palau A, Perucho M, Esteller M and
Buschbeck M: First Barcelona conference on epigenetics and cancer.
Epigenetics. 9:468–475. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu G, Liu YJ, Lian WJ, Zhao ZW, Yi T and
Zhou HY: Reduced BMP6 expression by DNA methylation contributes to
EMT and drug resistance in breast cancer cells. Oncol Rep.
32:581–588. 2014.PubMed/NCBI
|
|
28
|
Minning C, Mokhtar NM, Abdullah N,
Muhammad R, Emran NA, Ali SA, Harun R and Jamal R: Exploring breast
carcinogenesis through integrative genomics and epigenomics
analyses. Int J Oncol. 45:1959–1968. 2014.PubMed/NCBI
|
|
29
|
Vizoso M and Esteller M: German-Catalan
workshop on epigenetics and cancer. Epigenetics. 8:998–1003. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Esteller M: Epigenetics in cancer. N Engl
J Med. 358:1148–1159. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Guerrero-Preston R, Hadar T, Ostrow KL,
Soudry E, Echenique M, Ili-Gangas C, Pérez G, Perez J,
Brebi-Mieville P, Deschamps J, et al: Differential promoter
methylation of kinesin family member 1a in plasma is associated
with breast cancer and DNA repair capacity. Oncol Rep. 32:505–512.
2014.PubMed/NCBI
|