Introduction
Gastric cancer remains the fourth most common
malignancy and the second leading cause of cancer-related mortality
worldwide, although its incidence is gradually on the decrease in
most parts of the world (1). The
currently available chemotherapeutic agents have only limited
benefits for most gastric cancer patients. Therefore,
identification of novel targets and an understanding of their
molecular mechanisms is crucial for the treatment of gastric cancer
patients.
Histone deacetylases (HDACs) are a class of enzymes
that remove the acetyl groups from core histones, as well as
non-histone proteins, such as p53 (2). By modifying the status of
deacetylation, HDACs induce transcriptional repression through
chromatin condensation and alteration of protein activity (3). Among the 18 HDAC family members, HDAC1
accounts for more than half of cellular HDAC activity, and cannot
be compensated by other HDACs (4).
HDAC1 acts at all levels of gene expression including microRNA
(miRNA or miR) regulation (5).
Overexpression of HDAC1 is significantly associated with higher
lymphatic metastases and decreased 3-year survival rates in gastric
cancer patients (6). Many HDAC
inhibitors have been developed, several of which have shown promise
and are under investigation in clinical trials (3,7).
However, the underlying mechanisms for the anti-metastatic
activities of HDAC inhibitors, particularly for those targeting
specific HDAC1 isoform in gastric cancer, remain unclear.
miRNAs are a class of short-length (21–23
nucleotides) non-coding RNAs that regulate gene expression, and are
involved in various cell functions that are essential for
pathological processes in gastric cancer (8). miRNAs exert functions as tumor
suppressors or oncogenes during carcinogenesis, tumor growth and
metastatic dissemination (9).
miR-34a is a well-known tumor suppressor that regulates the
expression of Bcl-2 (10). miR-34a
is widely suppressed in cancer cells, and the highly enriched
miR-34a-responsive genes regulate cell proliferation, apoptosis and
angiogenesis (10). Therefore,
miR-34a has become a promising target for developing novel
anticancer agents. However, it is difficult to directly manipulate
miRNAs themselves, thus targeting the upstream regulators of miRNAs
is a more efficient and operable approach. It has been demonstrated
that the expression of miRNAs is altered by epigenetic drugs
including HDAC inhibitors (11–13).
In addition, the expression of miR-34a is positively regulated by
p53 (14) and HDAC1 suppresses p53
activity via deacety-lation (15).
Therefore, HDAC1 may play a role in the metastasis of gastric
cancer cells by negatively regulating miR-34a.
Materials and methods
Cell culture
Human gastric cancer SGC-7901 (metastatic carcinoma
of lymph node) and MGC-803 (poorly differentiated mucinous
adenocarcinoma) cells were purchased from the Type Culture
Collection Cell Bank (Chinese Academy of Sciences Committee,
Shanghai, China). The cells were routinely cultured in RPMI-1640
medium supplemented with 10% fetal bovine serum (FBS), 100 U/ml
penicillin, and 100 µg/ml streptomycin at 37°C in a
humidified atmosphere of 5% CO2. The culture media and
supplements were purchased from HyClone Co. (Logan, UT, USA).
Antibodies and related reagents
Primary antibodies (Abs) against HDAC1, Bcl-2 and
glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were purchased
from Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA; a primary
Ab against CD44 was purchased from Cell Signaling Technology,
Danvers, MA, USA; and primary Abs against Ras homolog family member
A (RhoA), active RhoA Ab, LIM domain kinase 1 (LIMK-1) and
phosphorylated LIMK-1 (p-LIMK-1) were purchased from Abcam,
Cambridge, MA, USA. A secondary horseradish peroxidase-conjugated
Ab was obtained from the Zhongshan Golden Bridge Biotechnology Co.,
Ltd., Beijing, China. A primary Ab against β-tubulin, a
biotin-conjugated secondary Ab and a streptavidin-biotin complex
(SABC)-Cy3 kit were purchased from Boster Biological Technology
Co., Ltd., Wuhan, China. The fluorescein isothiocyanate
(FITC)-conjugated phalloidin was purchased from Sigma, Shanghai,
China.
Transfection of RNAs
RNAs used in the present study included a
double-strand siRNA targeting human HDAC1 (sense,
5′-CCGGUCAUGUCCAAAGUAATT-3′ and antisense,
5′-ACGUGACACGUUCGGAGAATT-3′); a double-strand siRNA targeting CD44
(sense, 5′-CUCCCAGUAUGACAC AUAUTT-3′ and antisense,
5′-AUAUGUGUCAUACUGGGA GTT-3′); miR-34a mimics (sense,
5′-UGGCAGUGUCUUAGC UGGUUGU-3′ and antisense, 5′-AACCAGCUAAGACACU
GCCAUU-3′); a non-specific scrambled double-strand RNA (sense,
5′-UUCUCCGAACGUGUCACGUTT-3′ and antisense,
5′-ACGUGACACGUUCGGAGAATT-3′) serving as negative control siRNAs and
miRNA mimics; a miR-34a antagomiR (5′-ACAACCAGCUAAGACACUGCCA-3′);
and a scrambled control antagomiR (5′-CAGUACUUUUGUGU AGUACAA-3′)
(GenePharma Co., Ltd., Shanghai, China). Cells were grown to 60–70%
confluence, and incubated with RNAs at a final concentration of 0.1
µM using Lipofectamine™ 2000 (Invitrogen, Beijing, China) in
a serum-free medium for 48 h and then subjected to assays.
miRNA microarray analysis
Total RNA was extracted using a TRIzol (Invitrogen)
and RNeasy Mini kit (Qiagen, Hilden, Germany), and RNA
concentrations quantified with the NanoDrop NP-1000. The samples
were labeled using the miRCURY™ Hy3™/Hy5™ Power labeling kit and
hybridized on the miRCURY™ LNA Array (v.18.0) (both from Exiqon,
Vedbaek, Denmark). Slides were washed using a wash buffer (Exiqon),
dried and scanned on an Axon GenePix 4000B microarray scanner
(Molecular Devices, Downingtown, PA, USA). GenePix pro V6.0
(Molecular Devices) was used to measure the raw intensity. After
background subtraction and normalization, the results were
subjected to unsupervised hierarchical clustering (Cluster
3.0).
Reverse transcription-quantitative PCR
(RT-qPCR)
The RT-qPCR of mature miRNAs was performed using
TaqMan miRNA assays. Total RNA was harvested as above, and
complementary DNA was synthesized with a GeneAmp PCR System 9700
(Applied Biosystems, Foster City, CA, USA) using an MMLV reverse
transcriptase (Epicentre, Madison, WI, USA). RT-qPCR was performed
by an ABI PRISM 7900 system (Applied Biosystems). Experiments were
performed in triplicate, and data were presented as fold changes
calculated by the 2−ΔΔCt method. The levels of miRNAs
were normalized against U6 controls. The two reverse transcription
primers: 5′-GTCGTATCCAGTGCGTGTCGTGGAGTCGGCAATTG
CACTGGATACGACACAACCAG-3′, and 5′-CGCTTCACG AATTTGCGTGTCAT-3′ were
designed for miR-34a and U6, respectively. Two pairs of RT-qPCR
primers were designed for miR-34a-5p (forward,
5′-AGGGGGTGGCAGTGTCTTAG-3′ and reverse, 5′-GTGCGTGTCGTGGAGTCG-3′);
and U6 (forward, 5′-GCTTCGGCAGCACATATACTAAAAT-3′ and reverse,
5′-CGCTTCACGAATTTGCGTGTCAT-3′). The primers were obtained from
Invitrogen (Shanghai, China).
Immunoblotting assay
An immunoblotting assay was performed as previously
described (16,17). Briefly, the cells were lysed and
protein concentrations were quantified. Cell lysates were separated
by SDS-PAGE and transferred onto polyvinylidene difluoride (PVDF)
membranes (Millipore, Bedford, MA, USA). After blocking with 5%
non-fat milk, blots were probed overnight at 4°C with primary Abs
(dilutions: anti-HDAC1, 1:200; anti-CD44, 1:200; anti-Bcl-2, 1:200;
anti-RhoA, 1:1,000; anti-LIMK-1, 1:1,000; anti-P-LIMK-1, and
1:1,000; anti-GAPDH, 1:2,000), and then incubated with secondary
Abs (1:1,000 dilution). The signal was detected by BeyoECL plus
reagent (Beyotime Institute of Biotechnology, Jiangsu, China) and
visualized using a ChemiDoc™ XRS+ System (Bio-Rad Laboratories,
Inc., Hercules, CA, USA). The density of each band was measured
using Image Lab™ software (Bio-Rad Laboratories, Inc.).
Cell migration and invasion assays
Cell migration and invasion assays were performed as
previously described (18). For the
migration assays, 2.5×104 cells suspended in 500
µl serum-free RPMI-1640 medium were seeded in BD Falcon cell
culture inserts (8-µm pore; BD Biosciences, San Jose, CA,
USA). Medium supplemented (750 µl) with 10% FBS was added
into the wells of the BD Falcon TC companion plate (BD Biosciences)
as the chemoattractant. After incubation for 24 h, the
non-migrating cells were removed from the upper surface of the
membrane by cotton swabs. The cells on the lower surface were fixed
in 4% paraformaldehyde, stained with Giemsa reagent (Jiancheng
Biotechnology, Nanjing, China) and air dried. The cells were then
counted at a magnification of ×100 in five random fields, using an
IX71 inverted phase contrast fluorescence microscope (Olympus,
Tokyo, Japan) and photographed with a digital camera. For the
invasion assays, the method was similar to that of the migration
assays. The cells were placed into the BD BioCoat Matrigel invasion
chambers (8-µm pore; BD Biosciences), which were coated with
the Matrigel matrix and rehydrated for 2 h with pre-warmed culture
medium at 37°C, in a humidified atmosphere of 5% CO2.
After incubation for 48 h, the non-invading cells were removed. The
cells attached to the lower surface were then fixed, stained, air
dried, counted and photographed.
Cell adhesion assay
Plates (96-well) were coated with fibro-nectin (80
µg/ml; Sigma, St. Louis, MO, USA) overnight at 4°C, and
blocked with 2.0% bovine serum albumin at 37°C for 1 h in a
humidified incubator. The cells were seeded at a concentration of
1×104 cells/well and allowed to adhere for 1 h. The
non-adherent cells were then washed away with phosphate-buffered
solution and the adherent cells were subjected to CCK-8 assays to
read optical density (OD) values using a CCK-8 kit (Dojindo
Molecular Technologies, Beijing, China).
Immunofluorescence assay
The cells were fixed with 4% paraformaldehyde and
then permeabilized in 0.1% Triton X-100. For β-tubulin staining,
the cells were blocked in 10% normal goat serum, incubated with the
anti-β-tubulin Ab for 90 min, followed by incubation with the
biotin-conjugated secondary Ab for 30 min, and then stained with
the SABC-Cy3 for 30 min. For F-actin staining, the cells were
stained with FITC-conjugated phalloidin for 40 min. The cells were
then counterstained with 4′,6-diamidino-2-phenylindole (DAPI) for
15 min, and images were captured with an IX71 inverted phase
contrast fluorescence microscope (Olympus).
Gelatin zymography
A gelatin zymography assay was performed as
previously described (18). Cells
(30.0×104) were seeded in 6-well plates and starved with
serum-free medium for 24 h. The conditioned medium was collected
and ultracentrifuged. The respective centrifuged medium (40
µl) was separated by 10% SDS-PAGE gels containing 2 mg/ml
gelatin (Sigma) on ice under non-reducing conditions. After the
electrophoresis, the gels were incubated in the zymogram renaturing
buffer (2.5% Triton X-100 in deionized water) twice with gentle
agitation for 40 min at room temperature. The zymogram renaturing
buffer was decanted and replaced with zymogram-developing buffer
(50 mM Tris-HCl, 5 mM CaCl2, 0.02% Brij-35, 0.2 M NaCl,
pH 7.6). The gels were equilibrated for 30 min at room temperature
with gentle agitation, and incubated with a fresh
zymogram-developing buffer at 37°C for 42 h. Subsequently, the gels
were stained with 0.1% (w/v) Coomassie blue R-250 for 30 min at
room temperature, followed by destaining with an appropriate
Coomassie blue R-250 destaining solution [methanol, acetic acid,
water (30:10:60)]. Clear bands against the blue background
represented areas of gelatinolysis. Those bands were photographed
using a ChemiDoc™ XRS+ System (Bio-Rad Laboratories, Inc.).
RhoA-GTP pull-down assay
Active RhoA was pulled down from whole cell lysates
using a RhoA Activation Assay kit (NewEast Biosciences, Malvern,
PA, USA). Briefly, the cells were grown to 80–90% confluence, lysed
in the presence of protease inhibitors, and centrifuged. The
supernatant was incubated with anti-active RhoA Ab (1:1,000
dilution) and protein A/G Agarose bead slurry at 4°C for 1 h. The
beads were washed and resuspended in SDS-PAGE sample buffer, and
GTP-bound RhoA was immunoblotted.
Statistical analysis
Experiments were performed in triplicate and
repeated at least three times. Data are presented as mean values ±
standard deviation. Comparisons were made using one-way analysis of
variance followed by Tukey’s post-hoc test with SPSS statistical
software (version 21.0). P<0.05 was considered to indicate a
statistically significant result.
Results
Depletion of HDAC1 results in the
upregulation of miR-34a in gastric cancer cells
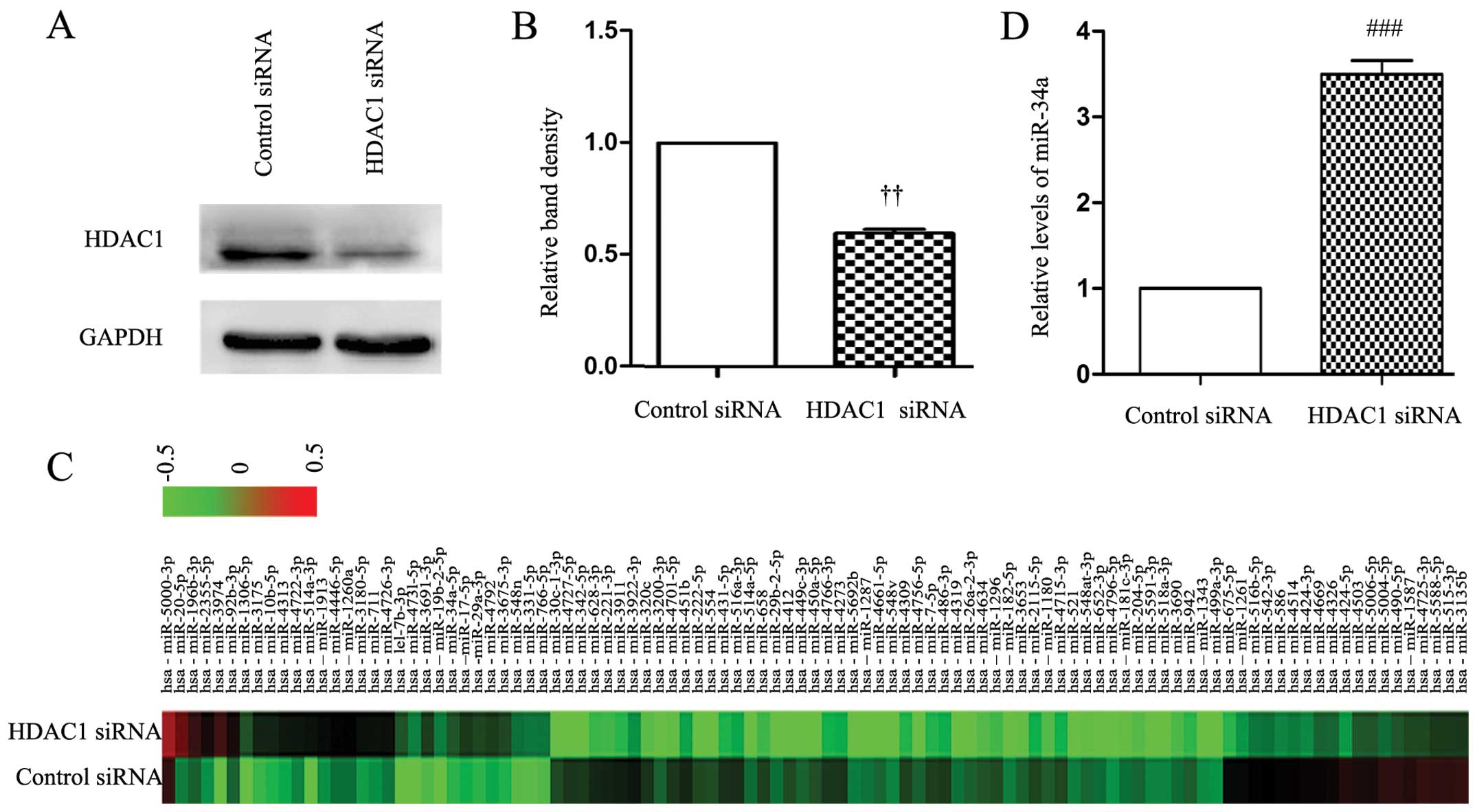
First, we used a specific siRNA to knock down the
expression of HDAC1 in SGC-7901 cells. As shown in Fig. 1A and B, the expression of HDAC1 was
significantly downregulated 48 h after HDAC1 siRNA transfection,
while the control siRNA had no effect on the expression of HDAC1.
We then examined the global miRNA expression in cells transfected
with control and HDAC1 siRNAs using a miRCURY™ LNA Array (Fig. 1C and Table I). The miRNA array data supported
our hypothesis that the expression of miR-34a was significantly
upregulated by knockdown of HDAC1. Consistently, the RT-qPCR
analysis confirmed that HDAC1 siRNA transfection significantly
increased the cellular expression level of miR-34a by ~4-fold
compared to the control siRNA (Fig.
1D).
 | Table IAlteration of miRNA expression
induced by HDAC1 siRNA in gastric cancer cells. |
Table I
Alteration of miRNA expression
induced by HDAC1 siRNA in gastric cancer cells.
| Upregulated
miRNAs | Control siRNA | HDAC1 siRNA | Fold-change | Downregulated
miRNAs | Control siRNA | HDAC1 siRNA | Fold-change |
|---|
| hsa-miR-20a-5p | 0.18565 | 4.47384 | 24.097710 | hsa-miR-342-5p | 0.46203 | 0.01744 | 0.037751 |
| hsa-let-7b-3p | 0.01477 | 0.32267 | 21.849670 |
hsa-miR-449c-3p | 0.26160 | 0.01163 | 0.044449 |
|
hsa-miR-196b-3p | 0.27637 | 2.22093 | 8.036038 | hsa-miR-222-5p | 0.37764 | 0.02035 | 0.053885 |
| hsa-miR-10b-5p | 0.08228 | 0.65988 | 8.020125 | hsa-miR-182-5p | 0.18565 | 0.01163 | 0.062632 |
| hsa-miR-29a-3p | 0.07384 | 0.38953 | 5.275415 | hsa-miR-4319 | 0.21941 | 0.01453 | 0.066246 |
| hsa-miR-17-5p | 0.09283 | 0.4564 | 4.916623 | hsa-miR-675-5p | 0.93882 | 0.07558 | 0.080507 |
| hsa-miR-92b-3p | 0.26793 | 1.29651 | 4.838949 | hsa-miR-320c | 0.50211 | 0.04070 | 0.081053 |
| hsa-miR-34a-5p | 0.07384 | 0.31686 | 4.291196 | hsa-miR-424-5p | 1.52954 | 0.12500 | 0.081724 |
|
hsa-miR-196a-3p | 0.40084 | 1.57849 | 3.937913 |
hsa-miR-450a-5p | 0.34599 | 0.02907 | 0.084019 |
| hsa-miR-10a-5p | 0.10759 | 0.40988 | 3.809508 | hsa-miR-221-3p | 0.66667 | 0.06105 | 0.091570 |
|
hsa-miR-24-1-5p | 1.12236 | 4.18605 | 3.729673 |
hsa-miR-26a-2-3p | 0.28059 | 0.03198 | 0.113962 |
| hsa-miR-1246 | 17.35230 | 64.6047 | 3.723113 | hsa-miR-652-3p | 0.22785 | 0.03198 | 0.140342 |
| hsa-miR-382-5p | 0.06329 | 0.23547 | 3.720349 | hsa-miR-424-3p | 1.09494 | 0.15698 | 0.143366 |
| hsa-miR-30b-5p | 0.14768 | 0.53198 | 3.602243 | hsa-miR-431-5p | 0.52321 | 0.08140 | 0.155570 |
| hsa-miR-320a | 0.52743 | 1.87209 | 3.549488 |
hsa-miR-181c-3p | 0.22152 | 0.03488 | 0.157475 |
| hsa-miR-133b | 0.41772 | 1.44477 | 3.458686 | hsa-miR-1296 | 0.36076 | 0.06105 | 0.169217 |
| hsa-miR-340-5p | 0.31013 | 1.06105 | 3.421334 | hsa-miR-204-5p | 0.25105 | 0.04360 | 0.173686 |
|
hsa-miR-193a-3p | 0.58861 | 1.99419 | 3.387972 | hsa-miR-7-5p | 0.41983 | 0.07849 | 0.186952 |
| hsa-miR-518b | 0.09916 | 0.32558 | 3.283523 |
hsa-miR-499a-3p | 0.13713 | 0.02616 | 0.190787 |
| hsa-miR-361-3p | 0.39662 | 1.28488 | 3.239547 | hsa-miR-486-3p | 0.44937 | 0.09302 | 0.207009 |
| hsa-miR-18b-3p | 0.98734 | 3.15116 | 3.191562 | hsa-miR-493-5p | 0.22363 | 0.06105 | 0.272982 |
|
hsa-miR-199a-5p | 0.29325 | 0.90698 | 3.092856 | hsa-miR-659-3p | 0.79325 | 0.23547 | 0.296836 |
| | | | hsa-miR-564 | 3.03797 | 0.92151 | 0.303331 |
| | | | hsa-miR-29b-3p | 0.20464 | 0.06395 | 0.312515 |
| | | | hsa-miR-96-5p | 0.14557 | 0.04942 | 0.339484 |
Knockdown of HDAC1 inhibits the abilities
of gastric cancer cells to migrate, invade and adhere by regulating
miR-34a
It has been reported that a high expression of HDAC1
is positively correlated with metastasis in gastric cancer
(6), thus we examined whether HDAC1
depletion affects the ability of SGC-7901 and MGC-803 cells to
migrate, invade and adhere. Transfection of HDAC1 siRNA led to
markedly reduced number of SGC-7901 cells (Fig. 2A and B) and MGC-803 cells (Fig. 2C and D) that had migrated and
invaded through the Transwell membranes. Quantitative analysis
confirmed that the numbers of migrated and invaded SGC-7901 cells
were highly significantly lower by 86.7 and 54.5%, respectively, in
HDAC1 siRNA-transfected cells, compared with controls (Fig. 2E and F), and the number of those
MGC-803 cells was significantly lower by 73.4 and 61.1% (Fig. 2G and H). In miR-34a
mimics-transfected cells, the numbers of migrated and invaded cells
were markedly decreased compared with the controls, while
co-transfection with HDAC1 siRNA and miR-34a mimics further
decreased those numbers (Fig.
2E–H). By contrast, transfection of a miR-34a antagomiR
significantly increased the numbers of migrated and invaded cells
compared with the controls. In addition, when cells were
co-transfected with HDAC1 siRNA and miR-34a antagomiR, the number
of migrated cells was significantly lower than cells transfected
with miR-34a antagomiR alone, and significantly higher than those
transfected with HDAC1 siRNA alone (Fig. 2E–H). HDAC1 siRNA or miR-34a mimics
significantly reduced, while miR-34a antagomiR significantly
increased, the number of adhered cells as expressed by OD values,
compared with the controls (Fig.
2I). Co-transfection with HDAC1 siRNA and miR-34a antagomiR
resulted in a significantly lower number of adhered cells than
miR-34 antagomiR alone, and a significantly higher number of
adhered cells than HDAC1 siRNA alone (Fig. 2I).
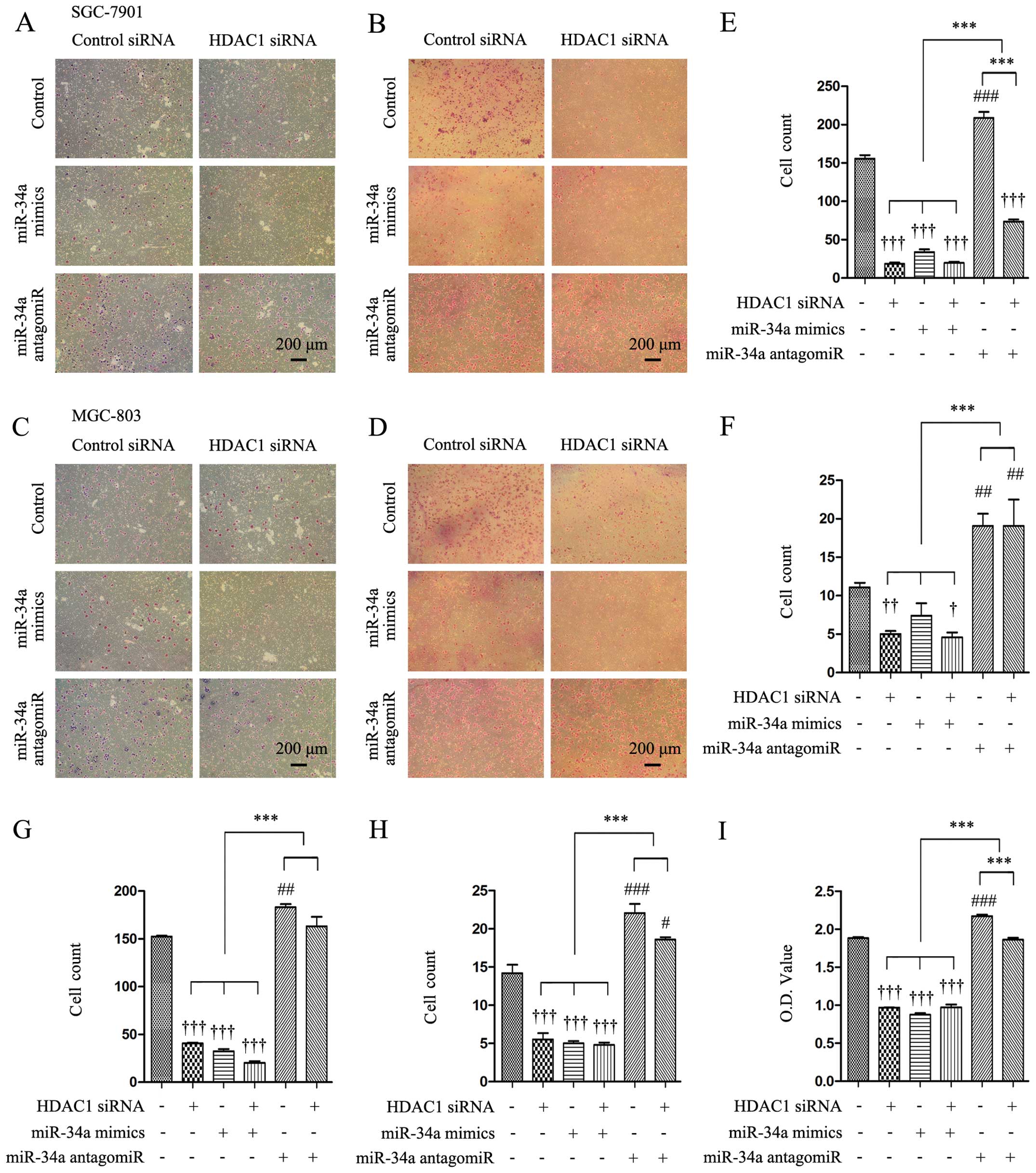 | Figure 2Knockdown of HDAC1 inhibits the
abilities of cells to migrate, invade and adhere via miR-34a.
SGC-7901 and MGC-803 cells were transfected with control or HDAC1
siRNAs in the presence of control or miR-34a mimics or miR-34a
antagomiR for 48 h, and then subjected to migration, invasion and
adhesion assays as described in Materials and methods. (A-D)
Representative images were captured from the migrated (A and C) and
invaded (B and D) cells (magnification, ×100). (E–H) The number of
migrated (E and G) and invaded (F and H) cells were counted. (I)
The adhered cells were subjected to a CCK-8 assay to measure OD
(optical density) values. †P<0.05,
††P<0.01 and †††P<0.001 indicate a
significant reduction, and #P<0.05,
##P<0.01 and ###P<0.001, a significant
increase, from cells transfected with control siRNA and control
antagomiR. ***P<0.001, a significant difference.
HDAC1, histone deacetylase 1. |
HDAC1 and miR-34a axis regulates cellular
cytoskeleton
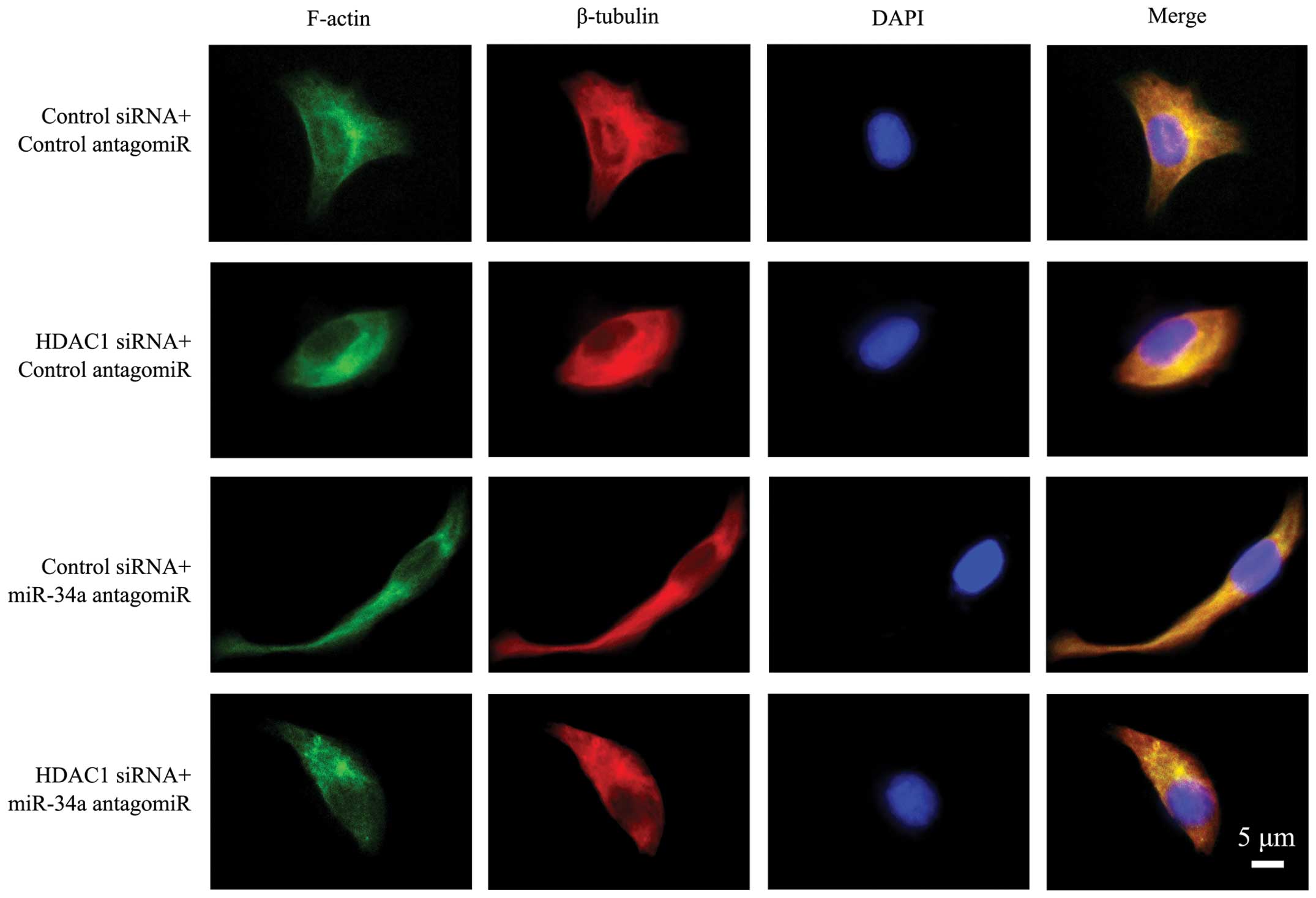
The above cells were immunostained with phalloidin
for the presence of microfilaments, an anti-β-tubulin Ab for the
presence of microtubules, and DAPI for the nuclei. As shown in
Fig. 3, the cells transfected with
control siRNA and antagomiR presented clear and intact
microfilaments, and intact microtubules with well-formed dendrites.
In cells depleted of HDAC1, microfilaments and microtubules were
impaired and blurred, leading to a rounder cell appearance without
obvious pseudopodia. The cells transfected with miR-34a antagomiR
had a more stretched arrangement of microfilaments and
microtubules, with obvious spread-out and long dendrites.
Co-transfection with miR-34a antagomiR partially restored the
impaired microfilaments and microtubules in cells transfected with
HDAC1 siRNA. However, the cells still showed a poorly organized
cytoskeleton and weakened dendrites (Fig. 3).
HDAC1/miR-34a regulates the expression of
CD44 in gastric cancer cells
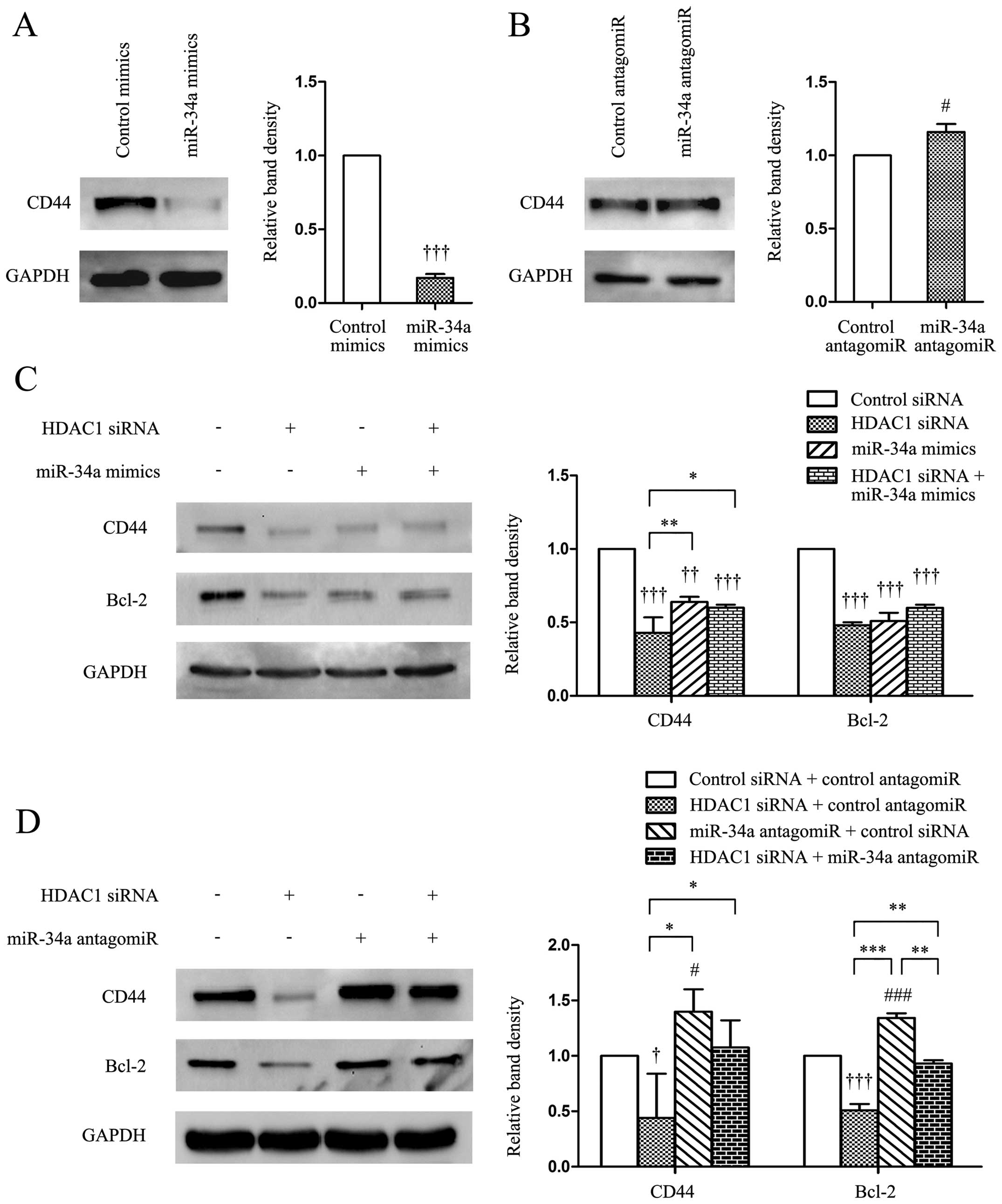
CD44 is a cell-surface adhesion glycoprotein
involved in cell adhesion, homing, migration and apoptosis
resistance and the development of anti-CD44 tumor-specific agents
has become a realistic therapeutic approach (19). In the present study, we demonstrated
that the transfection of miR-34a mimics significantly
downregulated, while miR-34a antagomiR upregulated, the expression
of CD44 in gastric cancer cells, compared with control mimics
(Fig. 4A and B). Transfection of
HDAC1 siRNA significantly reduced the expression of CD44 and Bcl-2
(Fig. 4C and D), with the latter
being a well-studied target protein of miR-34 (10). Co-transfection of miR-34a mimics and
HDAC1 siRNA did not significantly alter the expression levels of
CD44 and Bcl-2, compared with miR-34a mimics alone, whereas it
slightly reduced the expression of CD44, and significantly reduced
the expression of Bcl-2, compared with miR-34a antagomiR alone
(Fig. 4C and D).
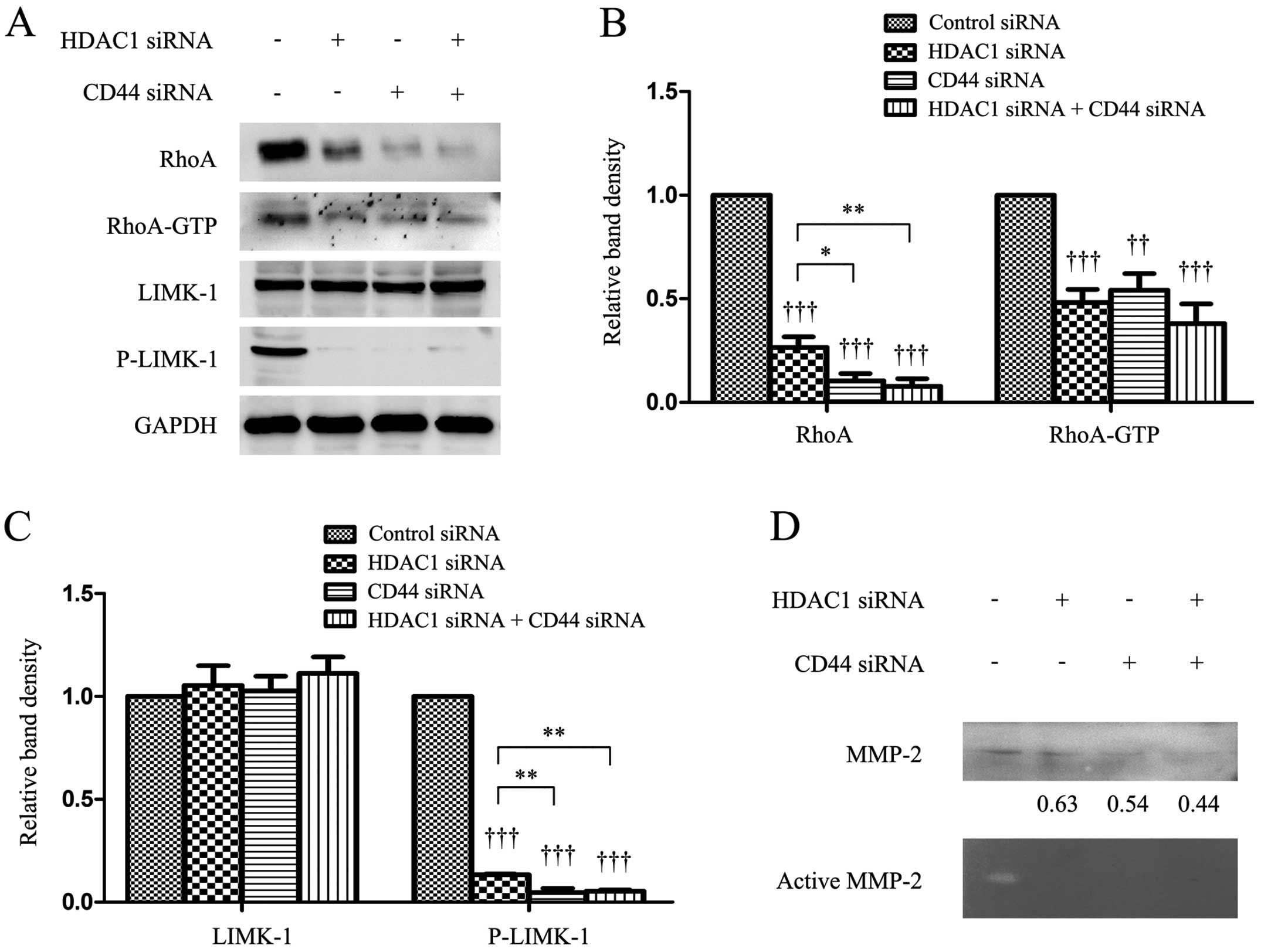
HDAC1 and CD44 regulates proteins
responsible for the cellular cytoskeleton
Transfection of HDAC1 or CD44 siRNA downregulated
the expression of RhoA and P-LIMK-1, and reduced RhoA activity as
evidenced by a lower level of GTP-bound RhoA. However, it had no
effect on the expression of LIMK-1, compared with the controls
(Fig. 5A–C). The simultaneous
depletion of HDAC1 and CD44 resulted in further downregulation of
RhoA and P-LIMK. However, the level of GTP-bound RhoA in the double
knockdown cells was only slightly lower than cells depleted of
HDAC1 or CD44 alone (Fig. 5A–C).
The immunoblotting results showed that the level of supernatant
MMP-2 protein was reduced by the depletion of HDAC1 or CD44, and
the results were supported by gelatin zymography assays, which
showed that depletion of HDAC1 or CD44 reduced the level of active
MMP-2 (Fig. 5D).
Discussion
The present study has, to the best of our knowledge,
demonstrated for the first time that HDAC1 depletion inhibits the
metastatic abilities of gastric cancer cells by regulating the
miR-34a/CD44 pathway. Depletion of HDAC1 significantly upregulated
the expression of miR-34a, and inhibited the abilities of gastric
cancer cells to migrate, invade and adhere, impaired the
organization of microfilaments and microtubules and impaired the
formation of cellular pseudopodia. These effects were restored by a
specific miR-34a antagomiR. The HDAC1 and miR-34a axis regulates
the cellular cytoskeleton by mediating the expression and/or
activation of CD44 and the downstream proteins including RhoA, LIMK
and MMP-2.
Given that HDACs are overexpressed in cancer,
inhibition of HDACs arrests cell cycling and often drives cancer
but not normal cells, into cell death pathways, attention has been
given to using HDAC inhibitors for cancer treatments (5). HDAC has also exhibited a profound
impact on the expression of microRNAs, resulting in changed
expression of ~40% of these microRNAs (20). Pan-HDAC inhibitors have been
demonstrated to regulate the expression of miRNAs in cells of colon
carcinoma, lymphoma and breast cancer (21). In accordance, in the present study
we have shown that depletion of HDAC1 by siRNA resulted in an
altered expression of 101 differentially expressed miRNAs matching
the 4-fold threshold in gastric cancer cells.
miR-34, a transcriptional target of p53, is a potent
tumor suppressor that inhibits a broad range of cancer cells by
repressing a plethora of oncogenes that control cell proliferation,
senescence, apoptosis and metastasis (10,22).
The miR-34 family comprises three processed miRNAs that are encoded
by two different genes: miR-34a is encoded by its own transcript,
whereas miR-34b and miR-34c share a common primary transcript
(10). miR-34a is often found
inactivated in cancer cells (10).
It has been reported that inhibition of HDAC1 mimics the miR-34a
phenotype (22). In the present
study, we have further demonstrated that depletion of HDAC1
upregulated the expression of miR-34a, but had little effect on the
expression of miR-34b or miR34c, suggesting that over-expressed
HDAC1 exerts its actions on gastric cancer cells by dysregulating
miR-34a. The results are also supported by a study that Vorinostat,
an HDAC inhibitor, restored the expression of miR-34a in pancreatic
cancer stem cells (23).
The present study has also demonstrated that the
antimetastatic effects of HDAC1 depletion are mediated by
regulating the expression of CD44 via miR-34a. Consistently, the
expression level of HDAC1 was positively correlated with that of
CD44 in chronic lymphocytic leukemia (7) and HDAC1 inhibitors reduced the
expression of CD44 via indirect mechanisms in hepatocellular
carcinoma cells (24). CD44 is a
direct and functional target of miR-34a as CD44 knockdown
phenocopies miR-34a overexpression in inhibiting the metastasis of
prostate cancer cells (25).
CD44 is a cell-surface adhesion glycoprotein
involved in cellular adhesion, migration and apoptosis resistance,
and is highly expressed in numerous cancer cells (26). CD44 indirectly links to actin by
interacting with ERM proteins (ezrin, radixin and moesin) (27). Knockdown of CD44 disturbs the
organization of actin cytoskeleton and the formation of cellular
pseudopodia (28). CD44 interacts
with RhoGEF to activate RhoA, which modulates cell motility through
the actin cytoskeleton, and promotes the lymphatic metastasis of
human gastric cancer cells (29).
The activated RhoA also activated LIMK-1, which is a key regulator
of cytoskeletal organization involved in the migration and
proliferation of cancer cells (30). In the present study, we have shown
that depletion of CD44 also reduced the level of supernatant MMP-2
protein and the activity of MMP-2, thus attenuating the cell
invasion ability since MMP-2 selectively increases the degradation
of extracellular matrix components, and facilitates tumor invasion
and metastasis (31).
In conclusion, the proposed mechanism by which
depletion of HDAC1 inhibits metastatic abilities of gastric cancer
cells via the miR-34a/CD44 pathway is shown in Fig. 6. HDAC1 downregulates the expression
of miR-34a, which in turn dysregulates CD44 mRNA, leading to the
over expression of the CD44 protein. CD44 indirectly links to actin
by interacting with ERM proteins (27), and activating RhoA by interacting
with RhoGEF. Activated RhoA guides the polymerization of tubulins
and actins by activating LIMK, which mediates the accumulation of
microfilaments and microtubules and cellular pseudopodia (32,33).
CD44 also regulates the expression and activation of MMP-2
(34), which indirectly activates
RhoA.
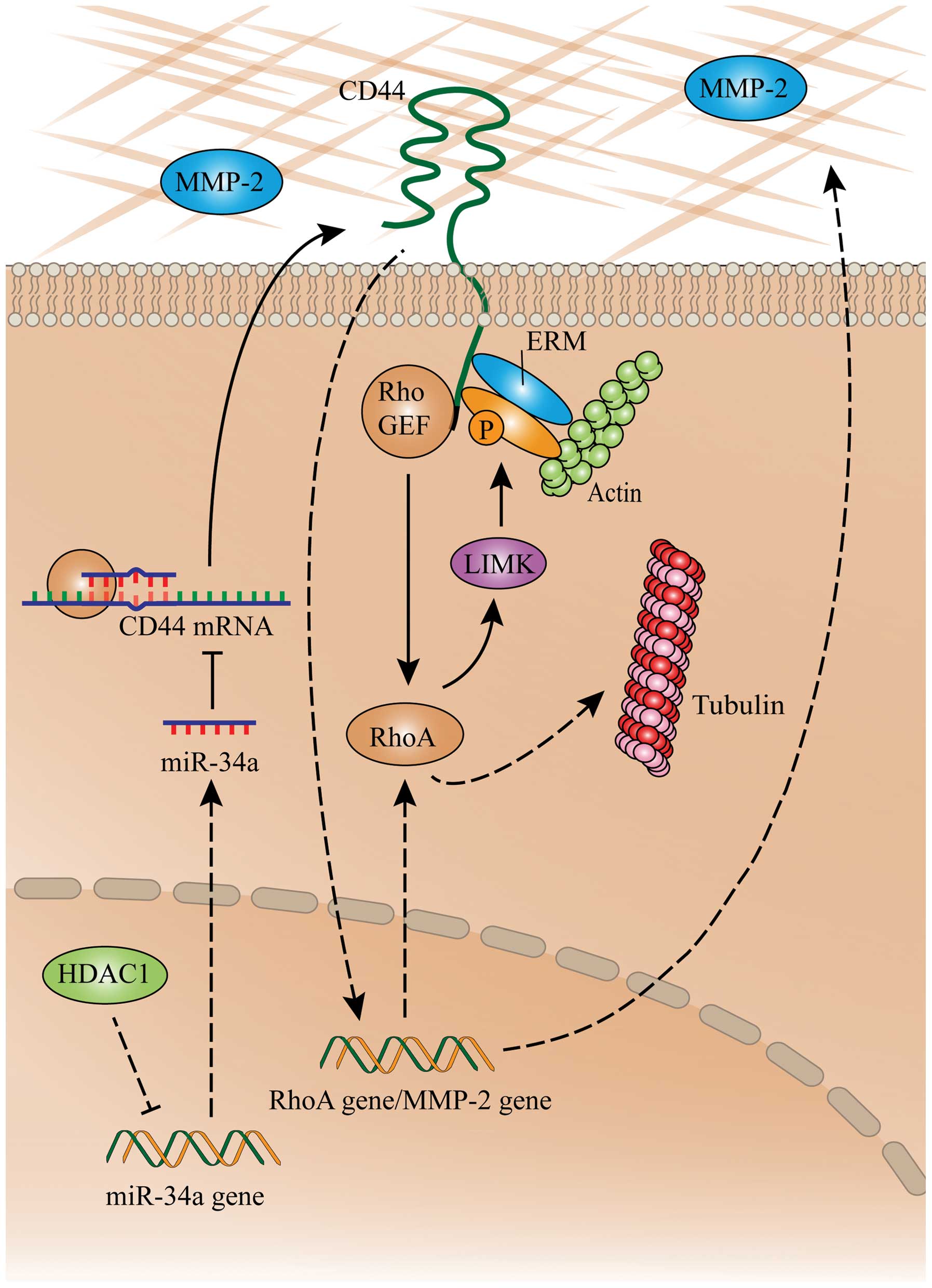 | Figure 6A schematic summary on the
HDAC1/miR-34a/CD44 pathway in gastric cancer cells. ‘→’ indicates
positive regulation or activation; ‘⊥’, negative regulation or
blockade. Solid lines indicate a direct regulation, while dashed
lines, an indirect regulation. ERM, ezrin, radixin and moesin;
HDAC1, histone deacetylase 1; LIMK, LIM domain kinase; miR,
microRNA; MMP-2, matrix metalloproteinase-2; RhoA, ras homolog
family member A. |
Acknowledgments
This study was supported by grants from the National
Natural Scientific Foundation (81272467), and the Ministry of
Health (201002015).
Abbreviations:
|
ERM proteins
|
ezrin, radixin and moesin
|
|
HDAC
|
histone deacetylase
|
|
LIMK
|
LIM domain kinase
|
|
miRNA or miR
|
microRNA
|
|
MMP
|
matrix metalloproteinase
|
|
P-LIMK
|
phosphorylated LIMK
|
|
RhoA
|
ras homolog family member A
|
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Choudhary C, Kumar C, Gnad F, Nielsen ML,
Rehman M, Walther TC, Olsen JV and Mann M: Lysine acetylation
targets protein complexes and co-regulates major cellular
functions. Science. 325:834–840. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Venugopal B and Evans TR: Developing
histone deacetylase inhibitors as anti-cancer therapeutics. Curr
Med Chem. 18:1658–1671. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dovey OM, Foster CT and Cowley SM: Histone
deacetylase 1 (HDAC1), but not HDAC2, controls embryonic stem cell
differentiation. Proc Natl Acad Sci USA. 107:8242–8247. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Delcuve GP, Khan DH and Davie JR:
Targeting class I histone deacetylases in cancer therapy. Expert
Opin Ther Targets. 17:29–41. 2013. View Article : Google Scholar
|
|
6
|
Weichert W, Röske A, Gekeler V, Beckers T,
Ebert MP, Pross M, Dietel M, Denkert C and Röcken C: Association of
patterns of class I histone deacetylase expression with patient
prognosis in gastric cancer: A retrospective analysis. Lancet
Oncol. 9:139–148. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang JC, Kafeel MI, Avezbakiyev B, Chen C,
Sun Y, Rathnasabapathy C, Kalavar M, He Z, Burton J and Lichter S:
Histone deacetylase in chronic lymphocytic leukemia. Oncology.
81:325–329. 2011. View Article : Google Scholar
|
|
8
|
Li T, Lu YY, Zhao XD, Guo HQ, Liu CH, Li
H, Zhou L, Han YN, Wu KC, Nie YZ, et al: MicroRNA-296–5p increases
proliferation in gastric cancer through repression of
Caudal-related homeobox 1. Oncogene. 33:783–793. 2014. View Article : Google Scholar
|
|
9
|
Garzon R, Marcucci G and Croce CM:
Targeting microRNAs in cancer: Rationale, strategies and
challenges. Nat Rev Drug Discov. 9:775–789. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hermeking H: The miR-34 family in cancer
and apoptosis. Cell Death Differ. 17:193–199. 2010. View Article : Google Scholar
|
|
11
|
Buurman R, Gürlevik E, Schäffer V, Eilers
M, Sandbothe M, Kreipe H, Wilkens L, Schlegelberger B, Kühnel F and
Skawran B: Histone deacetylases activate hepatocyte growth factor
signaling by repressing microRNA-449 in hepatocellular carcinoma
cells. Gastroenterology. 143:811–20.e1. 152012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang Y, Toh HC, Chow P, Chung AY, Meyers
DJ, Cole PA, Ooi LL and Lee CG: MicroRNA-224 is up-regulated in
hepatocellular carcinoma through epigenetic mechanisms. FASEB J.
26:3032–3041. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mims A, Walker AR, Huang X, Sun J, Wang H,
Santhanam R, Dorrance AM, Walker C, Hoellerbauer P, Tarighat SS, et
al: Increased anti-leukemic activity of decitabine via
AR-42-induced upregulation of miR-29b: A novel epigenetic-targeting
approach in acute myeloid leukemia. Leukemia. 27:871–878. 2013.
View Article : Google Scholar :
|
|
14
|
Chakraborty S, Mazumdar M, Mukherjee S,
Bhattacharjee P, Adhikary A, Manna A, Chakraborty S, Khan P, Sen A
and Das T: Restoration of p53/miR-34a regulatory axis decreases
survival advantage and ensures Bax-dependent apoptosis of non-small
cell lung carcinoma cells. FEBS Lett. 588:549–559. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
LeBoeuf M, Terrell A, Trivedi S, Sinha S,
Epstein JA, Olson EN, Morrisey EE and Millar SE: Hdac1 and Hdac2
act redundantly to control p63 and p53 functions in epidermal
progenitor cells. Dev Cell. 19:807–818. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhai B, Hu F, Jiang X, Xu J, Zhao D, Liu
B, Pan S, Dong X, Tan G, Wei Z, et al: Inhibition of Akt reverses
the acquired resistance to sorafenib by switching protective
autophagy to autophagic cell death in hepatocellular carcinoma. Mol
Cancer Ther. 13:1589–1598. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhao D, Zhai B, He C, Tan G, Jiang X, Pan
S, Dong X, Wei Z, Ma L, Qiao H, et al: Upregulation of HIF-2α
induced by sorafenib contributes to the resistance by activating
the TGF-α/EGFR pathway in hepatocellular carcinoma cells. Cell
Signal. 26:1030–1039. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wei Z, Jiang X, Liu F, Qiao H, Zhou B,
Zhai B, Zhang L, Zhang X, Han L, Jiang H, et al: Downregulation of
Skp2 inhibits the growth and metastasis of gastric cancer cells in
vitro and in vivo. Tumour Biol. 34:181–192. 2013. View Article : Google Scholar
|
|
19
|
Naor D, Nedvetzki S, Golan I, Melnik L and
Faitelson Y: CD44 in cancer. Crit Rev Clin Lab Sci. 39:527–579.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Scott GK, Mattie MD, Berger CE, Benz SC
and Benz CC: Rapid alteration of microRNA levels by histone
deacetylase inhibition. Cancer Res. 66:1277–1281. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Izzotti A, Cartiglia C, Steele VE and De
Flora S: MicroRNAs as targets for dietary and pharmacological
inhibitors of mutagenesis and carcinogenesis. Mutat Res.
751:287–303. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhao J, Lammers P, Torrance CJ and Bader
AG: TP53-independent function of miR-34a via HDAC1 and
p21CIP1/WAF1. Mol Ther. 21:1678–1686. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nalls D, Tang SN, Rodova M, Srivastava RK
and Shankar S: Targeting epigenetic regulation of miR-34a for
treatment of pancreatic cancer by inhibition of pancreatic cancer
stem cells. PLoS One. 6:e240992011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zeng SS, Yamashita T, Kondo M, Nio K,
Hayashi T, Hara Y, Nomura Y, Yoshida M, Hayashi T, Oishi N, et al:
The transcription factor SALL4 regulates stemness of EpCAM-positive
hepatocellular carcinoma. J Hepatol. 60:127–134. 2014. View Article : Google Scholar
|
|
25
|
Liu C, Kelnar K, Liu B, Chen X,
Calhoun-Davis T, Li H, Patrawala L, Yan H, Jeter C, Honorio S, et
al: The microRNA miR-34a inhibits prostate cancer stem cells and
metastasis by directly repressing CD44. Nat Med. 17:211–215. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang Y, Xia H, Ge X, Chen Q, Yuan D, Chen
Q, Leng W, Chen L, Tang Q and Bi F: CD44 acts through RhoA to
regulate YAP signaling. Cell Signal. 26:2504–2513. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zöller M: CD44: Can a cancer-initiating
cell profit from an abundantly expressed molecule? Nat Rev Cancer.
11:254–267. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sumoza-Toledo A and Santos-Argumedo L: The
spreading of B lymphocytes induced by CD44 cross-linking requires
actin, tubulin, and vimentin rearrangements. J Leukoc Biol.
75:233–239. 2004. View Article : Google Scholar
|
|
29
|
Lin MT, Lin BR, Chang CC, Chu CY, Su HJ,
Chen ST, Jeng YM and Kuo ML: IL-6 induces AGS gastric cancer cell
invasion via activation of the c-Src/RhoA/ROCK signaling pathway.
Int J Cancer. 120:2600–2608. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
McConnell BV, Koto K and
Gutierrez-Hartmann A: Nuclear and cytoplasmic LIMK1 enhances human
breast cancer progression. Mol Cancer. 10:752011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Murphy G and Nagase H: Progress in matrix
metalloproteinase research. Mol Aspects Med. 29:290–308. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hanna S and El-Sibai M: Signaling networks
of Rho GTPases in cell motility. Cell Signal. 25:1955–1961. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Struckhoff AP, Rana MK and Worthylake RA:
RhoA can lead the way in tumor cell invasion and metastasis. Front
Biosci. 16:1915–1926. 2011. View
Article : Google Scholar
|
|
34
|
Kim Y, Lee YS, Choe J, Lee H, Kim YM and
Jeoung D: CD44-epidermal growth factor receptor interaction
mediates hyaluronic acid-promoted cell motility by activating
protein kinase C signaling involving Akt, Rac1, Phox, reactive
oxygen species, focal adhesion kinase, and MMP-2. J Biol Chem.
283:22513–22528. 2008. View Article : Google Scholar : PubMed/NCBI
|