Introduction
Cholangiocarcinoma is a common primary hepatobiliary
malignancy that often arises from longstanding bile duct
inflammation, injury, and reparative biliary epithelial cell
proliferation (1,2). Prostaglandin E2 (PGE2), a key product
of cyclooxygenase-2, participates in the development of
inflammatory reactions and in oncogenesis (3,4). PGE2
was able to promote tumor cell proliferation, migration, invasion
and anti-apoptosis. In previous studies, PGE2 significantly
promoted proliferation and invasion by upregulating the expression
of survivin (5), focal adhesion
kinase (4), Snail (6,7), YB-1
(8), c-Myc (9) and β1-integrin (10) in hepatocellular carcinoma cells, and
by upregulating the expression of MMP2 (11) and the FUSE-binding protein 1
(12) in cholangiocarcinoma cells.
It was also reported that, PGE2 promoted cell proliferation and
invasion in breast (13), colon
(14), ovarian (15) cancer and several other types of
tumors.
The activity of PGE2 is mediated by four E
prostanoid receptors (EPR), known as EP1R, EP2R, EP3R and EP4R.
These constitute the G protein-coupled receptors (GPCRs). Although
previous studies have examined the relationship between tumor
growth, invasion and EP1R, EP2R, EP4R but few studies have focused
on the role EP3R. The EP3 receptor is more complicated (12,16).
EP3R mRNA has several splice variants that generate multiple
isoforms. Five isoforms of the human EP3 receptors: EP3-4, EP3-5,
EP3-6, EP3-7 and EP3-8 have been identified. Evidence of different
signal transduction pathways and the regulation of gene expression
among different EP receptors has also been demonstrated in a number
of studies (17). Thus, the
specific target of PGE2 in regulating cancer cell proliferation via
EP receptors have yet to be clarified.
It was reported that β-catenin is highly correlated
with cancer progression and poor prognosis. For example, the
expression level of β-catenin has been shown to correlate with
progression in colorectal (18),
cervical (19) and prostate cancer
(20). The activation of β-catenin
was also involved in various stages of hepatic carcinoma (21–24)
and cholangiocarcinoma (25,26).
β-catenin leads to the activation of target genes such as c-Myc
(27), Snail (28) and cyclin D1 (29). Although β-catenin has been regarded
as a useful biomarker of cancer progression, the detailed
mechanisms for its tumor-promoting function remain to be
elucidated.
In a previous study, EP3-4R, EP3-5R, EP3-6R and
EP3-7R were observed in human CCLP1 and HuCCT1 cholangiocar-cinoma
cells (12). Overexpression of the
stable transfection of EP3-4, EP3-5, EP3-6 and EP3-7 receptors to
CCLP1, HuCCT1 and HEK 293T cells showed that the EP3-4 receptor is
the most significant receptor in promoting the proliferation,
migration and invasion of the cholangiocarcinoma cells. The aim of
the present study was to assess the effects of the EP3R isoforms
4–7 on promoting cholangiocarcinoma cell growth and invasion and
the signaling pathway whereby PGE2 upregulates β-catenin protein.
Our results revealed that PGE2 upregulated β-catenin protein via
the EP3-4 receptor and involved the Src EGFR, AKT and GSK-3β
pathway. Upregulated β-catenin eventually caused increased c-Myc
and Snail expression.
Materials and methods
Antibodies and reagents
Human CCLP1 and HuCCT1 chol-angiocarcinoma cell
lines were obtained from the Department of Transplantation
Pathology, University of Pittsburgh Medical Center (UPMC;
Pittsburgh, PA, USA). HEK 293T cells were obtained from the
American Type Culture Collection (ATCC; Manassas, VA, USA).
Dulbecco’s modifed Eagle’s medium (DMEM), RPMI-1640 medium, and
Lipofectamine™ 2000 were purchased from Invitrogen-Life
Technologies (Carlsbad, CA, USA). PGE2, EP3 receptor selective
antagonist L-798106, EP3 receptor agonist sulprostone, anti-EP3
antibody were purchased from Cayman Chemical Co. (Ann Arbor, MI,
USA). Src inhibitor PP2 and G418 were purchased from Sigma-Aldrich
(St. Louis, MO, USA). Anti-phosphorylated EGFR (Tyr1173) and
anti-GAPDH antibodies were purchased from SAB Signalway Antibody,
(Nanjing, China). Anti-phosphorylated AKT (Ser473),
anti-phosphorylated GSK-3β (Ser9), anti-phos-phorylated β-catenin
(Ser33/37Thr41), anti-EGFR, anti-AKT, anti-GSK-3β, anti-β-catenin,
anti-Snail, anti-c-Myc antibodies, PI3K/AKT inhibitor LY294002, and
EGFR inhibitor AG1478 were purchased from Cell Signaling Technology
(Danvers, MA, USA). The protein assay was purchased from Bio-Rad
Laboratories (Hercules, CA, USA). Electrochemiluminescence (ECL)
reagents were purchased from Amersham Biosciences (Piscataway, NJ,
USA). The Transwell unit was purchased from Costar Corning
(Cambridge, MA, USA), while Matrigel was purchased from BD
Biosciences, (Discovery Labware, Bedford, MA, USA). The cell
proliferation assay reagent CCK-8 was purchased from Dojindo
Laboratories (Kumamoto, Japan). Primer sequences were determiend
using the PrimeScript™ RT reagent kit (Takara, Dalian, China).
FastStart Essential DNA Green Master Mix was purchased from Roche
Life Science (Indianapolis, IN, USA). PcDNA3.1, pcDNA-EP3-4,
pcDNA-EP3-5, pcDNA-EP3-6 and pcDNA-EP3-7 plasmids were purchased
from Jinsite Science and Technology Co. (Nanjing, China).
Cell culture
The cells were maintained at 37°C in a humidified 5%
CO2 incubator. CCLP1 and HEK 293T cells were cultured in
DMEM and HuCCT1 cells in RPMI-1640 medium containing 10% fetal
bovine serum (FBS).
Overexpression of EP3 receptor
The CCLP1, HuCCT1 and HEK 293T cells were exposed to
a mixture of Lipofectamine 2000 and pcDNA-EP3/pcDNA3.1 control
vector for 4 h. Following removal of the transfection mixtures,
fresh medium with 10% fetal bovine serum was added. On the second
day, the medium was changed, and the cells were incubated with
medium containing G418 sulfate for 14 days. Subsequent cultures of
selected cells were routinely grown in the presence of selective
pressure. Western blotting and quantitative PCR (qPCR) analyses
were performed in the selected cells permanently transfected with
EP3 receptors or control plasmids. The selected cells with the
successful increase in EP3 proteins and mRNA expression were used
for subsequent experiments.
Cell proliferation assay
Cell proliferation was determined using the CCK-8
kit. This kit contains the WST-8 reagent, a tetrazolium salt that
can be reduced by mitochondrial dehydrogenases in viable cells to
produce an orange-colored formazan dye. Briefly, 100 µl of
cell suspension was plated in each well of 96-well plates. After a
24-h culture to allow reattachment, the cells were incubated with
sulprostone at the indicated concentrations (0, 0.1, 1, 5 and 10
µM) and time periods. The cell proliferation reagent, WST-8
(10 µl), was subsequently added to each well. The incubation
was continued from 30 min to 4 h at 37°C and absorbance at 450 nm
was measured using an automatic ELISA plate reader. These
experiments were repeated in six individual wells.
Cell migration assay
Cells in 100 µl serum-free medium in the
presence or absence of sulprostone were seeded in the upper
chamber. Regular medium containing 10% FBS was added in the lower
chamber as the chemoattractant. After incubation at 37°C for 12 h,
the cells were fixed with ethanol and stained with 0.1% crystal
violet for 30 min. After washing the cells with PBS, the cells on
the upper surface of the filter were mechanically removed with a
cotton swab. The invading cells on the lower surface were
solubilized with 300 µl 10% acetic acid. The absorbance was
measured at 570 nm. These experiments were repeated three times,
and three wells were used for each treatment.
Cell invasion assay
The Matrigel was diluted with serum-free medium at
the indicated concentrations and then 40 µl of diluted
Matrigel was added to the upper chamber, and incubated at 37°C for
4 h to solidify the gel. Cells in 100 µl serum-free medium
in the presence or absence of sulprostone were seeded in the upper
chamber. Regular medium containing 10% FBS was added in the lower
chamber as the chemoattractant. After 24 h of incubation at 37°C,
the cells were fixed with ethanol and stained with 0.1% crystal
violet for 30 min. After washing the cells with PBS, the cells on
the upper surface of the filter were mechanically removed with a
cotton swab. The invading cells on the lower surface were
solubilized with 300 µl 10% acetic acid. The absorbance was
measured at 570 nm. These experiments were repeated three times,
and three wells were used for each treatment.
Western blotting
At the end of each treatment, the cells were washed
twice with ice-cold PBS and then sonicated on ice in a lysis buffer
(50 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.5% sodium deoxycholate, 1%
Nonidet P-40, 0.1% SDS, 1 mM PMSF, sodium orthovanadate, sodium
fluoride and aprotinin). Cell lysates were centrifuged at 12,000 ×
g for 10 min at 4°C, and the supernatants were collected for
western blotting. Protein concentration was measured using a
protein assay. After boiling for 5 min in the loading buffer with
10% 2-mercaptoethanol, the samples containing 30 µg protein
were separated by SDS-PAGE and transferred onto nitrocellulose
membranes. The membranes were blocked with 5% non-fat milk-TBST
buffer for 1 h at room temperature and incubated with the
corresponding primary antibodies overnight at 4°C with gentle
agitation. The membranes were washed in TBST and incubated for 2 h
with secondary antibodies at room temperature. The signals were
detected using ECL reagent and analyzed using Image Lab 4.0
analysis software from Bio-Rad.
RNA isolation and qPCR
Total RNA from the cultured cells was isolated using
the TRIzol reagent according to the manufacturer’s instructions.
Reverse transcription was carried out with PrimeScript™ RT reagent
kit according to the standard protocol. The primer sequences used
were: EP3-4R, 5′-TGC ATCCAGCTCCACCTCCT-3′ (forward) and 5′-GCAAAT
TCAGGGAAGCAGGAATTGC-3′ (reverse); EP3-5R, 5′-TGT
TCGCCTGGCTTCACTGAACC-3′ (forward) and 5′-AGA
GCAGCTGGAGACAGCATTTGC-3′ (reverse); EP3-6R,
5′-TGTTCGCCTGGCTTCACTGAACC-3′ (forward) and
5′-TGTGATCCTGGCAGAAAGGCAGG-3′ (reverse); EP3-7R,
5′-TGTGATCCTGGCAGAAAGGCAGG-3′ (forward) and
5′-TTTGGGAGGTGGGTGTTTCTGTGA′ (reverse); c-Myc,
5′-AGGCTATTCTGCCCATTT-3′ (forward) and 5′-TCGTAGTCGAGGTCATAGTTC-3′
(reverse); Snail, 5′-AAGGACCACCGCATCTCTAC-3′ (forward) and 5′-CC
AGCTCCTTGAAGCAGAAGA-3′ (reverse); β-catenin, 5′-GC
CAAGTGGGTGGTATAGAG-3′, (forward) and 5′-TGGGAT GGTGGGTGTAAG-3′
(reverse); GAPDH, 5′-TTCCAGGAG CGAGATCCCT-3′ (forward) and
5′-CACCCATGACGAA CATGGG-3′ (reverse). qPCR analysis was performed
on a Roche LightCycler Nano instrument using FastStart Essential
DNA Green Master Mix, and GAPDH was used as the endogenous control.
The qPCR conditions were: pre-incubation at 95°C for 10 min (1
cycle) followed by denaturation of 40 cycles at 95°C for 20 sec,
annealing at 60°C for 20 sec and extension at 72°C for 20 sec.
Treatments and conditions were performed in triplicate to calculate
the statistical significance.
Statistical analysis
Date are presented as the means ± SD. Statistical
analysis was performed with the Student’s t-test. P<0.05 was
considered statistically significant.
Results
Isoforms of EP3 receptors are
overexpressed in cholangiocarcinoma and HEK 293T cell
The CCLP1, HuCCT1 and HEK 293T cells were
transfected with EP3-4R, EP3-5R, EP3-6R, EP3-7R or the pcDNA
control plasmids and then selected by G418. The qPCR analysis
results showed that mRNAs of EP3-4R, EP3-5R, EP3-6R and EP3-7R were
increased significantly in pcDNA-EP3R-transfected cells
respectively (Fig. 1A). Western
blot analysis results showed that EP3 receptors were overexpressed
in the pcDNA-EP3R transfected cells (Fig. 1B). The selected cells with increased
EP3 proteins and mRNAs were used for subsequent experiments.
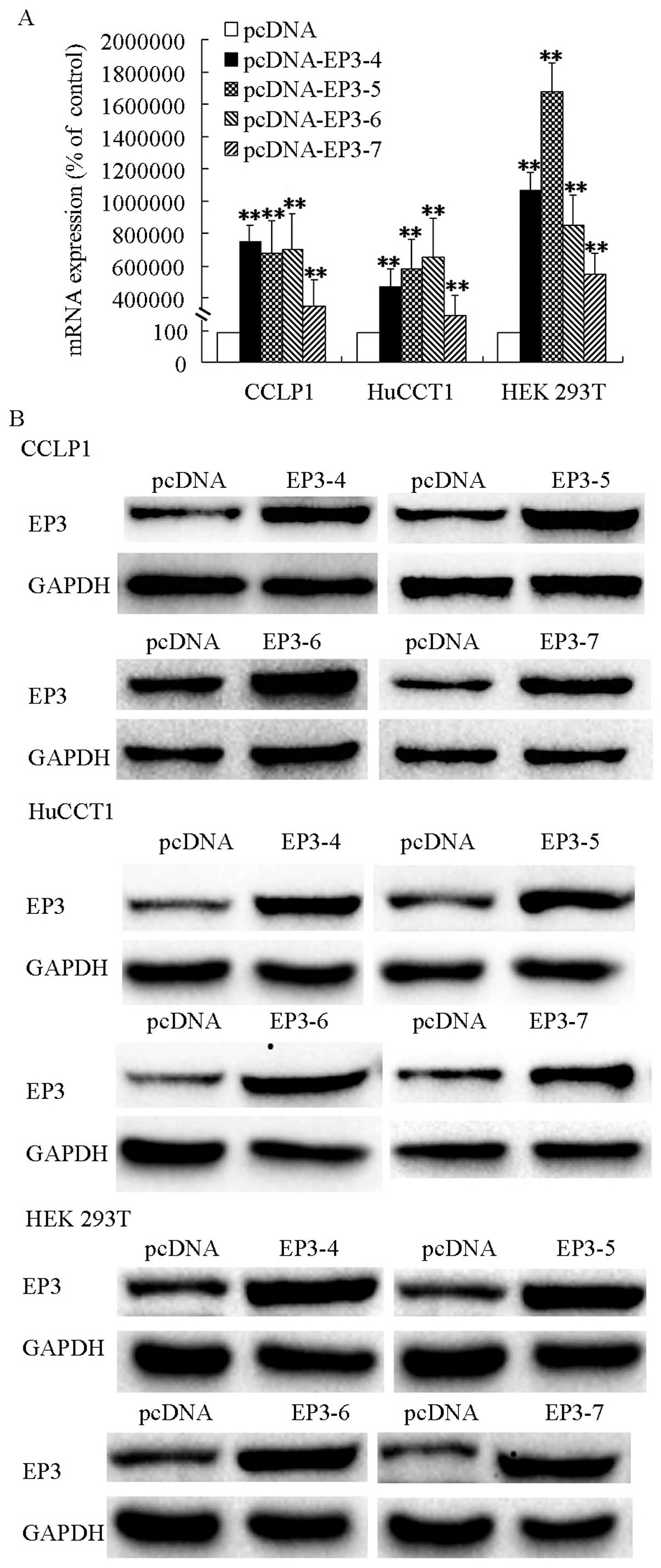 | Figure 1EP3 receptor isoforms mRNAs and
proteins were overexpressed in pcDNA-EP3-transfected cells. (A) EP3
receptor isoforms mRNAs were overexpressed in CCLP1, HuCCT1 and HEK
293T cells. Cells were trans-fected with the EP3-4, EP3-5, EP3-6,
EP3-7 or the pcDNA control plasmids. CCLP1 cells transfected with
EP3R were selected by 500 µg/ml G418. HuCCT1 cells were
selected using 1,000 µg/ml G418 and HEK 293T cells by 200
µg/ml G418. The expression of the EP3R isoforms mRNAs were
confirmed by qPCR analysis. Levels of GAPDH served as the control.
(B) EP3 receptor isoforms proteins were overexpressed in CCLP1,
HuCCT1 and HEK 293T cells. The cells were transfected with the
EP3-4, EP3-5, EP3-6, EP3-7 or the pcDNA control plasmids. The
expression of the EP3R isoforms proteins were determined by western
blotting with anti-EP3R antibody, and GAPDH as the loading control
with anti-GAPDH antibody. **P<0.01, compared with the
control. Data are presented as the mean ± SD of three independent
experiments. |
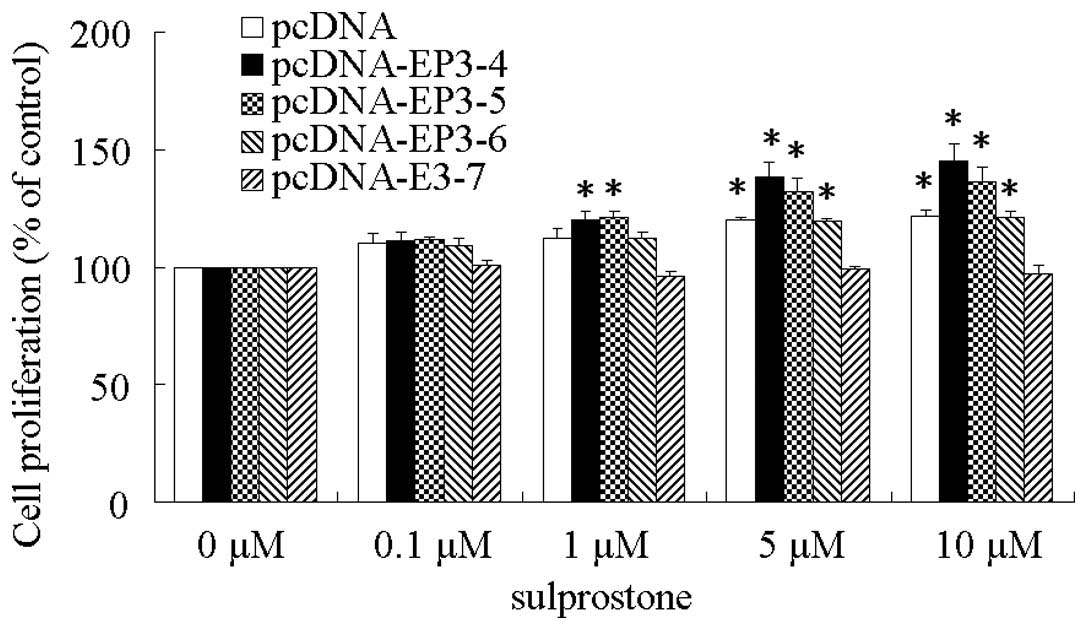
EP3 receptor isoforms play a key role in
the proliferation of cholangiocarcinoma cells
The EP3R or control plasmids pcDNA3.1 transfected
CCLP1 cells were examined for their response to treatment with the
EP3 agonist, sulprostone. After treatment with different
concentrations of sulprostone for 24 h, the cell proliferation rate
was detected by WST-8 analysis. The results showed that the
proliferation of CCLP1 cells treated with sulprostone was increased
compared with the control in pcDNA-, pcDNA-EP3-4-, pcDNA-EP3-5- and
pcDNA-EP3-6-transfected cells. Of these, the effect of
pcDNA-EP3-4-transfected cells was the most significant.
pcDNA-EP3-7-transfected cells did not exert any effect on cell
proliferation (Fig. 2). Similar
results were observed in the HuCCT1 and HEK 293T cells (data not
shown).
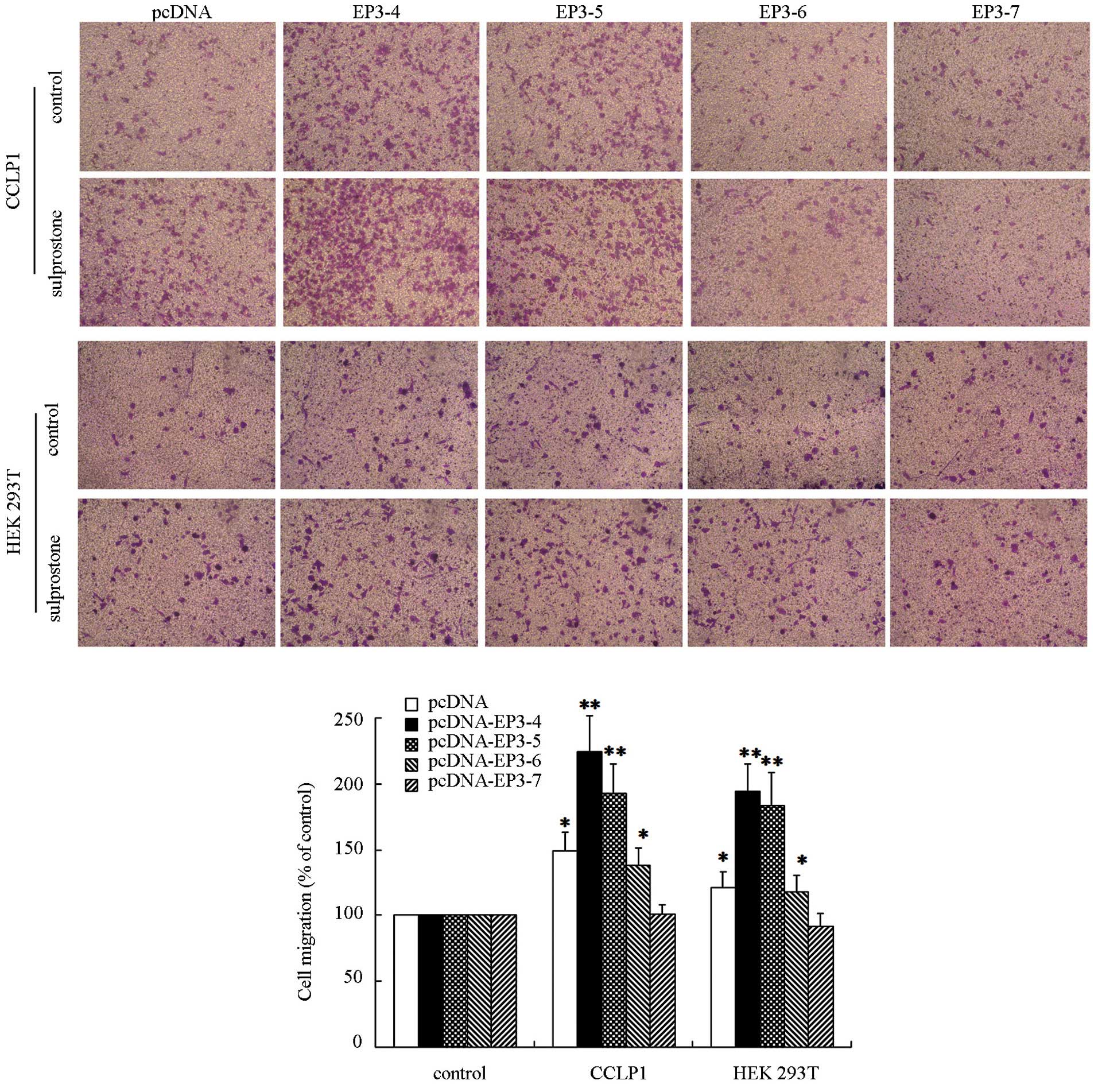
EP3 receptor isoforms mediate the
migration of cholangiocarcinoma cells
The EP3Rs or control plasmids pcDNA3.1-transfected
CCLP1 cells were examined to determine their response to treatment
with sulprostone. Following treatment with 10 µM sulprostone
for 12 h, the cell migration rate was detected by Transwell
analysis. The results showed that the migration of CCLP1 cells
treated with sulpros-tone was increased compared with the control
in pcDNA-, pcDNA-EP3-4-, pcDNA-EP3-5- and pcDNA-EP3-6-trans-fected
cells. Of these, the effect in pcDNA-EP3-4-transfected cells was
the most significant. The pcDNA-EP3-7-transfected cells did not
significantly affect the cell migration rate in the sulprostone
treatment group compared with the control (Fig. 3). Similar results were observed in
HuCCT1 (data not shown) and HEK 293T cells (Fig. 3).
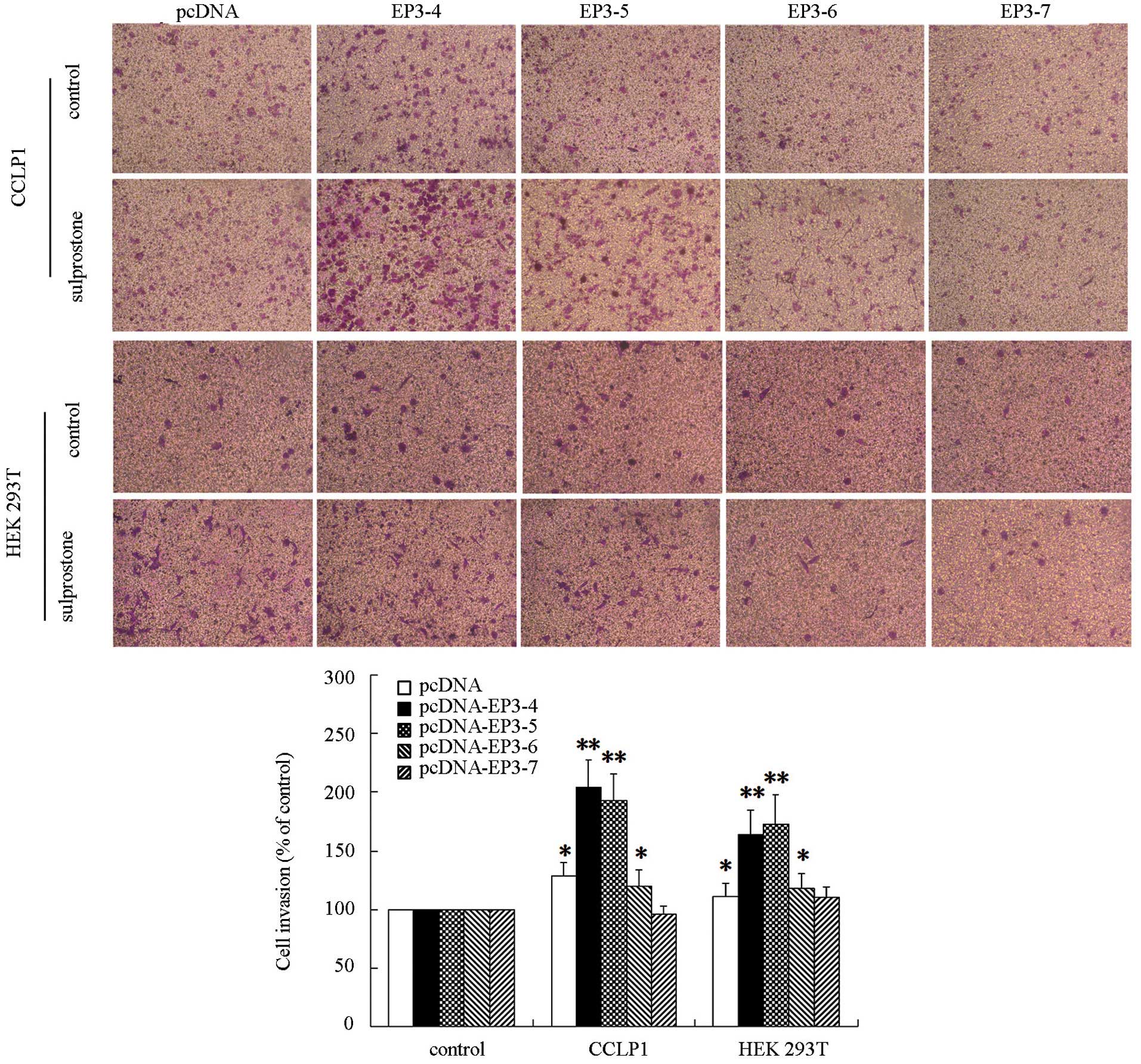
EP3 receptor isoforms are responsible for
invasion in cholangiocarcinoma cells
The EP3R or control plasmid pcDNA3.1-transfected
CCLP1 cells were examined to determine their response to treatment
with sulprostone. Following treatment with 10 µM sulprostone
for 24 h, the cell invasion rate was detected by Transwell
analysis. The results showed that the invasion of CCLP1 cells
treated with sulprostone was increased compared with the control in
pcDNA-, pcDNA-EP3-4-, pcDNA-EP3-5- and pcDNA-EP3-6-transfected
cells. Of these, the effect in pcDNA-EP3-4-transfected cells was
the most significant. The pcDNA-EP3-7-transfected cells did not
significantly affect the cell invasion rate in the sulprostone
treatment group compared with the control (Fig. 4). Similar results were observed in
HuCCT1 (data not shown) and HEK 293T cells (Fig. 4).
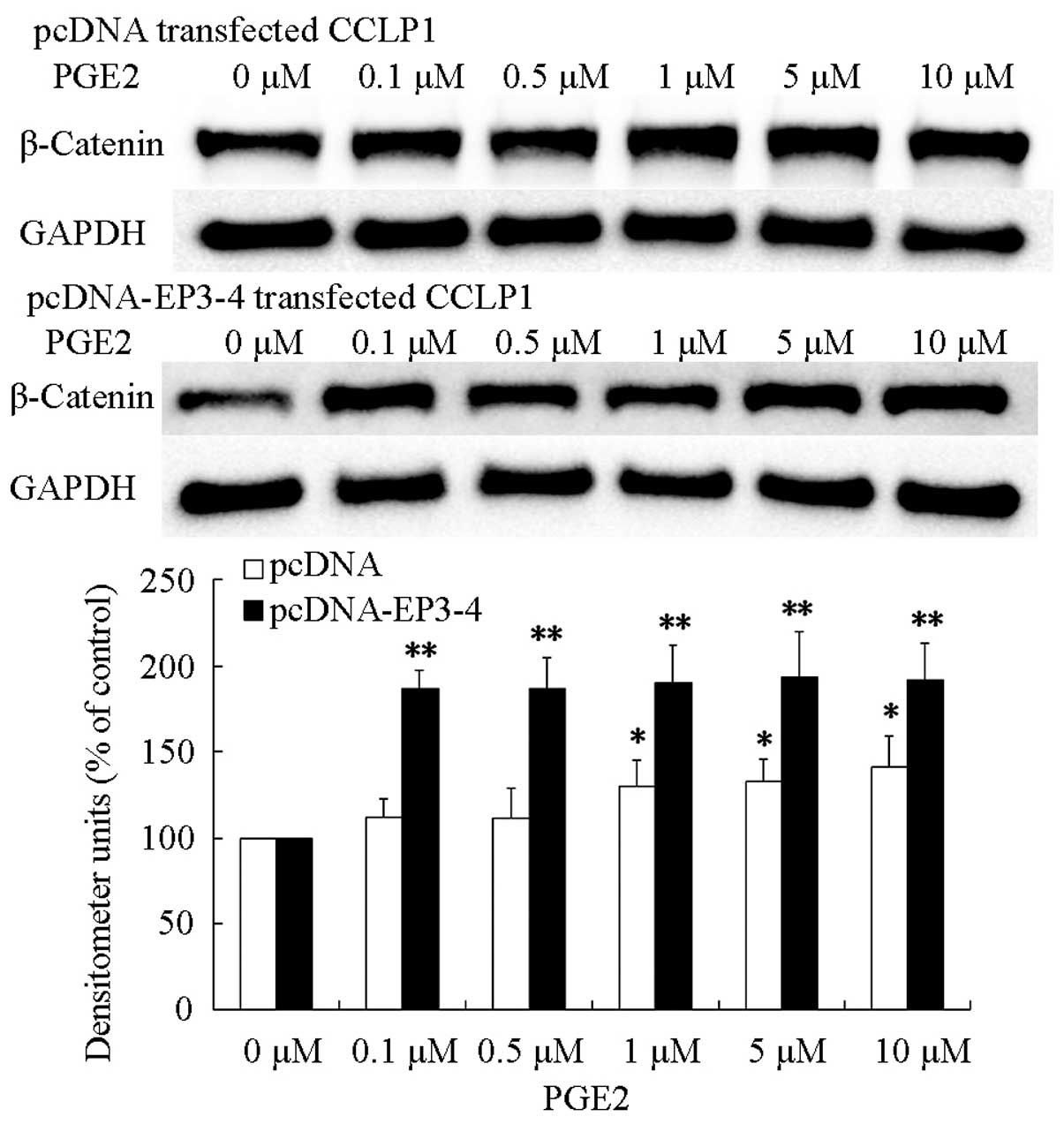
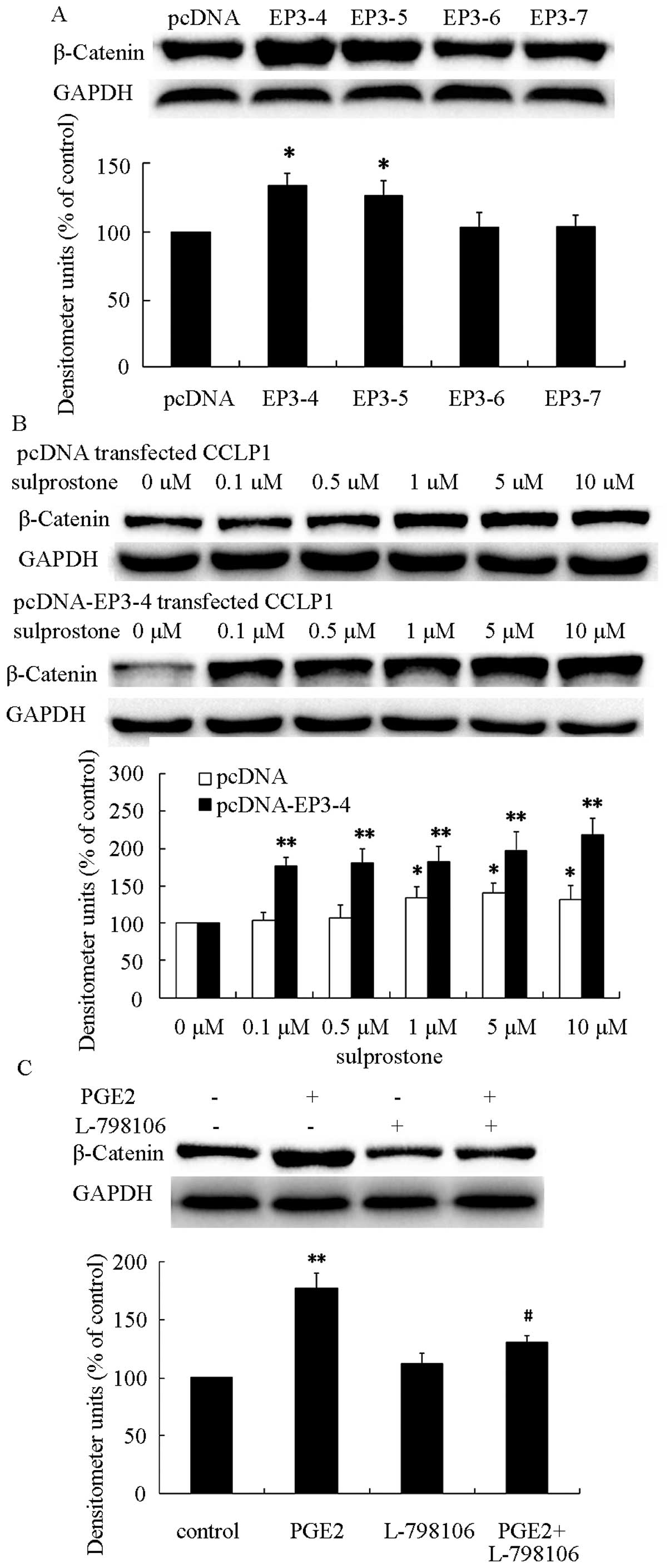
EP3-4 receptor pathway is involved in
upregulation of PGE2-induced β-catenin
PGE2 enhanced the malignancy of cancer cells
(8). Additionally, β-catenin has
been reported to correlated with malignancy of cancer (25). We hypothesized that PGE2 promoted
cholangiocarcinoma progression through the upregulation of
β-catenin protein level. To evaluate this hypothesis, we treated
pcDNA- and pcDNA-EP3-4-transfected CCLP1 cells with PGE2 with the
indicated concentrations for 24 h. The results showed that
treatment of pcDNA- and pcDNA-EP3-4-transfected CCLP1 cells with
PGE2 greatly increased the level of β-catenin protein and the
effect on pcDNA-EP3-4-transfected CCLP1 cells was more significant
than that in pcDNA-transfected CCLP1 cells (Fig. 5). These findings indicated that PGE2
was able to upregulate the β-catenin protein level in CCLP1
cells.
To investigate the role of EP3R isoforms in the
upregulation of β-catenin, the protein was determined in pcDNA-,
pcDNA-EP3-4-, pcDNA-EP3-5-, pcDNA-EP3-6- and
pcDNA-EP3-7-transfected CCLP1 cells. As shown in Fig. 6A, β-catenin was significantly
increased in pcDNA-EP3-4-transfected CCLP1 cells. Previous findings
suggested that sulprostone promotes cholangiocarcinoma cell
progression via the EP3-4 receptor, and PGE2 upregulated the
β-catenin protein level. Thus, we hypothesized that PGE2 was able
to upregulate β-catenin protein via the EP3-4 receptor. To evaluate
this hypothesis, the pcDNA- and pcDNA-EP3-4-transfected CCLP1 cells
were treated with the indicated concentrations of sulprostone for
24 h. The results showed that treatment of pcDNA- or
pcDNA-EP3-4-transtected CCLP1 cells with sulprostone greatly
increased the level of β-catenin protein in a
concentration-dependent manner (Fig.
6B). Pretreatment of pcDNA-EP3-4-transfected CCLP1 cells with
EP3 selective antagonist L-798106 inhibited the upregulation of
β-catenin induced by PGE2 (Fig.
6C). These results suggested that the EP3-4 receptor plays an
important role in PGE2-induced β-catenin upregulation.
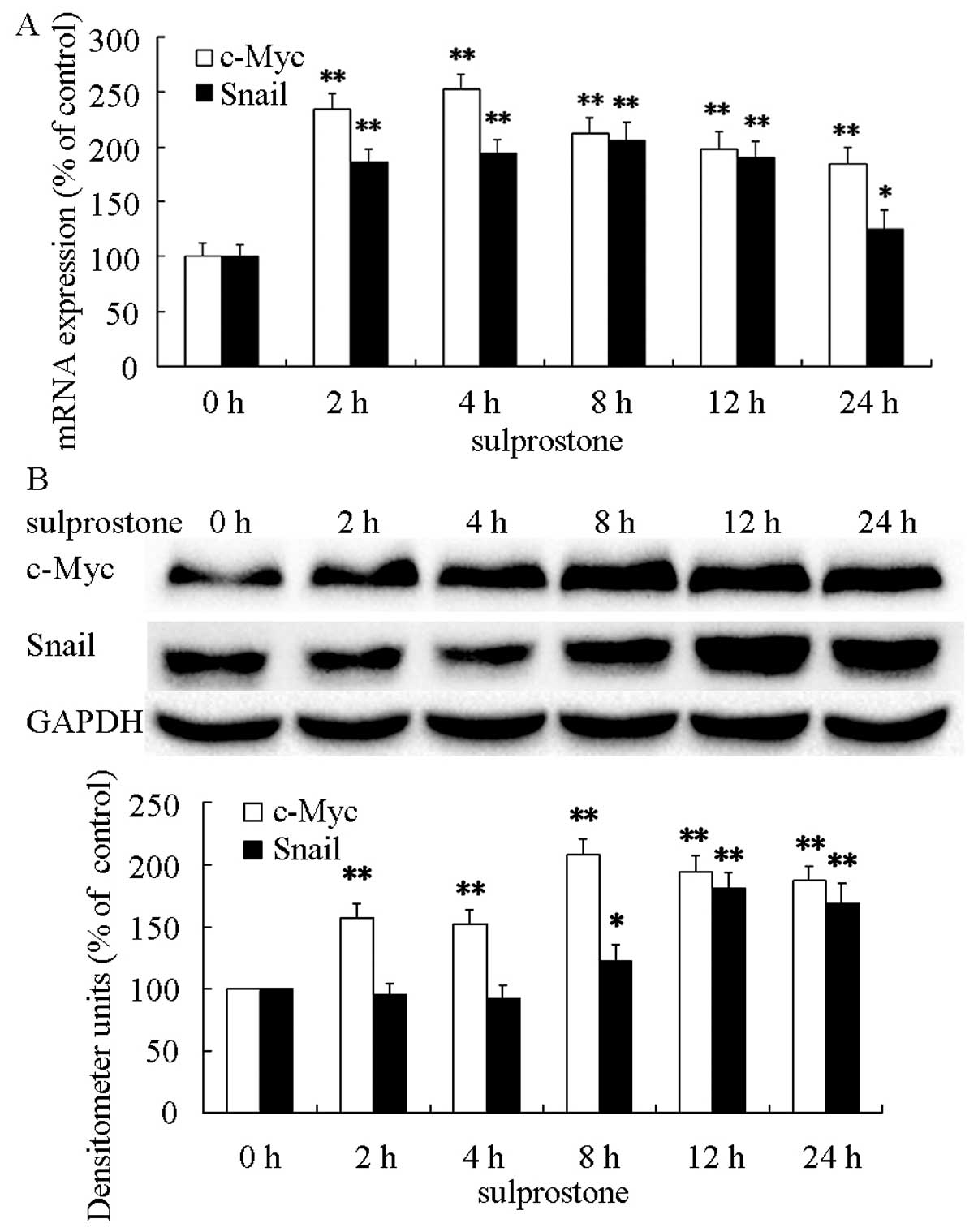
PGE2-associated β-catenin induces the
upregulation of downstream c-Myc and Snail expression in
cholangiocarcinoma cells
β-catenin was accumulated in the nucleus induced by
PGE2 in cholangiocarcinoma cells (26). The accumulation of β-catenin in the
nucleus induced transcription of important downstream target genes,
such as c-Myc and Snail (27,28).
The protein and mRMA levels were determined after pcDNA-EP3-4
transtected CCLP1 cells treated with 5 µM sulprostone for
the indicated time periods. The results showed that treatment of
pcDNA-EP3-4-transfected CCLP1 cells with sulprostone increased the
levels of c-Myc and Snail mRNAs and the levels of c-Myc and Snail
proteins (Fig. 7).
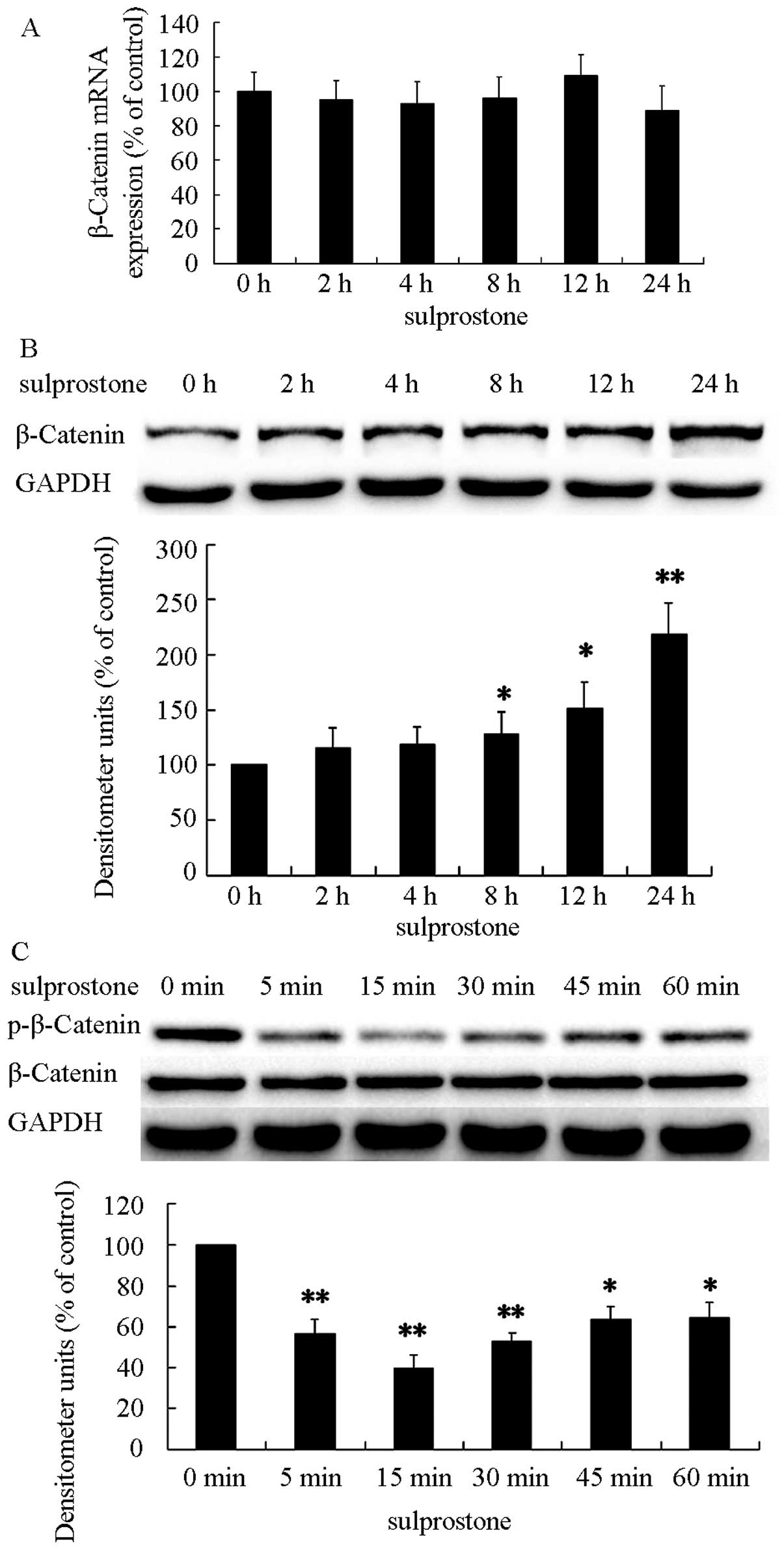
The mechanism of the upregulation of
β-catenin protein induced by PGE2
There are two main ways associated with the increase
of β-catenin protein: upregulation of mRNA and downregulation of
ubiquitin-dependent degradation. The degradation of β-catenin
depends on β-catenin phosphorylation. Previous findings suggest
that, PGE2 and sulprostone increased the level of β-catenin protein
in a concentration-dependent manner. To confirm the β-catenin
protein upregulation, the pcDNA-EP3-4-transfected CCLP1 cells were
treated with 5 µM sulprostone for the indicated time
periods. The levels of β-catenin and phosphorylated β-catenin
proteins were analyzed by western blotting and β-catenin mRNAs by
qPCR. The results showed that treatment of pcDNA-EP3-4-transfected
CCLP1 cells with sulprostone increased the level of β-catenin
protein and decreased the level of phosphorylated β-catenin protein
in a time-dependent manner (Fig. 8B and
C). However, the change of β-catenin mRNA was not observed in
the experiment (Fig. 8A). These
results indicated that the degradation of β-catenin is the main
reason for the upregulation of β-catenin induced by PGE2.
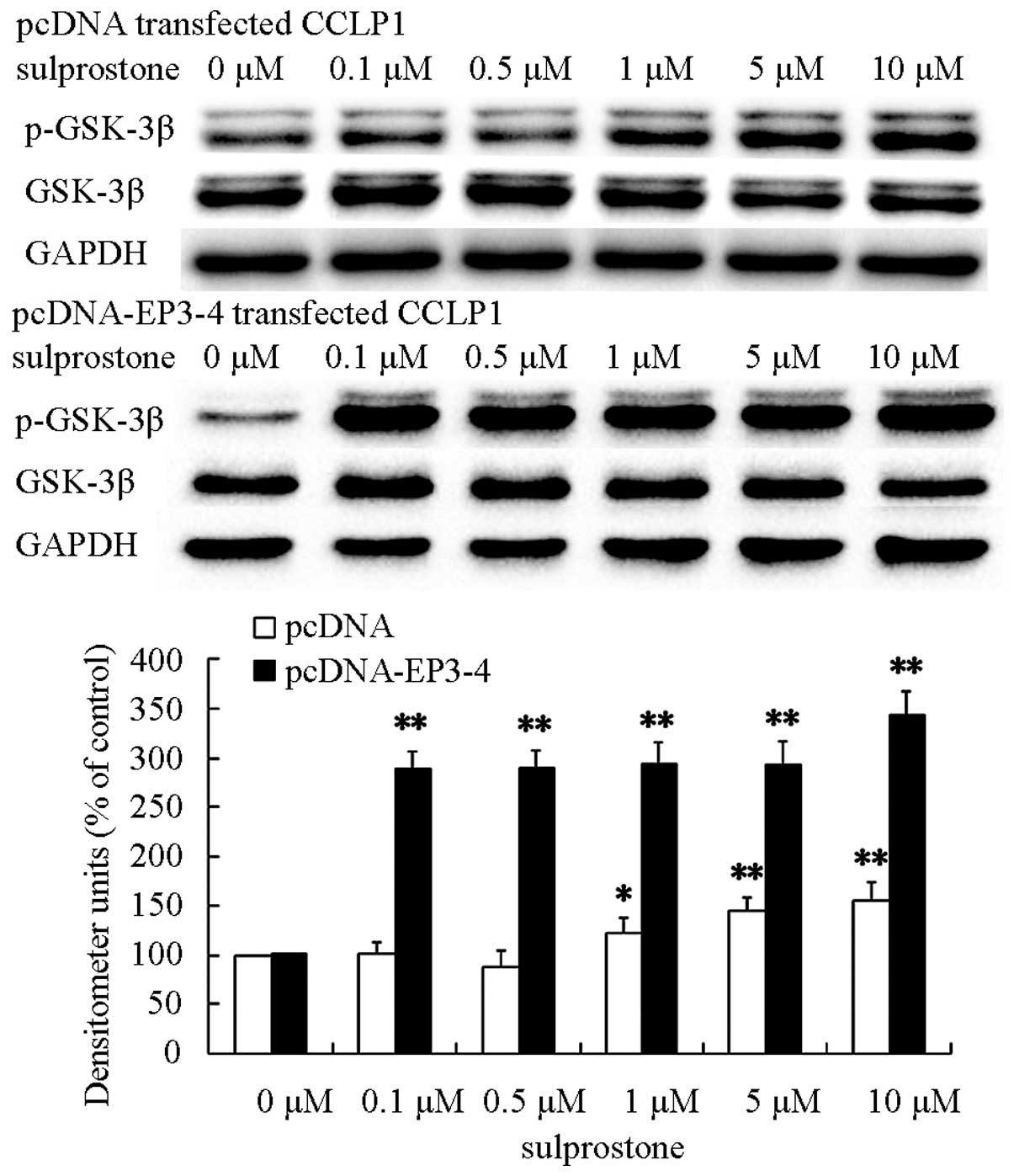
PI3K/AKT and GSK-3β are involved in the
upregulation of β-catenin induced by PGE2
GSK-3β associates in a complex with β-catenin, then
β-catenin is phosphorylated and targeted for degradation (30). The phosphorylation of GSK-3β on
serine 9 inhibits its kinase activity. Therefore, the
phosphorylation of GSK-3β results in the loss of phosphorylation of
β-catenin and the accumulation of β-catenin protein. Previous
findings suggested that loss phosphorylation of β-catenin was
induced by PGE2. Thus, we postulated that PGE2 could upregulate
β-catenin protein via GSK-3β. To evaluate this hypothesis, the
pcDNA- and pcDNA-EP3-4-transfected CCLP1 cells were treated at the
indicated concentrations of sulprostone for 30 min. The levels of
GSK-3β and phosphorylated- GSK-3β proteins were determined by
western blotting. The results showed that treatment of pcDNA- and
pcDNA-EP3-4-transfected CCLP1 cells with sulprostone increased the
levels of phosphorylated GSK-3β proteins (Fig. 9).
It has been reported that AKT is the kinase
responsible for the phosphorylation of GSK-3β (31,32).
We postulated that PGE2 upregulated β-catenin protein and
inactivated GSK-3β via AKT. To evaluate this hypothesis, the pcDNA-
and pcDNA-EP3-4-transfected CCLP1 cells were treated at the
indicated concentrations of sulprostone for 30 min. The levels of
AKT and phosphorylated AKT proteins were analyzed by western
blotting. The results showed that treatment of pcDNA- and
pcDNA-EP3-4-transfected CCLP1 cells with sulprostone increased the
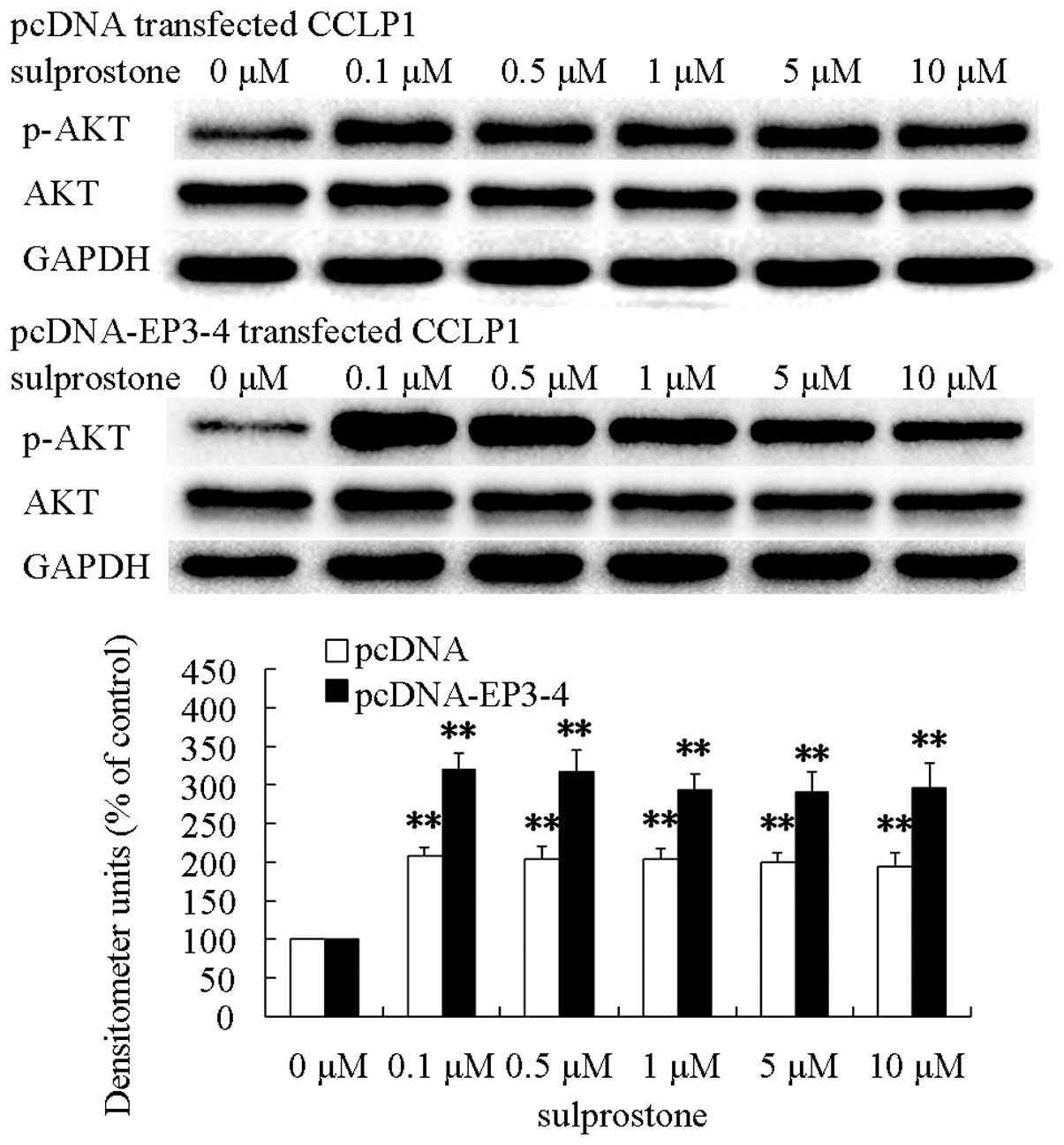
levels of phosphorylated-AKT proteins (Fig. 10).
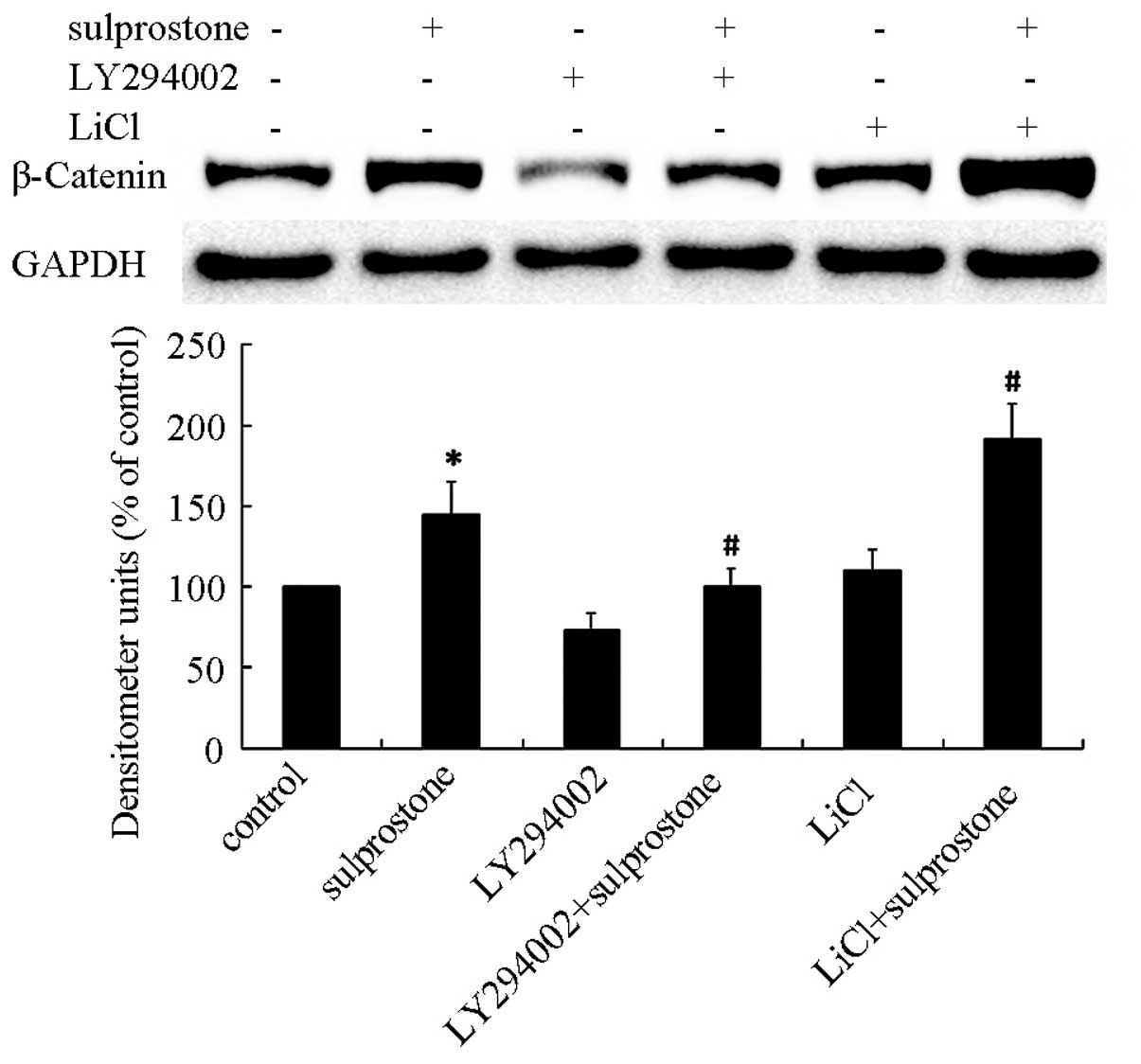
The pcDNA-EP3-4-transfected CCLP1 cells were
pretreated with the PI3K/AKT inhibitor LY-294002 or GSK-3β
inhibitor LiCl, and then treated with sulprostone. The β-catenin
expression level was determined by western blotting. It was also
shown that pretreatment of pcDNA-EP3-4-transfected CCLP1 cells with
LY-294002 inhibited the upregulation of β-catenin protein and with
LiCl promoted the upregulation of β-catenin protein induced by
sulprostone (Fig. 11).
These results indicated that the upregulation of
β-catenin induced by PGE2 is mediated through the activation of
PI3K/AKT and GSK-3β.
EGFR and Src are involved in AKT
activation
EGFR and Src have been reported to be downstream of
the EP receptors, and are important for progression of cancer
(7,8,33).
Therefore, we hypothesized that upregulation of β-catenin induced
by sulprostone was mediated by EGFR and Src. To evaluate this
hypothesis, the pcDNA- and pcDNA-EP3-4-transfected CCLP1 cells were
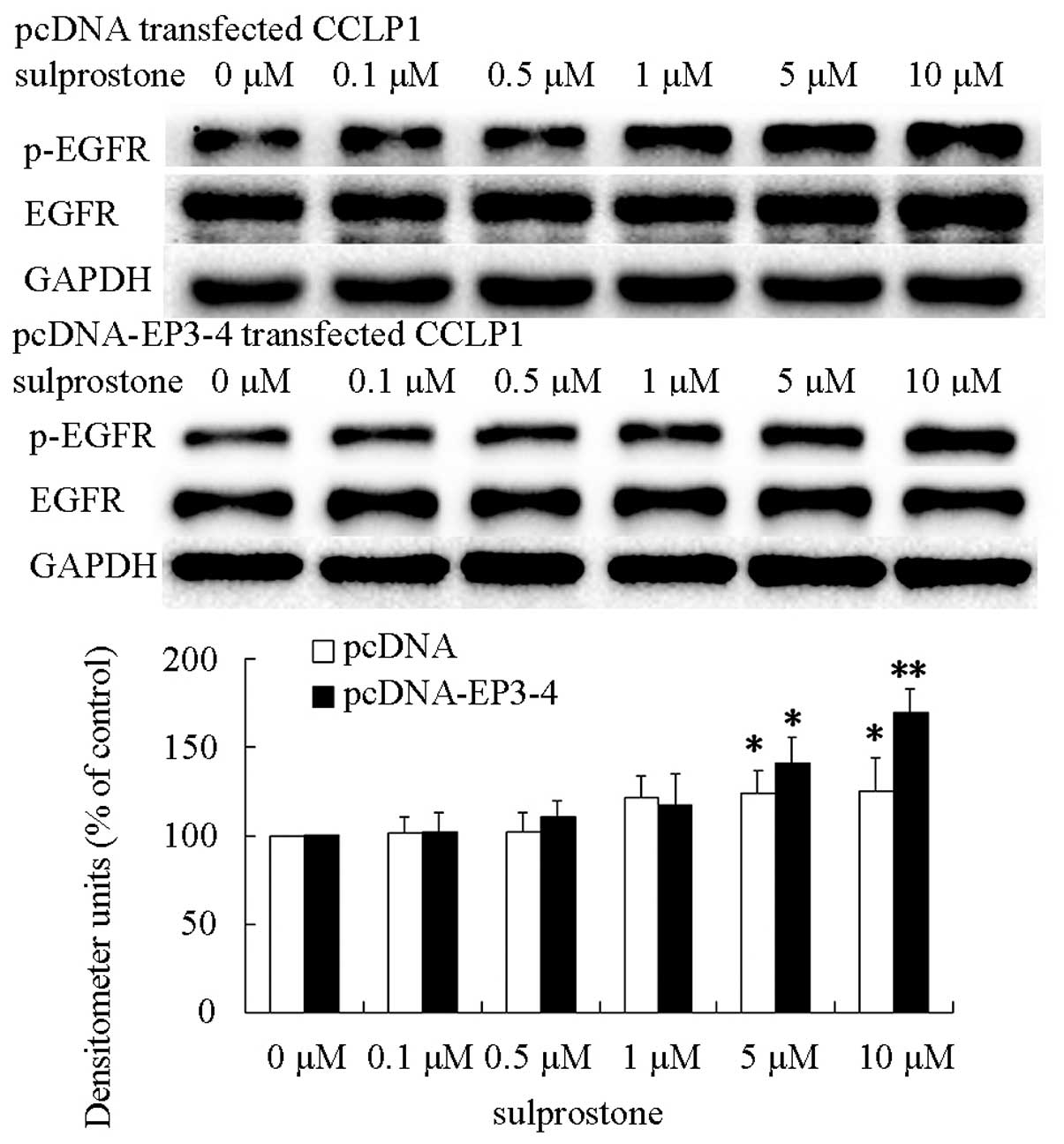
treated with the indicated concentrations of sulprostone for 45
min. The levels of EGFR and phosphorylated-EGFR proteins were
analyzed by western blotting. The results showed that treatment of
pcDNA- and pcDNA-EP3-4-transfected CCLP1 cells with sulprostone
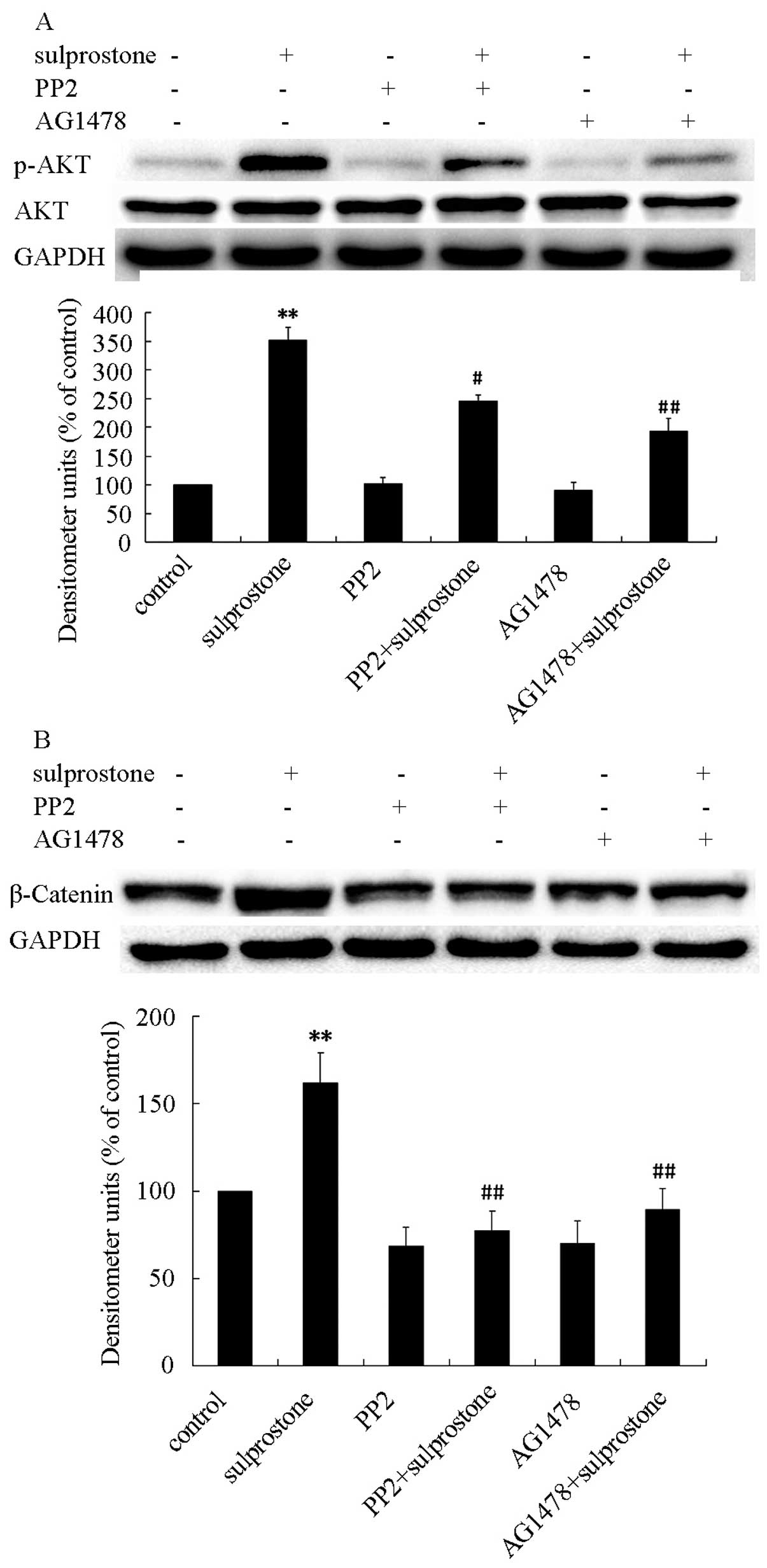
increased the levels of phosphorylated-EGFR proteins (Fig. 12). The pcDNA-EP3-4-transfected
CCLP1 cells were pretreated with Src inhibitor PP2 or EGFR
inhibitor AG1478, and then treated with sulprostone. The
phosphorylation of AKT was determined by western blotting after 30
min of treatment with sulprostone and β-catenin was determined
after 24 h of treatment with sulprostone. Pretreatment of
pcDNA-EP3-4-transfected CCLP1 cells with PP2 or AG1478 inhibited
the phosphorylation of AKT and the upregulation of β-catenin
protein (Fig. 13). These results
indicated that PGE2 upregulates β-catenin expression is mediated
through the activation of Src and EGFR.
Discussion
Prostaglandin E2 (PGE2) appears to participate in
the development of inflammatory reactions and in oncogenesis
(3,4). Our previous results revealed that PGE2
significantly enhanced the cell proliferation, migration and
invasion in cholangiocar-cinoma cells, and several signaling
pathways were identified such as the upregulation of MMP-2
expression through the CREB pathway (11), upregulaion of FBP1 expression
through the cAMP/PKA pathway (12)
and stimulation of Erk through the phosphorylation of EGFR
(34). PGE2 significantly promoted
hepatocellular carcinoma cell proliferation and invasion by
upregulating the expression of Snail (6,7), YB-1
(8), c-Myc (9), β1-integrin (10) and other factors. Additionally, PGE2
promoted cell proliferation and invasion in other types of cancer
(13–15).
The action of PGE2 is mediated by four G
protein-coupled receptors (GPCRs): EP1, EP2, EP3 and EP4 (35), which activate multiple signal
transduction pathways leading to downstream responses. The EP1
receptor mainly couples to Gαq protein and upregulates the level of
intracellular Ca2+. The EP2 and EP4 receptors couple to
Gαs protein, activate adenylate cyclase (AC) and increase the
production of cyclic AMP (cAMP). However, the EP3 receptor which
couples to G protein is more complicated (12,16).
Among EP receptors, EP3 is unique in that alternative mRNA splicing
leads to multiple isoforms including EP3 subtype isoforms 4, 5, 6,
7, and 8 (also known as EP3–4, -5, -6, -7 and-8). In a previous
study (12), EP3-4, -5, -6 and-7
receptors were observed in CCLP1 and HuCCT1 cells. Those results
indicated that, EP3-4 and EP3-5 receptors significantly promoted
cholangiocarcinoma cell proliferation, migration and invasion,
whereas no significant increase in EP3-6 and EP3-7 receptors was
discernable. EP3 isoforms may mediate different intracellular
responses to PGE2, contributing to the different roles played by
the progression of cancer. EP3 isoform receptors that promote
cancer progression may be coupled with Gαs and stimulate the
AC-cAMP-PKA pathway (12). EP3
isoform receptors that exhibit no significant increase in cancer
progression may be coupled with Gαi and downregulate the
AC-cAMP-PKA pathway (36). All the
EP3 isoforms share a common N-terminal sequence, which includes
hormone binding and membrane spanning regions. However, each
isoform has a unique amino acid composition in the C-terminal
region which regulates the intracellular location and plays a key
role in G-protein coupling. These unique sequences in the
C-terminal domains may allow each EP3 isoform to localize to
different areas within the cell and trigger different intracellular
signaling pathways in response to PGE2. Evidence of different
signal transduction pathways and the regulation of gene expression
among different EP3 receptor isoforms has also been previously
demonstrated (12,17). There are EP1, EP2, EP4 and four EP3
isoforms receptors involved in cholangiocarcinoma progression, and
some receptors play a positive role while others do not. The reason
that some EP3 isoforms couple with Gαi and do not promote
cholangiocarcinoma cell growth and invasion may due to the fact
that they are negative regulatory receptors that downregulate the
function of other EP receptors.
β-catenin is critically responsible for epithelial
cell-cell adhesion (37), and plays
a key role in several developmental and pathologic processes
including early embryonic development (38), fibrosis (39–42),
cystic disease (43), renal failure
(44) and cancer. For example, the
expression level of β-catenin has been shown to correlate with
progression in colorectal (18),
cervical (19), prostate (20), renal cell carcinoma (45,46),
hepatocellular carcinoma (47,48),
cholangiocarcinoma (26) and
several other types of tumors. PGE2 and β-catenin are involved in
cholangiocarcinoma progression, and our studies confirm that PGE2
upregulates the β-catenin protein through the EP3-4 receptor. There
are two main ways leading to the increase of β-catenin protein,
upregulation of mRNA and downregulation of ubiquitin-dependent
protein degradation. The ubiquitin-dependent degradation is caused
by phosphorylation of β-catenin at Ser33, Ser37, and Thr41 or
coupled with Ebi which transfers the ubiquitin of
ubiquitin-conjugating enzymes UcbH5 to β-catenin. Our results show
that the down-regulation of ubiquitin-dependent degradation induced
by β-catenin phosphorylation is the main reason for the
upregulation of β-catenin induced by PGE2 instead of an increasing
in mRNA expression. The complex including Axin, APC, Amer1, CK1α
Glycogen synthase kinase 3β (GSK-3β) and β-catenin was separated
when β-catenin phosphorylated at Ser33, Ser37 and Thr41. As a
scaffold protein, Axin, a plurality of the protein interaction
site, can combine APC Amer1, CK1α GSK-3β and β-catenin together.
The main functions of APC and Amer1 enhanced the affinity of
β-catenin and the complex (49).
Based on the phosphorylation of β-catenin at Ser45 induced by CK1α,
GSK-3β can phosphorylate β-catenin at Thr41, Ser37, and Ser33,
which is discerned by β-TrCP and lead to β-catenin degradation.
When β-catenin phosphorylation was downregulated by PGE2 at Ser33,
Ser37 and Thr41, the ubiquitin-dependent degradation was inhibited,
and β-catenin accumulated in the cytoplasm forming a complex with
the transcription factor, lymphoid enhancing factor-1/T-cell
factor, which is subsequently transported into the cell nucleus
(50). This transcription complex
activates the expression of downstream target genes, including
c-Jun, c-Fos, c-Myc (27), Snail
(28), cyclin D1 (29) and survivin, resulting in abnormal
cell proliferation and cell carcinogenesis. Our results show that
the expression of c-Myc and Snail which is associated with hepatic
carcinoma progression observed in previous studies (6,9) also
induced an increase of the EP3 receptor agonist.
The phosphorylation of GSK-3β at Ser 9 inhibits its
kinase activity and results in the accumulation of β-catenin
protein (30). Our results indicate
that the phosphorylation of GSK-3β and AKT is responsible for the
upregulation of β-catenin induced by PGE2. Phosphatidylinositol
3-kinase (PI3K) is composed of the p85 regulatory subunit and the
p110 catalytic subunit. When PI3K is activated, the p85 subunit is
recruited and the p110 subunit is activated. Activated PI3K leads
to the consequent activation of PDK and AKT. AKT is the important
kinase responsible for the phosphorylation of GSK-3β (31,32).
EP3-4 receptor activated by PGE2 may couple to Gαs
protein and activate the AC/PKA pathway (12). Based on this pathway, treatment of
CCLP1 cells with the AC inhibitor SQ22536 and the PKA inhibitor H89
inhibit the effects of EP3 receptor agonist-induced β-catenin
expression. However, those results have not been observed in our
experiments (data not shown). This observation suggests that the
Gαs/AC/PKA pathway may not be responsible for the EP3-4
receptor-mediated β-catenin protein upregulation.
EP receptors may also modulate activation of the
epidermal growth factor receptor (EGFR) (33,34,51).
In our previous studies, the EP1, EP2 and EP4 receptors activated
the EGFR by forming the complex with EGFR and Src (6–8). The
EGFR is a transmembrane tyrosine kinase that belongs to the
HER/ErbB protein family. EGFR controls a variety of biological
responses such as cell proliferation and migration. These effects
are mediated via activation of the downstream molecules, including
the PI3K/AKT pathway (52). It was
reported that EGFR mediates Grb2 associated binder protein 1 (Gab1)
activation in cultured rat hepatocytes. Depletion of Gab1, using
siRNA, decreased AKT activation (53). In breast and lung cancer cell lines,
PGE2 was associated with activated AKT, through increases in the
EGFR-Gab1 complexes (54,55). It was also reported that the EP
receptor was able to bind to EGFR directly (33). Our results indicate that PGE2
stimulated EGFR leading to the upregulation of β-catenin. Thus that
the ligand combines its specific receptor is not the only manner in
which to conduct signaling. The combination and interreaction
between two receptors may also mediate the function of the ligand,
such as the binding and interaction of EPR and EGFR.
To the best of our knowledge, this is the first
study detailing the function of EP3 isoforms in human
cholangiocarcinoma cells. The results also show that PGE2
upregulated the β-catenin protein through the
EP3-4R/Src/EGFR/AKT/GSK-3β pathway. Our findings reveal the role of
EP3 isoforms in promoting cholangiocarcinoma cell growth and
invasion and a new theory that interaction between two receptors
can conduct cell signaling without the stimulation of a specific
ligand.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (81172003) and a project funded
by the Priority Academic Program Development of Jiangsu Higher
Education Institutions (PAPD).
References
|
1
|
Zou S, Li J, Zhou H, Frech C, Jiang X, Chu
JS, Zhao X, Li Y, Li Q, Wang H, et al: Mutational landscape of
intrahepatic cholangiocarcinoma. Nat Commun. 5:56962014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ali SM, Clark CJ, Mounajjed T, Wu TT,
Harmsen WS, Reid-Lombardo KM, Truty MJ, Kendrick ML, Farnell MB,
Nagorney DM, et al: Model to predict survival after surgical
resection of intrahepatic cholangiocarcinoma: the Mayo Clinic
experience. HPB (Oxford). 7:244–250. 2014.
|
|
3
|
Montrose DC, Nakanishi M, Murphy RC,
Zarini S, McAleer JP, Vella AT and Rosenberg DW: The role of PGE in
intestinal inflammation and tumorigenesis. Prostaglandins Other
Lipid Mediat. 116–117C:26–36. 2014.
|
|
4
|
Bai XM, Zhang W, Liu NB, Jiang H, Lou KX,
Peng T, Ma J, Zhang L, Zhang H and Leng J: Focal adhesion kinase:
Important to prostaglandin E2-mediated adhesion,
migration and invasion in hepatocellular carcinoma cells. Oncol
Rep. 21:129–136. 2009.
|
|
5
|
Bai XM, Jiang H, Ding JX, Peng T, Ma J,
Wang YH, Zhang L, Zhang H and Leng J: Prostaglandin E2
upregulates survivin expression via the EP1 receptor in
hepatocellular carcinoma cells. Life Sci. 86:214–223. 2010.
View Article : Google Scholar
|
|
6
|
Zhang M, Zhang H, Cheng S, Zhang D, Xu Y,
Bai X, Xia S, Zhang L, Ma J, Du M, et al: Prostaglandin E2
accelerates invasion by upregulating Snail in hepatocellular
carcinoma cells. Tumour Biol. 35:7135–7145. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cheng SY, Zhang H, Zhang M, Xia SK, Bai
XM, Zhang L, Ma J, Rong R, Wang YP, Du MZ, et al: Prostaglandin
E2 receptor EP2 mediates Snail expression in
hepatocellular carcinoma cells. Oncol Rep. 31:2099–2106.
2014.PubMed/NCBI
|
|
8
|
Zhang H, Cheng S, Zhang M, Ma X, Zhang L,
Wang Y, Rong R, Ma J, Xia S, Du M, et al: Prostaglandin
E2 promotes hepatocellular carcinoma cell invasion
through upregulation of YB-1 protein expression. Int J Oncol.
44:769–780. 2014.PubMed/NCBI
|
|
9
|
Xia S, Ma J, Bai X, Zhang H, Cheng S,
Zhang M, Zhang L, Du M, Wang Y, Li H, et al: Prostaglandin
E2 promotes the cell growth and invasive ability of
hepatocellular carcinoma cells by upregulating c-Myc expression via
EP4 receptor and the PKA signaling pathway. Oncol Rep.
32:1521–1530. 2014.PubMed/NCBI
|
|
10
|
Bai X, Wang J, Guo Y, Pan J, Yang Q, Zhang
M, Li H, Zhang L, Ma J, Shi F, et al: Prostaglandin E2 stimulates
β1-integrin expression in hepatocellular carcinoma through the EP1
receptor/PKC/NF-κB pathway. Sci Rep. 4:65382014. View Article : Google Scholar
|
|
11
|
Sun B, Rong R, Jiang H, Zhang H, Wang Y,
Bai X, Zhang M, Ma J, Xia S, Shu W, et al: Prostaglandin
E2 receptor EP1 phosphorylate CREB and mediates MMP2
expression in human cholangiocarcinoma cells. Mol Cell Biochem.
378:195–203. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ma J, Chen M, Xia SK, Shu W, Guo Y, Wang
YH, Xu Y, Bai XM, Zhang L, Zhang H, et al: Prostaglandin
E2 promotes liver cancer cell growth by the upregulation
of FUSE-binding protein 1 expression. Int J Oncol. 42:1093–1104.
2013.PubMed/NCBI
|
|
13
|
Cao J, Yang X, Li WT, Zhao CL and Lv SJ:
Silencing of COX-2 by RNAi modulates epithelial-mesenchymal
transition in breast cancer cells partially dependent on the PGE2
cascade. Asian Pac J Cancer Prev. 15:9967–9972. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dufour M, Faes S, Dormond-Meuwly A,
Demartines N and Dormond O: PGE2-induced colon cancer growth is
mediated by mTORC1. Biochem Biophys Res Commun. 451:587–591. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Qiu X, Cheng JC, Chang HM and Leung PC:
COX2 and PGE2 mediate EGF-induced E-cadherin-independent human
ovarian cancer cell invasion. Endocr Relat Cancer. 21:533–543.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wu T: Cyclooxygenase-2 and prostaglandin
signaling in cholangiocarcinoma. Biochim Biophys Acta.
1755:135–150. 2005.PubMed/NCBI
|
|
17
|
Kim SO, Dozier BL, Kerry JA and Duffy DM:
EP3 receptor isoforms are differentially expressed in
subpopulations of primate granulosa cells and couple to unique
G-proteins. Reproduction. 146:625–635. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lemieux E, Cagnol S, Beaudry K, Carrier J
and Rivard N: Oncogenic KRAS signalling promotes the Wnt/β-catenin
pathway through LRP6 in colorectal cancer. Oncogene. Dec 15–2014,
http://dx.doi.org/10.1038/onc.2014.416.
View Article : Google Scholar
|
|
19
|
Rath G, Jawanjal P, Salhan S, Nalliah M
and Dhawan I: Clinical significance of inactivated glycogen
synthase kinase 3beta in HPV-associated cervical cancer:
Relationship with Wnt/beta-catenin pathway activation. Am J Reprod
Immunol. 73:460–478. 2015. View Article : Google Scholar
|
|
20
|
Vatansever HS, Gumus B, Aydogdu O,
Sivrikoz ON, Türköz-Uluer E, Kivanç M, Ateşçi YZ and Bugdayci H:
The role of stem/progenitor cells and Wnt/β-catenin signaling
pathway in the patients with prostate cancer. Minerva Urol Nefrol.
66:249–255. 2014.PubMed/NCBI
|
|
21
|
Thompson MD and Monga SP: WNT/beta-catenin
signaling in liver health and disease. Hepatology. 45:1298–1305.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li P, Cao Y, Li Y, Zhou L, Liu X and Geng
M: Expression of Wnt-5a and β-catenin in primary hepatocellular
carcinoma. Int J Clin Exp Pathol. 7:3190–3195. 2014.
|
|
23
|
Ao R, Zhang DR, Du YQ and Wang Y:
Expression and significance of Pin1, β-catenin and cyclin D1 in
hepatocellular carcinoma. Mol Med Rep. 10:1893–1898.
2014.PubMed/NCBI
|
|
24
|
Gedaly R, Galuppo R, Daily MF, Shah M,
Maynard E, Chen C, Zhang X, Esser KA, Cohen DA, Evers BM, et al:
Targeting the Wnt/β-catenin signaling pathway in liver cancer stem
cells and hepatocellular carcinoma cell lines with FH535. PLoS One.
9:e992722014. View Article : Google Scholar
|
|
25
|
Tokumoto N, Ikeda S, Ishizaki Y, Kurihara
T, Ozaki S, Iseki M, Shimizu Y, Itamoto T, Arihiro K, Okajima M, et
al: Immunohistochemical and mutational analyses of Wnt signaling
components and target genes in intrahepatic cholangiocarcinomas.
Int J Oncol. 27:973–980. 2005.PubMed/NCBI
|
|
26
|
Lim K, Han C, Xu L, Isse K, Demetris AJ
and Wu T: Cyclooxygenase-2-derived prostaglandin E2
activates beta-catenin in human cholangiocarcinoma cells: Evidence
for inhibition of these signaling pathways by omega 3
polyunsaturated fatty acids. Cancer Res. 68:553–560. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lee KB, Ye S, Park MH, Park BH, Lee JS and
Kim SM: p63-Mediated activation of the β-catenin/c-Myc signaling
pathway stimulates esophageal squamous carcinoma cell invasion and
metastasis. Cancer Lett. 353:124–132. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang H, Zhang G, Zhang H, Zhang F, Zhou B,
Ning F, Wang HS, Cai SH and Du J: Acquisition of
epithelial-mesenchymal transition phenotype and cancer stem
cell-like properties in cisplatin-resistant lung cancer cells
through AKT/β-catenin/Snail signaling pathway. Eur J Pharmacol.
723:156–166. 2014. View Article : Google Scholar
|
|
29
|
Ripple MJ, Parker Struckhoff A,
Trillo-Tinoco J, Li L, Margolin DA, McGoey R and Del Valle L:
Activation of c-Myc and Cyclin D1 by JCV T-Antigen and β-catenin in
colon cancer. PLoS One. 9:e1062572014. View Article : Google Scholar
|
|
30
|
Manoukian AS and Woodgett JR: Role of
glycogen synthase kinase-3 in cancer: Regulation by Wnts and other
signaling pathways. Adv Cancer Res. 84:203–229. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Castellone MD, Teramoto H, Williams BO,
Druey KM and Gutkind JS: Prostaglandin E2 promotes colon
cancer cell growth through a Gs-axin-beta-catenin signaling axis.
Science. 310:1504–1510. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zheng WH, Kar S and Quirion R:
Insulin-like growth factor-1-induced phosphorylation of the
forkhead family transcription factor FKHRL1 is mediated by Akt
kinase in PC12 cells. J Biol Chem. 275:39152–39158. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Han C, Michalopoulos GK and Wu T:
Prostaglandin E2 receptor EP1 transactivates EGFR/MET
receptor tyrosine kinases and enhances invasiveness in human
hepatocellular carcinoma cells. J Cell Physiol. 207:261–270. 2006.
View Article : Google Scholar
|
|
34
|
Zhang L, Jiang L, Sun Q, Peng T, Lou K,
Liu N and Leng J: Prostaglandin E2 enhances mitogen-activated
protein kinase/Erk pathway in human cholangiocarcinoma cells:
Involvement of EP1 receptor, calcium and EGF receptors signaling.
Mol Cell Biochem. 305:19–26. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bos CL, Richel DJ, Ritsema T,
Peppelenbosch MP and Versteeg HH: Prostanoids and prostanoid
receptors in signal transduction. Int J Biochem Cell Biol.
36:1187–1205. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Orie NN and Clapp LH: Role of prostanoid
IP and EP receptors in mediating vasorelaxant responses to PGI2
analogues in rat tail artery: Evidence for Gi/o modulation via EP3
receptors. Eur J Pharmacol. 654:258–265. 2011. View Article : Google Scholar
|
|
37
|
De Langhe SP and Reynolds SD: Wnt
signaling in lung organogenesis. Organogenesis. 4:100–108. 2008.
View Article : Google Scholar
|
|
38
|
Wodarz A and Nusse R: Mechanisms of Wnt
signaling in development. Annu Rev Cell Dev Biol. 14:59–88. 1998.
View Article : Google Scholar
|
|
39
|
Cui L, Jia X, Zhou Q, Zhai X, Zhou Y and
Zhu H: Curcumin affects β-catenin pathway in hepatic stellate cell
in vitro and in vivo. J Pharm Pharmacol. 66:1615–1622. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ge WS, Wang YJ, Wu JX, Fan JG, Chen YW and
Zhu L: β-catenin is overexpressed in hepatic fibrosis and blockage
of Wnt/β-catenin signaling inhibits hepatic stellate cell
activation. Mol Med Rep. 9:2145–2151. 2014.PubMed/NCBI
|
|
41
|
Pulkkinen K, Murugan S and Vainio S: Wnt
signaling in kidney development and disease. Organogenesis.
4:55–59. 2008. View Article : Google Scholar
|
|
42
|
Nelson PJ, von Toerne C and Gröne HJ:
Wnt-signaling pathways in progressive renal fibrosis. Expert Opin
Ther Targets. 15:1073–1083. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wuebken A and Schmidt-Ott KM:
WNT/β-catenin signaling in polycystic kidney disease. Kidney Int.
80:135–138. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Terada Y, Tanaka H, Okado T, Shimamura H,
Inoshita S, Kuwahara M and Sasaki S: Expression and function of the
developmental gene Wnt-4 during experimental acute renal failure in
rats. J Am Soc Nephrol. 14:1223–1233. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hsu RJ, Ho JY, Cha TL, Yu DS, Wu CL, Huang
WP, Chu P, Chen YH, Chen JT and Yu CP: WNT10A plays an oncogenic
role in renal cell carcinoma by activating WNT/β-catenin pathway.
PLoS One. 7:e476492012. View Article : Google Scholar
|
|
46
|
Ueno K, Hirata H, Majid S, Tabatabai ZL,
Hinoda Y and Dahiya R: IGFBP-4 activates the Wnt/beta-catenin
signaling pathway and induces M-CAM expression in human renal cell
carcinoma. Int J Cancer. 129:2360–2369. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Lu D, Han C and Wu T: Microsomal
prostaglandin E synthase-1 promotes hepatocarcinogenesis through
activation of a novel EGR1/β-catenin signaling axis. Oncogene.
31:842–857. 2012. View Article : Google Scholar
|
|
48
|
Llovet JM and Bruix J: Molecular targeted
therapies in hepatocellular carcinoma. Hepatology. 48:1312–1327.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Liu C, Li Y, Semenov M, Han C, Baeg GH,
Tan Y, Zhang Z, Lin X and He X: Control of beta-catenin
phosphorylation/degradation by a dual-kinase mechanism. Cell.
108:837–847. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hoppler S and Kavanagh CL: Wnt signalling:
Variety at the core. J Cell Sci. 120:385–393. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Han C and Wu T: Cyclooxygenase-2-derived
prostaglandin E2 promotes human cholangiocarcinoma cell
growth and invasion through EP1 receptor-mediated
activation of the epidermal growth factor receptor and Akt. J Biol
Chem. 280:24053–24063. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Glaysher S, Bolton LM, Johnson P, Atkey N,
Dyson M, Torrance C and Cree IA: Targeting EGFR and PI3K pathways
in ovarian cancer. Br J Cancer. 109:1786–1794. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Aasrum M, Odegård J, Sandnes D and
Christoffersen T: The involvement of the docking protein Gab1 in
mitogenic signalling induced by EGF and HGF in rat hepatocytes.
Biochim Biophys Acta. 1833:3286–3294. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Mouradian M, Kikawa KD, Johnson ED, Beck
KL and Pardini RS: Key roles for GRB2-associated-binding protein 1,
phosphatidylinositol-3-kinase, cyclooxygenase 2, prostaglandin E2
and transforming growth factor alpha in linoleic acid-induced
upregulation of lung and breast cancer cell growth. Prostaglandins
Leukot Essent Fatty Acids. 90:105–115. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Cardoso AP, Pinto ML, Pinto AT, Oliveira
MI, Pinto MT, Gonçalves R, Relvas JB, Figueiredo C, Seruca R,
Mantovani A, et al: Macrophages stimulate gastric and colorectal
cancer invasion through EGFR Y(1086), c-Src, Erk1/2 and Akt
phosphorylation and smallGTPase activity. Oncogene. 33:2123–2133.
2014. View Article : Google Scholar
|