Introduction
Malignant melanoma is a potentially lethal form of
skin cancer. Although it accounts for <2% of all skin cancer
cases, it is responsible for ~75% of all mortality from skin cancer
(1). Two targeted therapeutic
agents (ipilimumab and vemurafenib) have shown promise in the
survival rates in patients with advanced melanoma (2–4).
However, the majority of patients who respond to the targeted
therapies eventually develop resistance and disease progression
(5). Novel agents need to be
developed to overcome the limitations of the current therapeutic
agents.
Medical plants have been considered a valuable
source of bioactive compounds for the treatment of many conditions,
including cancer (6).
Atractylodis macrocephalae rhizoma (Baizhu in
Chinese) is a traditional Chinese medicinal herb. The extracts of
Baizhu exhibited various pharmacological activities, such as
anti-inflammation (7),
anti-lipid-peroxidation (8) and
antitumor activities (9,10). In a previous study, we isolated
eight sesquiterpene compounds from Baizhu and evaluated
their anti-melanoma properties (11). The MTT data demonstrated that
atractylenolide I (AT-I) was one of the major active components,
which displayed cytotoxic action in melanoma cells (11). We also observed that AT-I inhibited
the activation of ERK in melanoma cells (11). However, the molecular mechanisms of
AT-I anti-melanoma properties remain to be elucidated.
P53 is a major tumor suppressor. Increased p53
activity is associated with cell cycle arrest, through increased
expression of p21 (12) and the
induction of apoptosis via the intrinsic and extrinsic pathways
(13). Glycogen synthase kinase-3β
(GSK3β) has been identified as a major regulator of p53
localization and expression (14,
15). Activation of GSK3β promoted
responses to p53 including increases in the p21 expression level
and caspase 3 activity (14,15).
Pharmacological inhibition of GSK3β activity produced marked
reductions in the activation of Bax and caspase 3 and in cell death
(14,15).
c-Jun has been reported to directly repress p53
transcription by binding to a variant AP-1 site in the p53 promoter
(16). In cells absent of c-Jun,
the expression of p53 and p21 is increased, and those cells exhibit
cell cycle arrest (16).
Overexpression of c-Jun in cells results in decreased levels of p53
and p21, and exhibits accelerated cell proliferation (16). In melanoma cells, activation of ERK
can inactivate GSK3β, which in turn increases c-Jun stability and
decreases p53 activity (17).
In the present study, the cell cycle-arrest and
apoptosis-promoting effects as well as the ERK/GSK3β
signaling-related mechanism of action of AT-I were
investigated.
Materials and methods
Reagents and antibodies
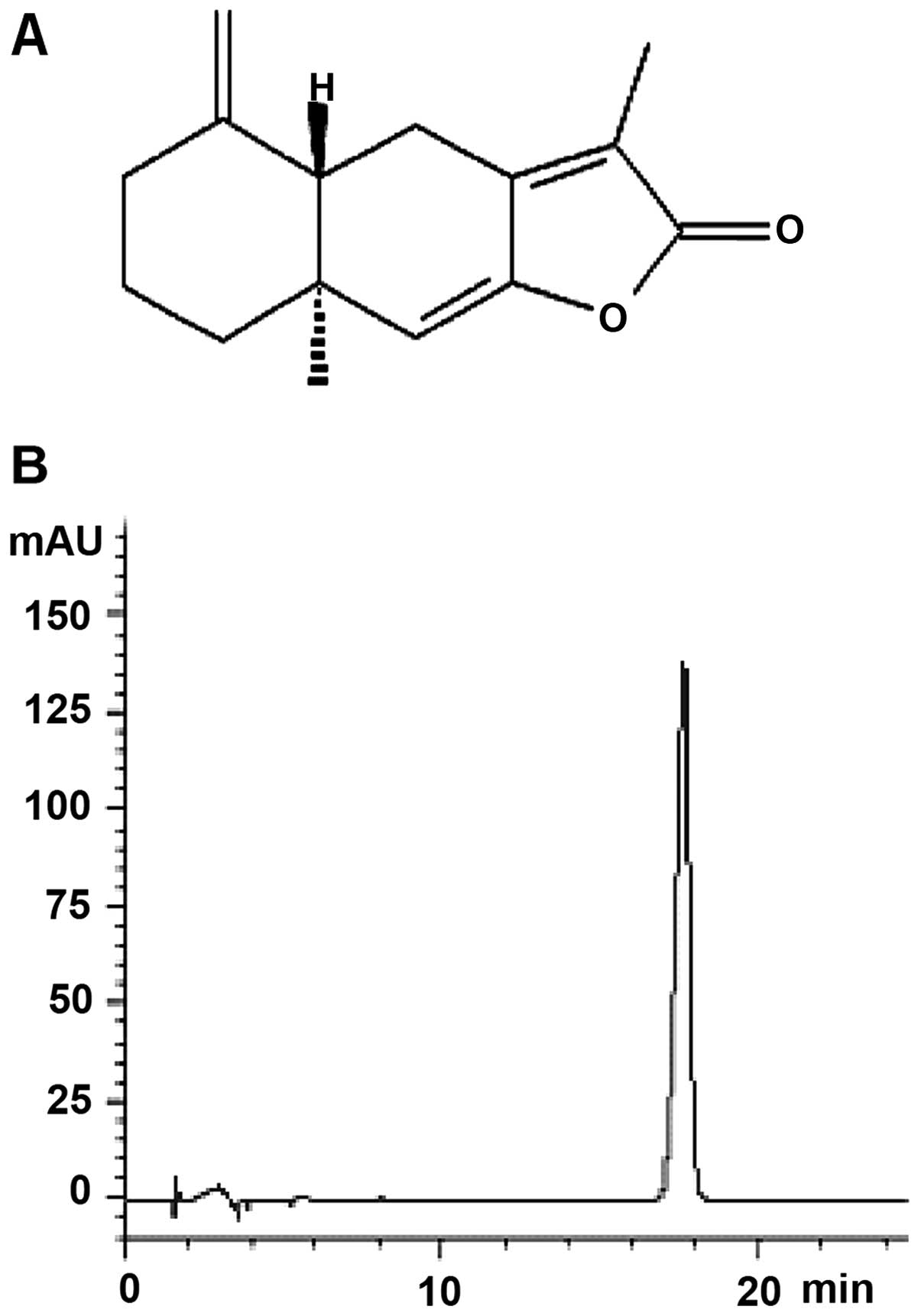
AT-I was isolated from Baizhu. The purity of
the isolated AT-I was determined to be >98% by HPLC (Fig. 1). Stock solutions of AT-I (100 mM)
were prepared in dimethyl sulfoxide (DMSO). Cleaved caspase-3
and-8, p21, cdk2, phospho-ERK (p-ERK, thr202/tyr204), ERK, p-GSK3β
(ser9), c-Jun and p-p53 (ser15) antibodies were obtained from Cell
Signaling Technology (Beverly, MA, USA). Caspase-3 and-8, p53,
β-actin antibodies and anti-mouse and anti-rabbit IgG antibodies
(horseradish peroxidase-conjugated) were purchased from Santa Cruz
Biotechnology (Dallas, TX, USA). Ribonuclease (RNase A), trypsin,
propidium iodide (PI) and lithium chloride (LiCl) were purchased
from Sigma-Aldrich (St. Louis, MO, USA). The Annexin V-FITC
Apoptosis Detection kit was obtained from BD Biosciences (San Jose,
CA, USA).
HPLC analysis
HPLC analysis was performed in an Agilent 1100
system equipped with a diode-array detector. Solvents for HPLC
analysis were HPLC grade. Experimental conditions were summarized
as follows: 1 mg of AT-I was prepared in 1 ml methanol. The
separation was performed on Synergi Fursion-RPC18 column (25×4.6
mm, 4 µm) with acetonitrile-water (40:60) as the mobile
phase. The column temperature was maintained at 30°C. The flow rate
was 1.0 ml/min and the detection wavelength was set at 220 nm.
Cell culture
Murine melanoma B16 cells (Shanghai Branch, Chinese
Academy of Sciences, Shanghai, China) were grown in RPMI-1640
medium supplemented with 10% fetal bovine serum (FBS) and 1%
penicillin/streptomycin (all from Gibco, Grand Island, NY, USA).
The cells were cultured at 37°C in a humidified atmosphere of 5%
CO2.
Determination of cell cycle
distribution
The distribution of cells in various phases was
determined from DNA content assessed by flow cytometry. Cells were
seeded at a density of 4×105 in 60-mm dish and grown
overnight. Various concentrations of AT-I (50, 75 and 100
µM) and/or 5 mM LiCl were added and the cells were incubated
for 48 h. Detached and adherent cells were collected and
centrifuged at 300 × g for 5 min at 4°C. Pellets were rinsed with
ice-cold phosphate-buffered saline (PBS) and fixed with ice-cold
70% ethanol overnight. The cells were then stained with staining
buffer (PBS containing 20 µg/ml of PI, 100 µg/ml
RNase A, and 0.1% Triton X-100) for 30 min at 37°C in the dark.
Stained cells were analyzed using a FACSCalibur™ flow cytometer (BD
Biosciences).
Apoptosis analysis
Early (Annexin V+PI−) and late
(Annexin V+PI+) phase apoptotic cells were
monitored using an Annexin V-FITC apoptosis detection kit. B16
cells (2.5×105) were grown in 35-mm dishes. Following
treatment with AT-I (50, 75 and 100 µM) and/or 5 mM LiCl for
72 h, adherent and floating cells were collected and washed with
cold PBS. The cells were resuspended in binding buffer and
incubated with Annexin V and PI staining solution following the
manufacturer's instructions. Samples of 10,000 stained cells were
analyzed using a flow cytometer (BD Biosciences).
Western blot analysis
The cells were treated as mentioned above and
collected. The proteins were extracted with RIPA lysis buffer [50
mM Tris-Cl, 1% v/v NP-40, 0.35% w/v sodium-deoxycholate, 150 mM
NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM phenylmethylsulfonyl fluoride
(PMSF), 1 mM NaF and 1 mM Na3VO4, pH adjusted
to 7.4] containing a protease inhibitor cocktail (Roche, Mannheim,
Germany) for 15 min at 4°C. After centrifugation at 12,000 × g for
15 min at 4°C, the supernatant was collected and regarded as whole
cell extract. The protein concentration was determined by a Bio-Rad
protein assay (Bio-Rad, Hercules, CA, USA). Equal amounts of
individual protein samples were separated by SDS-PAGE and then
electro-transferred onto nitrocellulose membranes (Amersham
Biosciences, Piscataway, NJ, USA). The membranes were blocked for
60 min with 5% skimmed milk in TBST buffer composed of 50 mM Tris
(pH 7.6), 150 mM NaCl and 0.1% Tween-20 and incubated with the
primary antibodies overnight at 4°C. β-actin was used as the
loading control. After incubation with secondary antibodies
(1:2,000), ECL detection reagents (Amersham Biosciences) were used
to detect signals.
Statistical analysis
Results were presented as the mean ± SD of three
independent experiments. Data analysis was performed by one-way
analysis of variance (ANOVA). For comparison of two groups, the
Student's t-test was used. P<0.05 was considered statistically
significant.
Results
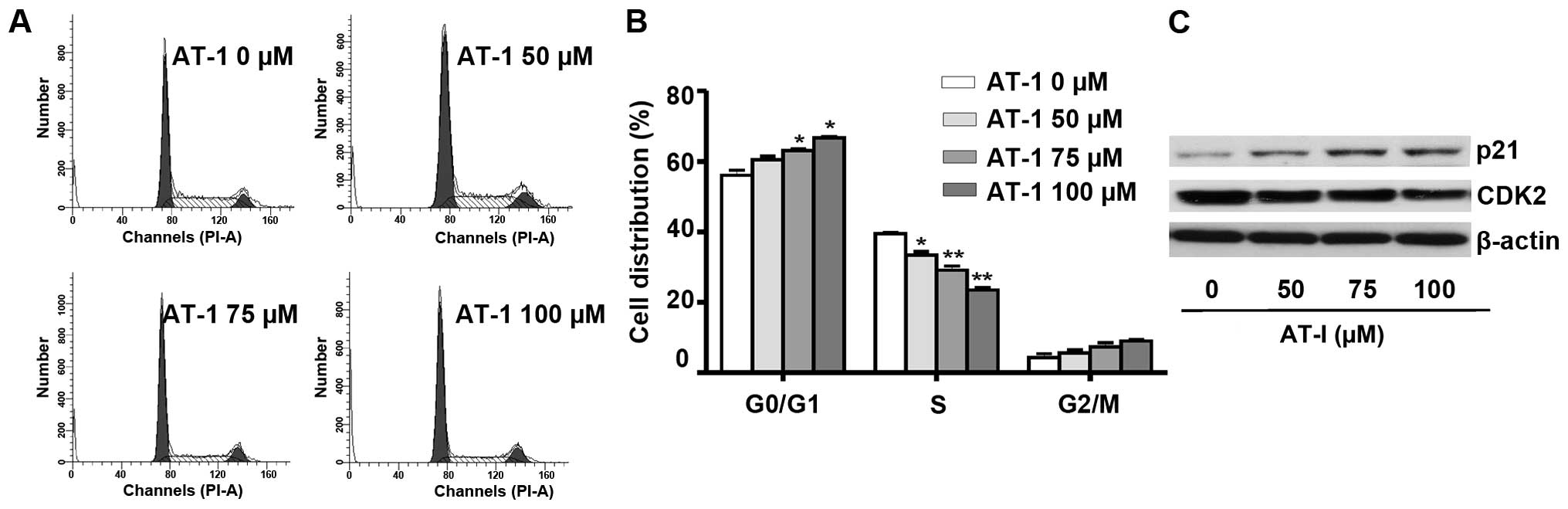
AT-I induces cell cycle arrest in B16
cells
Cell cycle distribution analysis demonstrated that
AT-I treatment for 48 h at the concentrations of 50, 75 and 100
µΜ caused a dose-dependent delay of cell cycle progression
from G1 to S phase (Fig. 2A).
Fig. 2B shows the quantified cell
distributions in different phases (P<0.01 or P<0.05). These
results suggested that AT-I treatment induced G1 phase arrest in
B16 cells. The cyclin E-CDK2 complex has a critical role in the
G1/S phase transition and it can be directly inhibited by p21
(18). As demonstrated by western
blot assays (Fig. 2C), AT-I
treatment markedly decreased the expression levels of CDK2 and
increased the expression levels of p21.
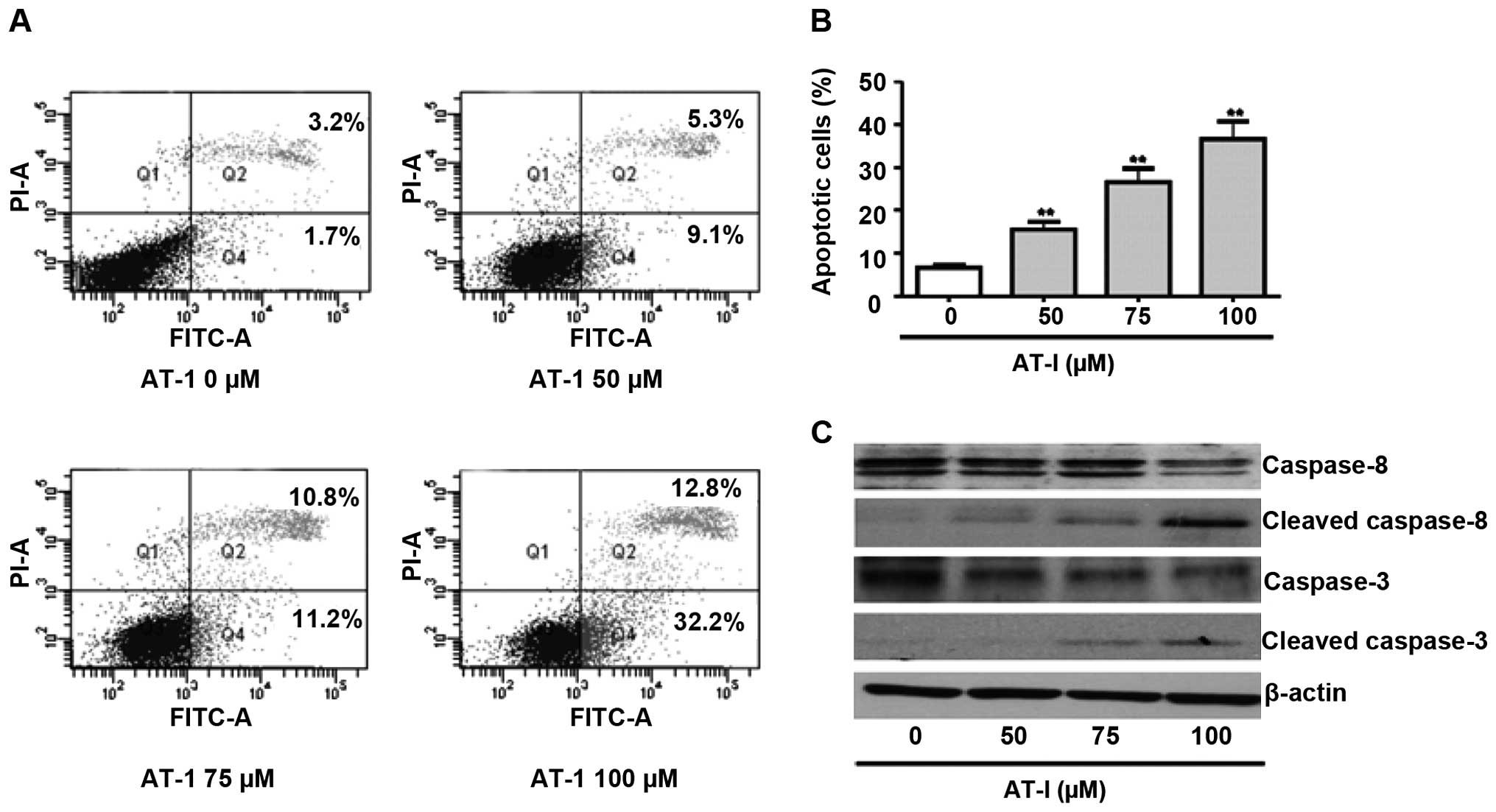
AT-I induces apoptosis in B16 cells
To investigate whether AT-I induced apoptosis, we
analyzed Annexin V/PI-stained B16 cells by flow cytometry.
Treatment with 100 µΜ AT-I for 48 h did not induce
significant apoptosis in B16 cells (data not shown), while AT-I
treatment for 72 h dose-dependently increased late (Q2 in Fig. 3A) and early stage apoptotic cells
(Q4 in Fig. 3A). Fig. 3B shows the statistical results of
total apoptotic cells from three independent experiments.
Caspases-3 and-8 are situated at pivotal junctions in apoptosis
pathways. Western blot assays demonstrated that treatment with AT-I
markedly activated caspase-3 and-8, as evidenced by decreased
expression levels of the procaspases and increased expression
levels of the cleaved caspases (Fig.
3C).
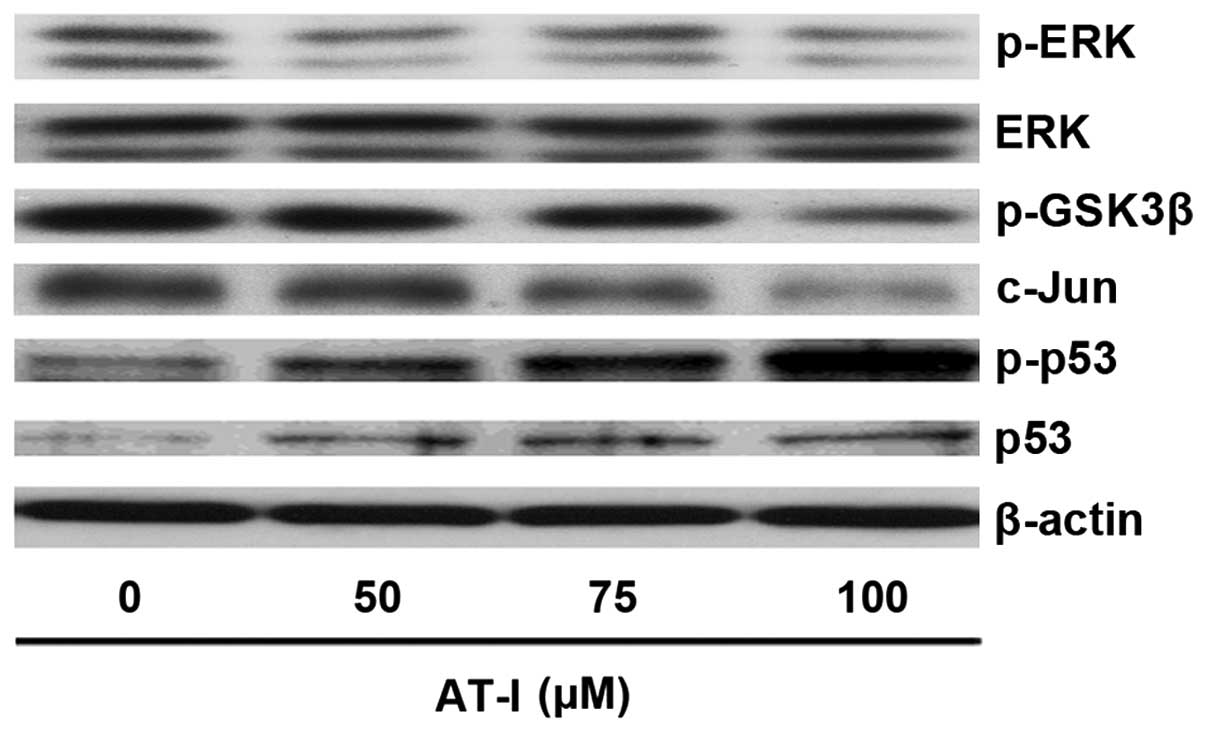
AT-I decreases ERK and c-Jun activation
but increases GSK3β and p53 activation in B16 cells
In melanoma, constitutive activation of ERK can
inactivate GSK3β, which in turn increases c-Jun stability and
decreases p53 activity (14,17,19).
As demonstrated by the western blot assays, AT-I treatment markedly
decreased the expression levels of p-ERK and c-Jun, while the
expression levels of phospho-p53 and p53 were significantly
increased, as compared with the medium control (Fig. 4). Phosphorylation of GSK-3β at ser9
leads to inactivation of GSK3β (20). Western blot assays showed that AT-I
treatment activated GSK3β, as evidenced by decreased expression
levels of p-GSK3β (ser9) (Fig.
4).
LiCl pretreatment reverses AT-I-induced
apoptosis and G1 phase arrest
LiCl is a GSK3β inhibitor (20). It has been reported that LiCl could
counteract cisplatin-induced apoptosis of cancer cells (21,22).
As demonstrated by the flow cytometric analysis, LiCl (5 mM)
treatment alone did not increase the apoptosis of B16 cells, while
pretreatment with LiCl (5 mM) significantly reversed AT-I (100
µM)-induced apoptosis (Fig
5A and B). The cell cycle distribution analysis showed that
LiCl (5 mM) pretreatment partially reversed AT-I (100
µM)-induced G1 phase arrest (Fig. 5C).
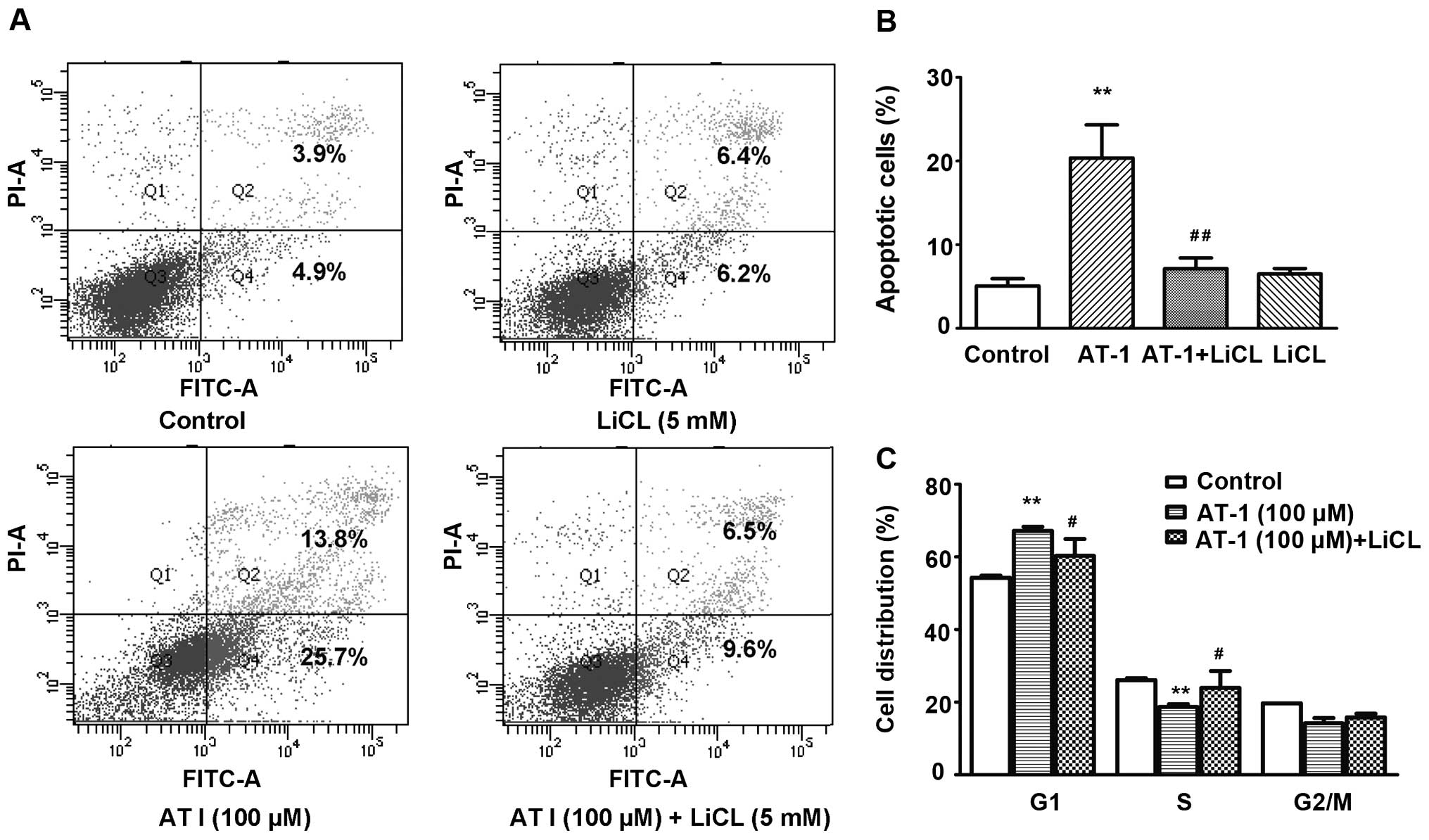 | Figure 5LiCl pretreatment reverses
AT-I-induced apoptosis and cell cycle arrest. Cells were pretreated
with or without 5 mM LiCl for 1 h, then treated with AT-1 (0, 100
µM) for 48 or 72 h. (A) Representative apoptosis analysis.
Cells were treated for 72 h, stained with FITC-Annexin V and PI and
then analyzed using a flow cytometer. Annexin
V+PI+ are the late stage apoptotic cells (Q2)
and Annexin V+PI− are the early stage
apoptotic cells (Q4). (B) Statistical analysis of apoptosis in
three independent experiments. **P<0.01, as compared
with the medium control group, ##P<0.01, as compared
with the AT-I alone treatment group. (C) Statistical analysis of
cell cycle distribution in three independent experiments. Cells
were treated for 48 h. After fixation, the cells were stained with
PI and then analyzed using a flow cytometer.
**P<0.01, as compared with the medium control group,
#P<0.05, as compared with the AT-I treatment alone
group. AT-I, atractylenolide I. |
Discussion
Malignant melanoma is a lethal skin cancer. Although
mutant BRAF-targeted therapy and immunotherapy show promising
clinical response, available chemotherapeutics often carry a low
response rate, tolerance, high price and/or toxicity (23). Novel agents need to be developed to
overcome the limitations of the current therapeutic agents. In the
present study, we reported that AT-I, isolated from the Chinese
medicinal herb Baizhu, induced G1 phase arrest and apoptosis
in B16 melanoma cells by regulating the ERK/GSK3β signaling
pathway.
Progression of cells from G1 to S phase requires the
coordination of a group of regulatory proteins. Among the
regulators, p53 is well characterized. As a transcriptional factor,
p53 can regulate the transcription of p21, which plays a crucial
role in G1 phase arrest (12,24).
P21 binds to the cyclin E-CDK2 complex and inhibits the kinase
activity of CDK, thereby inducing cell cycle arrest (25,26).
In the present study, we observed that the G1 phase-arresting
activity of AT-I was accompanied by increased expression levels of
phospho-p53, p53 and p21, and decreased expression levels of CDK2,
suggesting that the p53/p21 pathway may contribute to the G1
phase-arresting activity of AT-I.
Apoptosis is triggered through the extrinsic and
intrinsic pathways. The extrinsic pathway involves engagement of
particular death receptors that belong to the tumor necrosis factor
receptor (TNF-R) family and through the formation of the
death-inducing-signaling-complex (DISC), and leads to a cascade of
activation of caspases, including caspase-3 and-8 (27). It is well documented that caspase 8
may be activated by p53 (13). In
our investigations, AT-I treatment-induced apoptosis was associated
with p53, and caspase-3 and-8 activation. These findings reveal
that activation of the p53/caspase 8 pathway may be involved in the
apoptosis-promoting effect of AT-I.
It has been reported that c-jun directly represses
p53 transcription by binding to a variant AP-1 site in the p53
promoter (16). In the present
study, AT-1 treatment significantly decreased the expression levels
of c-Jun. c-Jun regulates the cell cycle progression via direct
transcriptional control of cyclin D1 (28). The present results show that AT-I
treatment decreased the mRNA levels of cylin D1 (data not shown).
p53 and c-Jun can be regulated by GSK3β (12,13,17).
Classically, GSK3β has been described as a key regulator of
glycogen metabolism and is also known to regulate other processes,
such as apoptosis, cell proliferation, cell motility and Wnt
signaling (28,29). Phosphorylation at tyrosine 216
enhances the enzymatic activity of GSK3β, while phosphorylation at
serine 9 significantly decreases the activity of GSK3β (30). In the present study, AT-I treatment
significantly activated GSK3β, which is evidenced by the decrease
of p-GSK3β (ser9). LiCl can inhibit GSK3β activity by increasing
GSK3β phosphorylation at serine 9 (20). Recent findings suggest that LiCl may
counteract the cisplatin-induced apoptosis of cancer cells
(21,22). In our investigations, pretreatment
with LiCl significantly reversed AT-I-induced apoptosis.
Additionally, AT-I-induced G1 phase arrest was partially reversed
by LiCl. AT-I-induced decreases of cyclin D1 mRNA were also
reversed by LiCl (data not shown). These findings suggest that
GSK3β signaling may be involved in the apoptosis-promoting and G1
phase-arrest effects of AT-I. In melanoma, the constitutive
activation of ERK has been reported to inactivate GSK3β (17). In the present study, AT-I treatment
dose-dependently inhibited ERK activity, suggesting that AT-I may
regulate GSK3 signaling through inactivation of ERK.
AT-I reduces the symptoms of patients with gastric
cancer cachexia without overt toxicity, slight nausea and dry mouth
are the only reported side effects (31,32).
In rats, AT-I can be rapidly absorbed with a T1/2α of
0.92 h and is eliminated gradually with a T1/2β of 9.74
h after intragastric (i.g.) administration (33), suggesting a good oral
bioavailability. We have shown that AT-I can induce cell
differentiation, inhibit cell migration and inhibit the
phosphorylation of Akt in melanoma cells (11). Moreover, in the present study we
found that AT-I induced G1 phase arrest and apoptosis and inhibited
ERK/GSK3β signaling in melanoma cells. Thus further investigations
are required to develop AT-I as a pharmaceutical agent for melanoma
prevention and/or treatment, although it does not exhibit potent
cytotoxic effect.
In conclusion, we have demonstrated the G1
phase-arresting and apoptosis-promoting effects and revealed the
ERK/GSK3β signaling-related mechanism of action of AT-I in B16
cells. The results of the present study shed light on the molecular
mechanisms of AT-1′s anti-melanoma properties.
Acknowledgments
This study was supported by the Research Grants
Council of Hong Kong (HKBU 262512), Food and Health Bureau of Hong
Kong (HMRF 11122521), the Science, Technology and Innovation
Commission of Shenzhen (JCYJ20120829154222473 and JCY
J20140807091945050) and the Hong Kong Baptist University
(FRG1/14-15/061 and FRG2/14-15/056).
References
|
1
|
Sladden MJ, Balch C, Barzilai DA, Berg D,
Freiman A, Handiside T, Hollis S, Lens MB and Thompson JF: Surgical
excision margins for primary cutaneous melanoma. Cochrane Database
Syst Rev. pp. CD0048352009, View Article : Google Scholar
|
|
2
|
Hodi FS, O'Day SJ, McDermott DF, Weber RW,
Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel
JC, et al: Improved survival with ipilimumab in patients with
metastatic melanoma. N Engl J Med. 363:711–723. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Robert C, Thomas L, Bondarenko I, O'Day S,
Weber J, Garbe C, Lebbe C, Baurain JF, Testori A, Grob JJ, et al:
Ipilimumab plus dacarbazine for previously untreated metastatic
melanoma. N Engl J Med. 364:2517–2526. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Flaherty KT, Puzanov I, Kim KB, Ribas A,
McArthur GA, Sosman JA, O'Dwyer PJ, Lee RJ, Grippo JF, Nolop K, et
al: Inhibition of mutated, activated BRAF in metastatic melanoma. N
Engl J Med. 363:809–819. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kudchadkar RR, Gonzalez R and Lewis K: New
targeted therapies in melanoma. Cancer Control. 20:282–288.
2013.PubMed/NCBI
|
|
6
|
Bauer BA: Herbal therapy: What a clinician
needs to know to counsel patients effectively. Mayo Clin Proc.
75:835–841. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Endo K, Taguchi T, Taguchi F, Hikino H,
Yamahara J and Fujimura H: Antiinflammatory principles of
Atractylodes rhizomes. Chem Pharm Bull (Tokyo). 27:2954–2958. 1979.
View Article : Google Scholar
|
|
8
|
Kiso Y, Tohkin M and Hikino H: Mechanism
of antihepatotoxic activity of atractylon, I: Effect on free
radical generation and lipid peroxidation. Planta Med. 51:97–100.
1985. View Article : Google Scholar
|
|
9
|
Mori H, Xu Q, Sakamoto O, Uesugi Y, Koda A
and Nishioka I: Mechanisms of antitumor activity of aqueous
extracts from Chinese herbs: Their immunopharmacological
properties. Jpn J Pharmacol. 49:423–431. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kang TH, Bang JY, Kim MH, Kang IC, Kim HM
and Jeong HJ: Atractylenolide III, a sesquiterpenoid, induces
apoptosis in human lung carcinoma A549 cells via
mitochondria-mediated death pathway. Food Chem Toxicol. 49:514–519.
2011. View Article : Google Scholar
|
|
11
|
Yan Ye, Chou GX, Hui Wang, Chu JH, Fong WF
and Yu ZL: Effects of sesquiterpenes isolated from largehead
atractylodes rhizome on growth, migration, and differentiation of
B16 melanoma cells. Integr Cancer Ther. 10:92–100. 2011. View Article : Google Scholar
|
|
12
|
el-Deiry WS, Tokino T, Velculescu VE, Levy
DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW and
Vogelstein B: WAF1, a potential mediator of p53 tumor suppression.
Cell. 75:817–825. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ehrhardt H, Häcker S, Wittmann S, Maurer
M, Borkhardt A, Toloczko A, Debatin KM, Fulda S and Jeremias I:
Cytotoxic drug-induced, p53-mediated upregulation of caspase-8 in
tumor cells. Oncogene. 27:783–793. 2008. View Article : Google Scholar
|
|
14
|
Watcharasit P, Bijur GN, Zmijewski JW,
Song L, Zmijewska A, Chen X, Johnson GV and Jope RS: Direct,
activating interaction between glycogen synthase kinase-3beta and
p53 after DNA damage. Proc Natl Acad Sci USA. 99:7951–7955. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Eom TY, Roth KA and Jope RS: Neural
precursor cells are protected from apoptosis induced by trophic
factor withdrawal or genotoxic stress by inhibitors of glycogen
synthase kinase 3. J Biol Chem. 282:22856–22864. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Schreiber M, Kolbus A, Piu F, Szabowski A,
Möhle-Steinlein U, Tian J, Karin M, Angel P and Wagner EF: Control
of cell cycle progression by c-Jun is p53 dependent. Genes Dev.
13:607–619. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lopez-Bergami P, Huang C, Goydos JS, Yip
D, Bar-Eli M, Herlyn M, Smalley KS, Mahale A, Eroshkin A, Aaronson
S, et al: Rewired ERK-JNK signaling pathways in melanoma. Cancer
Cell. 11:447–460. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sherr CJ and Roberts JM: CDK inhibitors:
Positive and negative regulators of G1-phase progression. Genes
Dev. 13:1501–1512. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shao J, Teng Y, Padia R, Hong S, Noh H,
Xie X, Mumm JS, Dong Z, Ding HF, Cowell J, et al: COP1 and GSK3β
cooperate to promote c-Jun degradation and inhibit breast cancer
cell tumorigenesis. Neoplasia. 15:1075–1085. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dihlmann S, Klein S and Doeberitz Mv Mv:
Reduction of beta-catenin/T-cell transcription factor signaling by
aspirin and indomethacin is caused by an increased stabilization of
phosphorylated beta-catenin. Mol Cancer Ther. 2:509–516.
2003.PubMed/NCBI
|
|
21
|
Gao Y, Liu Z, Zhang X, He J, Pan Y, Hao F,
Xie L, Li Q, Qiu X and Wang E: Inhibition of cytoplasmic GSK-3β
increases cisplatin resistance through activation of Wnt/β-catenin
signaling in A549/DDP cells. Cancer Lett. 336:231–239. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Novetsky AP, Thompson DM, Zighelboim I,
Thaker PH, Powell MA, Mutch DG and Goodfellow PJ: Lithium chloride
and inhibition of glycogen synthase kinase 3β as a potential
therapy for serous ovarian cancer. Int Gynecol Cancer. 23:361–366.
2013. View Article : Google Scholar
|
|
23
|
Grimaldi AM, Cassidy PB, Leachmann S and
Ascierto PA: Novel approaches in melanoma prevention and therapy.
Cancer Treat Res. 159:443–455. 2014. View Article : Google Scholar
|
|
24
|
el-Deiry WS, Harper JW, O'Connor PM,
Velculescu VE, Canman CE, Jackman J, Pietenpol JA, Burrell M, Hill
DE, Wang Y, et al: WAF1/CIP1 is induced in p53-mediated G1 arrest
and apoptosis. Cancer Res. 54:1169–1174. 1994.PubMed/NCBI
|
|
25
|
Toyoshima H and Hunter T: p27, a novel
inhibitor of G1 cyclin-Cdk protein kinase activity, is related to
p21. Cell. 78:67–74. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Peter M and Herskowitz I: Joining the
complex: Cyclin-dependent kinase inhibitory proteins and the cell
cycle. Cell. 79:181–184. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ashkenazi A and Dixit VM: Death receptors:
Signaling and modulation. Science. 281:1305–1308. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Doble BW and Woodgett JR: GSK-3: Tricks of
the trade for a multi-tasking kinase. J Cell Sci. 116:1175–1186.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jope RS and Johnson GV: The glamour and
gloom of glycogen synthase kinase-3. Trends Biochem Sci. 29:95–102.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jope RS, Yuskaitis CJ and Beurel E:
Glycogen synthase kinase-3 (GSK3): Inflammation, diseases, and
therapeutics. Neurochem Res. 32:577–595. 2007. View Article : Google Scholar :
|
|
31
|
Liu Y, Ye F, Qiu GQ, Zhang M, Wang R, He
QY and Cai Y: Effects of lactone I from Atractylodes macrocephala
Koidz on cytokines and proteolysis-inducing factors in cachectic
cancer patients. Di Yi Jun Yi Da Xue Xue Bao. 25:1308–1311. 2005.In
Chinese. PubMed/NCBI
|
|
32
|
Liu Y, Jia Z, Dong L, Wang R and Qiu G: A
randomized pilot study of atractylenolide I on gastric cancer
cachexia patients. Evid Based Complement Altemat Med. 5:337–344.
2008. View Article : Google Scholar
|
|
33
|
Wang C, Wang S, Chen Q and He L: A
capillary gas chromatography-selected ion monitoring mass
spectrometry method for the analysis of atractylenolide I in rat
plasma and tissues, and application in a pharmacokinetic study. J
Chromatogr B Analyt Technol Biomed Life Sci. 863:215–222. 2008.
View Article : Google Scholar : PubMed/NCBI
|