Introduction
Breast cancer is one of the most frequent cancers
and the second leading cause of cancer deaths in American women
(1). Approximately 70% of breast
cancers express estrogen receptors (ER) (1). Εstrogen receptor-α (ER-α) is a very
important prognostic marker for selecting an appropriate hormonal
therapy (2,3). Breast cancers that express ER and/or
the progesterone receptor (PR) are treated with targeted
anti-estrogen therapy such as tamoxifen (2,4).
Adjuvant tamoxifen therapy is effective to prolong disease-free and
overall survival of ER-positive breast cancer patients and induces
the arrest of tumor progression in 50% of patients with breast
cancer (5). However, although
anti-estrogen therapies targeting ER-α prevent disease recurrence
in patients with hormone-dependent breast cancer, de novo or
acquired resistance occurs during therapy (6,7). Thus,
we investigated the effects of new therapeutic drugs for treatment
of ER-positive breast cancer patients.
Acquired tamoxifen-resistant (TamR) cells developed
from the parental MCF7 breast cancer cells, elevates some epidermal
growth factor receptor (EGFR) ligands as well as the level of EGFR
and HER2 receptors compared with tamoxifen-responsive MCF7 cells
(8). The heterodimerization of
enhanced EGFR and HER2 triggers phosphorylation of downstream
kinases, including ERK1/2 MAP kinase, Akt, and PKC-α (9–11). In
addition, EGF suppresses the expression and activity of the ER-α
via the phosphatidylinositol-3-kinase (PI-3K)/Akt pathway in MCF7
cells (12). In contrast, stable
transfection of parental MCF-7 cells with a dominant-negative Akt
mutant recovers ER-α expression and activity by 70–80% (12). Generally, an inverse correlation
between EGFR and ER-α was maintained at relapse on tamoxifen
(13). The acquired expression of
EGFR during treatment plays an important role in the development of
tamoxifen resistance in human breast cancer (14).
In the present study, we evaluated the effect of
EGFR inhibitors, neratinib and gefitinib, in tamoxifen-sensitive
(TamS) and TamR cells. TamR cells showed mesenchymal phenotypes and
highly expressed mesenchymal marker proteins, including fibronectin
(FN), N-cadherin (N-cad), and Slug. In addition, the level of EGFR
expression was significantly increased in TamR cells.
Interestingly, neratinib, one of EGFR inhibitors, induced cell
death of TamR but not gefitinib. Therefore, we demonstrated that
neratinib treatment may be a promising therapeutic strategy to
overcome tamoxifen resistance.
Materials and methods
Reagents
Dulbecco's modified Eagle's medium (DMEM) was
purchased from Thermo Scientific (Hemel Hempstead, UK). Fetal
bovine serum (FBS) was purchased from Hyclone (Logan, UT, USA).
Phenol red-free DMEM, penicillin (100 U/ml) and 100 mg/ml
streptomycin were purchased from Life Technologies (Rockville, MD,
USA). Neratinib and gefitinib were purchased from Selleck Chemicals
(Houston, TX, USA). 4-Hydroxytamoxifen (4-OHT) was purchased from
Sigma-Aldrich (St. Louis, MO, USA). Rabbit monoclonal anti-PARP-1,
FN, ER-α, and Twist were purchased from Epitomics (Burlingame, CA,
USA). Epithelial-mesenchymal transition (EMT) antibody sampler
[E-cadherin (E-cad), N-cad, and Slug antibodies] was purchased from
Cell Signaling Technology (Beverly, MA, USA). The secondary
HRP-conjugated antibody and mouse monoclonal anti-β-actin antibody
were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA,
USA). The ECLprime reagents were purchased from Amersham
(Buckinghamshire, UK).
Establishment of TamR MCF7 breast cancer
cells
TamS and TamR breast cancer cell lines were kindly
provided by Professor Keun Wook Kang (Seoul National University,
Seoul, Korea). The TamR was established using methodology reported
previously (8). Briefly, to
establish TamR, MCF-7 cells were washed with PBS, and the culture
medium was changed to phenol red-free DMEM containing 10%
charcoal-stripped steroid-depleted FBS (both from Life
Technologies) and 0.1 mM 4-OHT. The cells were continuously exposed
to this treatment regimen for 2 weeks, and the 4-OHT concentration
was increased gradually up to 3 mM over a 9-month period.
Initially, cell growth rates were depressed. However, after
exposure to the medium for 9 months, the rate of cell growth
increased gradually, indicating the establishment of TamR
cells.
Soft agar colony formation assay
TamS and TamR cells were seeded at a density of
5×104 cells/well in 6-well plates in growth medium
containing 0.7% agar (1.5 ml/well) on top of a layer of growth
medium containing 1.4% agar (2 ml/well). Growth medium (500
μl) with 10% FBS was added on top of the agar. The cell
suspension was plated and cultured in a 37°C incubator for 2 weeks.
After 2 weeks, viable colonies were stained 0.01% crystal violet
and then were observed using a CK40 inverted microscope (Olympus,
Tokyo, Japan).
Flow cytometry analysis (FACS)
Apoptosis assays were performed with the Annexin
V-fluorescein isothiocyanate (FITC) apoptosis kit-I (BD Pharmingen,
San Diego, CA, USA), according to the manufacturers instructions.
Briefly, cells (1×106 cells/ml) were collected and
washed twice with PBS and then resuspended in 500 μl of
staining solution containing 5 μl FITC-conjugated Annexin V
and propidium iodide (PI). After incubation for 15 min at room
temperature (RT) in the dark, cells were immediately analyzed on a
flow cytometer. Apoptotic cells were double-stained with Annexin V
and PI and then they were analyzed using the FACS Vantage system
(Becton-Dickinson, San Diego, CA, USA). The percentage of cells
undergoing apoptosis was determined.
Cell viability
To measure the sensitivities to EGFR inhibitors,
neratinib or gefitinib, we analyzed using a Countess Automated Cell
Counter (Invitrogen, Carlsbad, CA, USA). Briefly, TamS and TamR
cells (5×104 cells/well) were seeded onto 6-well plates.
TamS and TamR cells were incubated in phenol red-free DMEM
containing 10% charcoal-stripped steroid-depleted FBS without 3
μM 4-OHT and 2.5 μM neratinib or gefitinib,
respectively for 24 h.
Western blotting
The cell lysates were used in the immunoblot
analysis for PARP-1, E-cad, N-cad, FN, Twist, Slug, EGFR, HER2, and
β-actin. The proteins were boiled for 5 min in Laemmli sample
buffer and then electrophoresed on 8% or 10% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis gels. The proteins were
transferred to PVDF membranes, and the membranes were blocked with
10% skim milk in TBS with 0.01% Tween-20 for 15 min. The blots were
incubated with anti-PARP-1, ER-α, E-cad, N-cad, FN, Twist, Slug,
EGFR, HER2, and β-actin antibodies (1:1,000 dilution) in 1% TBS/T
buffer (0.01% Tween-20 in TBS) at 4°C overnight. The blots were
washed 3 times in TBS with 0.01% Tween-20, and they were
subsequently incubated with anti-rabbit HRP-conjugated antibody
(1:5,000 dilution) in TBS/T buffer. After 1-h incubation at RT, the
blots were washed 3 times and ECLprime reagents were
applied for further development.
Microarray data analysis
We downloaded expression data from a public database
(GSE1378) and analyzed the clinical value of EGFR in
tamoxifen-treated breast cancer patients.
Real-time polymerase chain reaction
(RT-PCR)
Total RNA was extracted from the cells using TRIzol
reagent (Invitrogen), according to the manufacturers instructions.
Isolated RNA samples were then used for RT-PCR. Samples (1
μg total RNA) were reverse-transcribed into cDNA in
20-μl reaction volumes using a First Strand cDNA synthesis
kit for RT-PCR, according to the manufacturer's instructions (MBI
Fermentas, Hanover, MD, USA).
Gene expression was quantified by real-time PCR
using a SensiMix SYBR kit (Bioline Ltd., London, UK) and 100 ng of
cDNA per reaction. The sequences of the primer sets used for this
analysis are shown in Table I. An
annealing temperature of 60°C was used for all primers. PCRs were
performed in a standard 384-well plate format with an ABI 7900HT
Real-Time PCR detection system (Applied Biosystems, Foster City,
CA, USA). The raw threshold cycle (CT) value was first
normalized to the housekeeping gene for each sample to obtain
ΔCT. The normalized ΔCT was then calibrated
to the control cell samples to obtain ΔΔCT. All cDNA
samples were analyzed in 3 independent experiments.
 | Table IPrimer sequences for analysis of the
various gene mRNA expression. |
Table I
Primer sequences for analysis of the
various gene mRNA expression.
| Gene name | Primer sequences |
|---|
| ER-α | F: CGC TAC TGT GCA
GTG TGC AAT |
| R: CCT CAC AGG ACC
AGA CTC CAT AA |
| E-cad | F: GGC ACA AAG ATG
GGG GCT TC |
| R: TCA CCA CCT CCA
CAG CCA CC |
| N-cad | F: GCA GAT CGG ACC
GGA TAC TG |
| R: TGG GAA TCC GAC
GAA TGG |
| FN | F: CCA CCC CCA TAA
GGC ATA GG |
| R: GTA GGG GTC AAA
GCA CGA GTC ATC |
| Slug | F: CTG TGG TCC TTG
GAG GAG GT |
| R: GTA GGT GCC AGG
GTG GAA AT |
| Twist | F: CAG CTT GCC ATC
TTG GAG TC |
| R: GAC GAC AGC CTG
AGC AAC AG |
| EGFR | F: CAT GTC GAT CTT
CCA GA |
| R: GGG ACA GCT TGG
ATC ACA CT |
| HER2 | F: CAC TTC AAC CAC
AGT GGC AT |
| R: ATT CAC ATA CTC
GGG GA |
| GAPDH | F: ATT GTT GCC ATC
AAT GAC CC |
| R: AGT AGA GGC AGG
GAT GAT GT |
Statistical analysis
Statistical significance was determined using the
Student's t-test. Results are presented as means ± standard errors.
All P-values are two-tailed, and differences were considered
significant at P<0.05.
Results and Discussion
To establish a new therapeutic strategy for
treatment of TamR breast cancer, we analyzed differential
characteristics of TamS and TamR breast cancer cell lines. In a
previous study, breast cancer cell lines could be classified into
four distinct morphological groups referred to as round, mass,
grape-like, and stellate (15). As
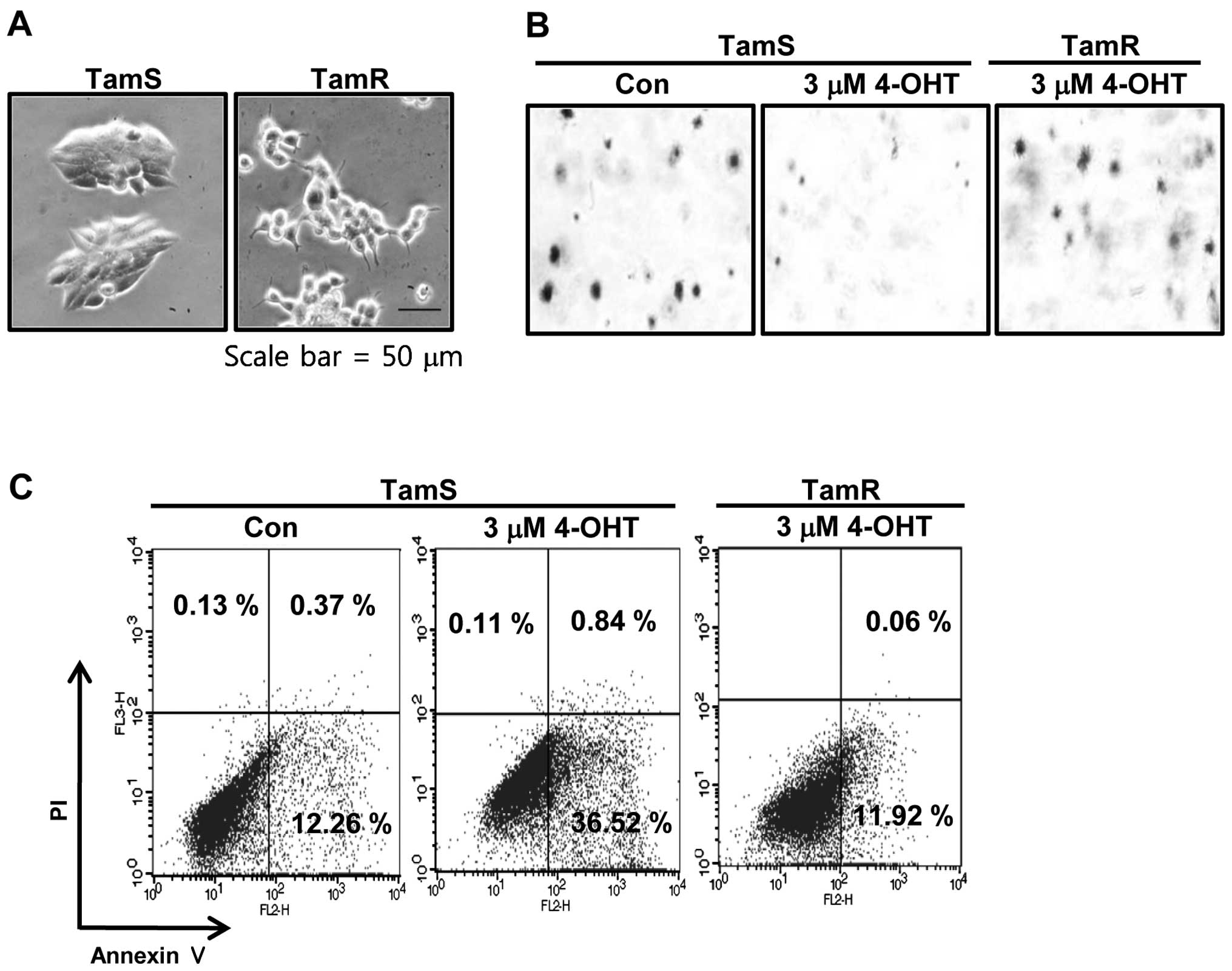
shown in Fig. 1A, we also observed
the morphological difference between TamS and TamR cells. TamS
cells stacked up to form colonies and TamR cells scattered, in
loosely-packed colonies, and had many branches. In addition, to
assess the tumorigenicity degree of TamS and TamR cells, we
examined the effect of tamoxifen on the ability of cells to form
colonies using soft agar colony formation assays. The colonies of
TamS cells were greatly decreased by 3 μM tamoxifen
treatment while TamR cells still maintained colonies (Fig. 1B). We also measured the apoptotic
cell death of TamS and TamR cells by tamoxifen. Cells were treated
with 3 μM tamoxifen for 24 h and examined by Annexin V/PI
staining. As shown in Fig. 1C, the
apoptotic cell population of TamS cells was significantly increased
by 3-fold of control level. However, the apoptotic cell population
of TamR cells was similar to control of TamS (Fig. 1C).
In a previous study, tamoxifen resistance was
associated with enhanced cell motility (16). Antiestrogens also promote cellular
invasion and motility in TamR breast cancer cells (17). In addition, TamR MCF7 cells promote
EMT-like behavior and inhibition of EGFR in these cells augments
cell-cell adhesion (18). Thus, we
also investigated the relationship between the morphological change
of TamR cells and EMT. Although the level of Twist expression did
not change, the expression levels of mesenchymal marker proteins
such as N-cad, FN, and Slug, was significantly increased in TamR
cells (Fig. 2A). In contrast, ER-α
and E-cad expression (epithelial marker protein) was decreased in
TamR cells (Fig. 2A). Our results
also showed that the mRNA expression of these proteins was similar
with the patterns of proteins expression (Fig. 2B). Therefore, we demonstrated that
tamoxifen resistance is associated with the severe reduction of
ER-α expression and the induction of EMT.
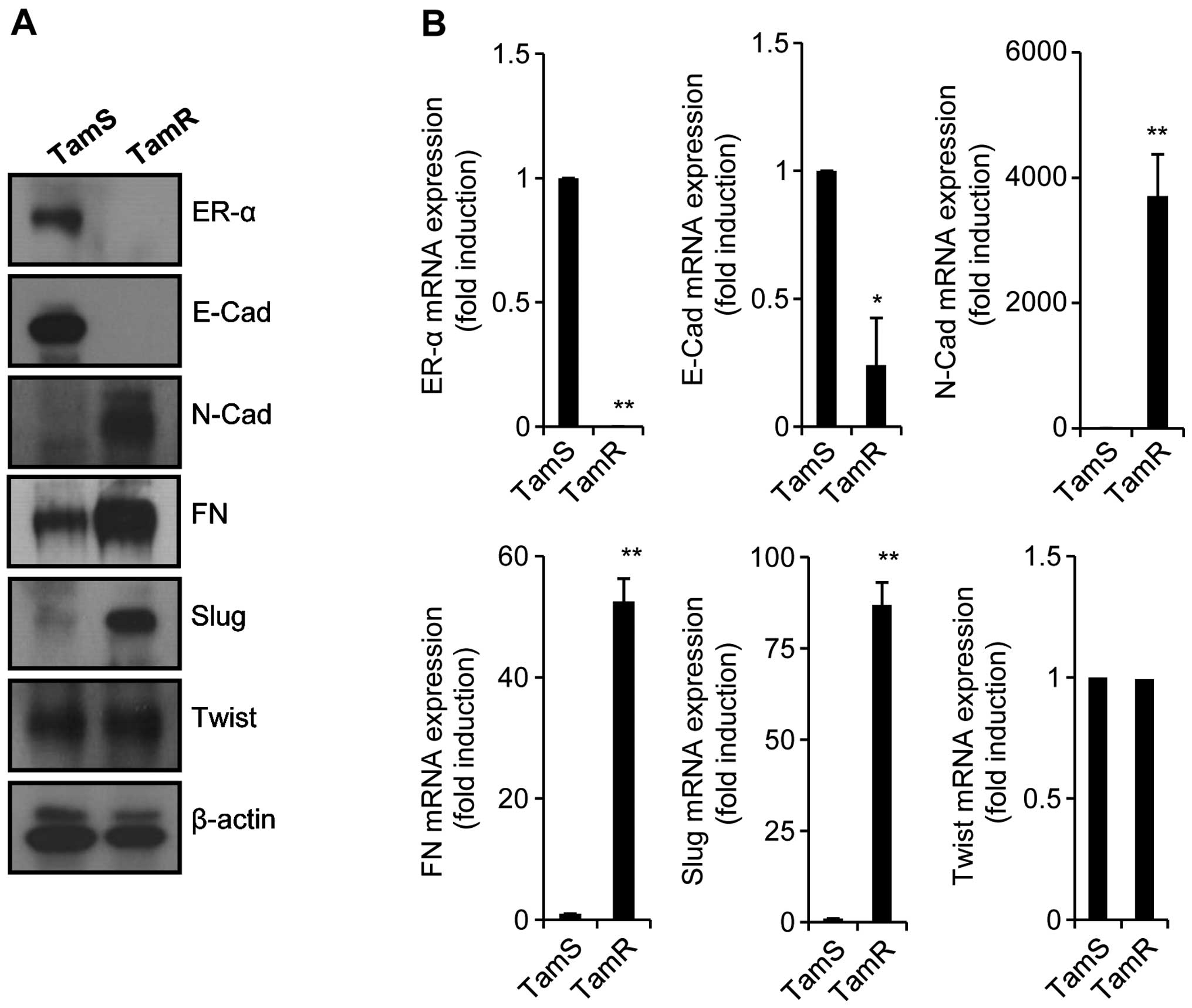 | Figure 2TamR cells highly express mesenchymal
marker proteins. Established TamS and TamR cells were seeded in a
6-well plate with or without 3 μM 4-OHT for 24 h. After 24
h, TamS and TamR cells were harvested for detection of protein
expressions (A) and mRNA expressions (B). ER-α, E-cad, N-cad, FN,
Slug, Twist, and β-actin expression was analyzed by western
blotting (A) and real-time PCR (B), respectively. Results are
representative of 3 independent experiments. Values shown are means
± standard errors. *P<0.05, **P<0.01
vs. TamS. TamR, tamoxifen-resistant; TamS, tamoxifen-sensitive;
4-OHT, 4-Hydroxytamoxifen; ER-α, estrogen receptor-α; E-cad,
E-cadherin; N-cad, N-cadherin; FN, fibronectin. |
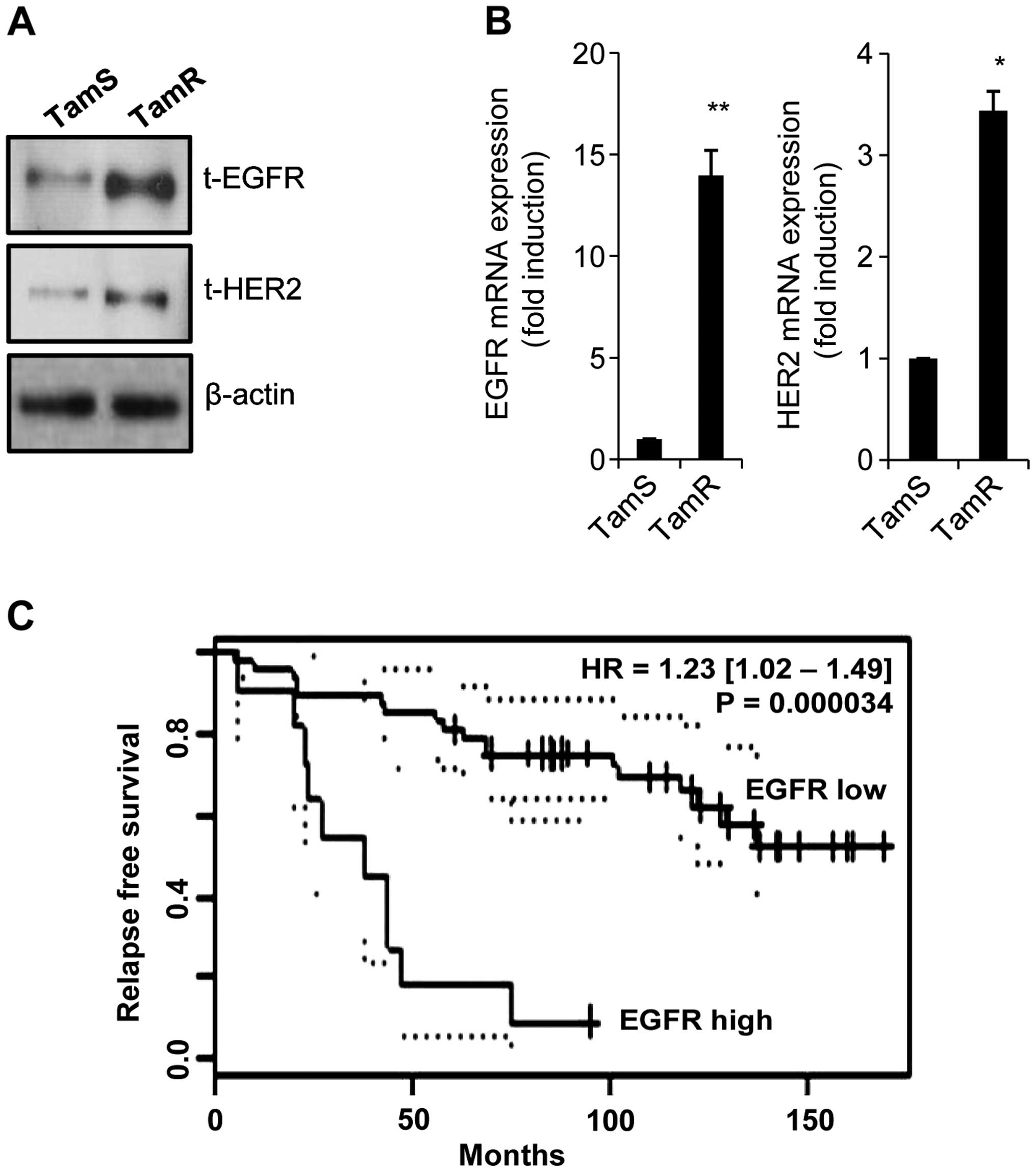
We observed that the levels of EGFR and HER2
proteins and mRNA expression were dramatically enhanced in TamR
cells (Fig. 3A and B). The levels
of EGFR and HER2 mRNA expression were increased by 14.0-fold and
3.4-fold of control level, respectively (Fig. 3A and B). Consistent with our data,
the induction of growth factor receptors, such as EGFR and HER2 has
been implicated in acquired resistance to endocrine therapy
(19,20). Snail overexpressed cells are
resistant to tamoxifen and increase the level of EGFR expression
(21). Next, we analysed the
clinical value of EGFR expression in tamoxifen-treated breast
cancer patients using public microarray datasets (GSE1378).
Interestingly, patients with high EGFR expression levels showed
significantly shorter relapse-free survival time (Fig. 3C, P=0.000034). These results suggest
that the level of EGFR expression plays an important role in
tamoxifen resistance.
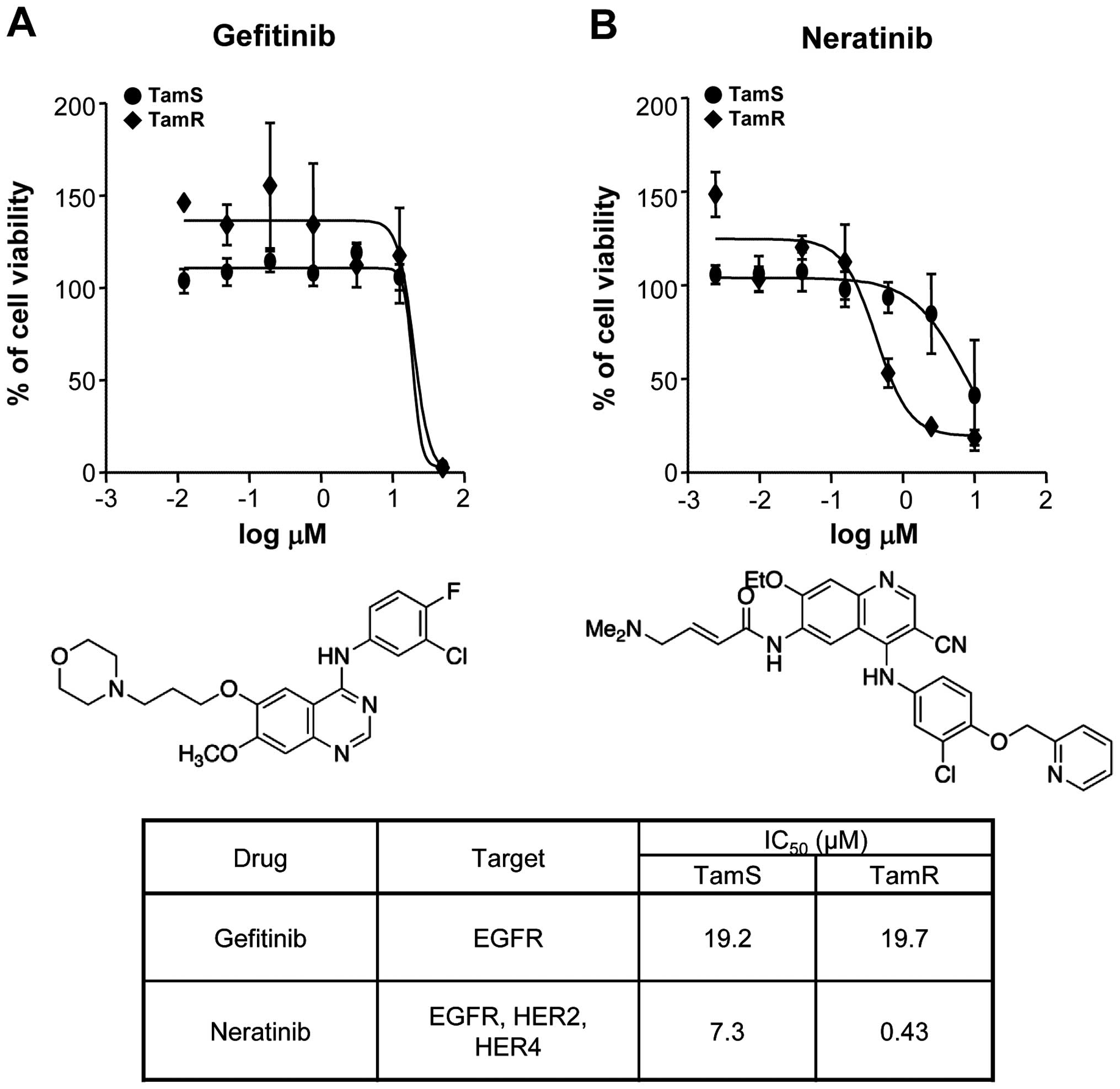
We investigated the effect of EGFR inhibitors,
neratinib and gefitinib, in TamS and TamR cells. Neratinib
(HKI-272, Pfizer; Puma Biotechnology) is a pan-HER receptor
tyrosine kinase inhibitor (EGFR, HER2 and HER4) and binds
irreversible to these kinases (22). Gefitinib (Iressa; AstraZeneca
Pharmaceuticals) is the first selective inhibitor of EGFR tyrosine
kinase domain and is responsible for activating anti-apoptotic
pathways in non-small cell lung cancers (23). As shown in Fig. 4A, gefitinib did not affect the
viabilities of TamS and TamR cells. The IC50 value of
gefitinib was ~20 μM concentration. However, neratinib
significantly decreased the viability of TamR cells at 0.43
μM concentration but the viability of TamS cells did not
change by 7.3 μM neratinib treatment (Fig. 4B). Therefore, we demonstrated that
neratinib prevents more effectively EGFR and HER2 signaling
pathways as well as triggers apoptotic cell death of TamR
cells.
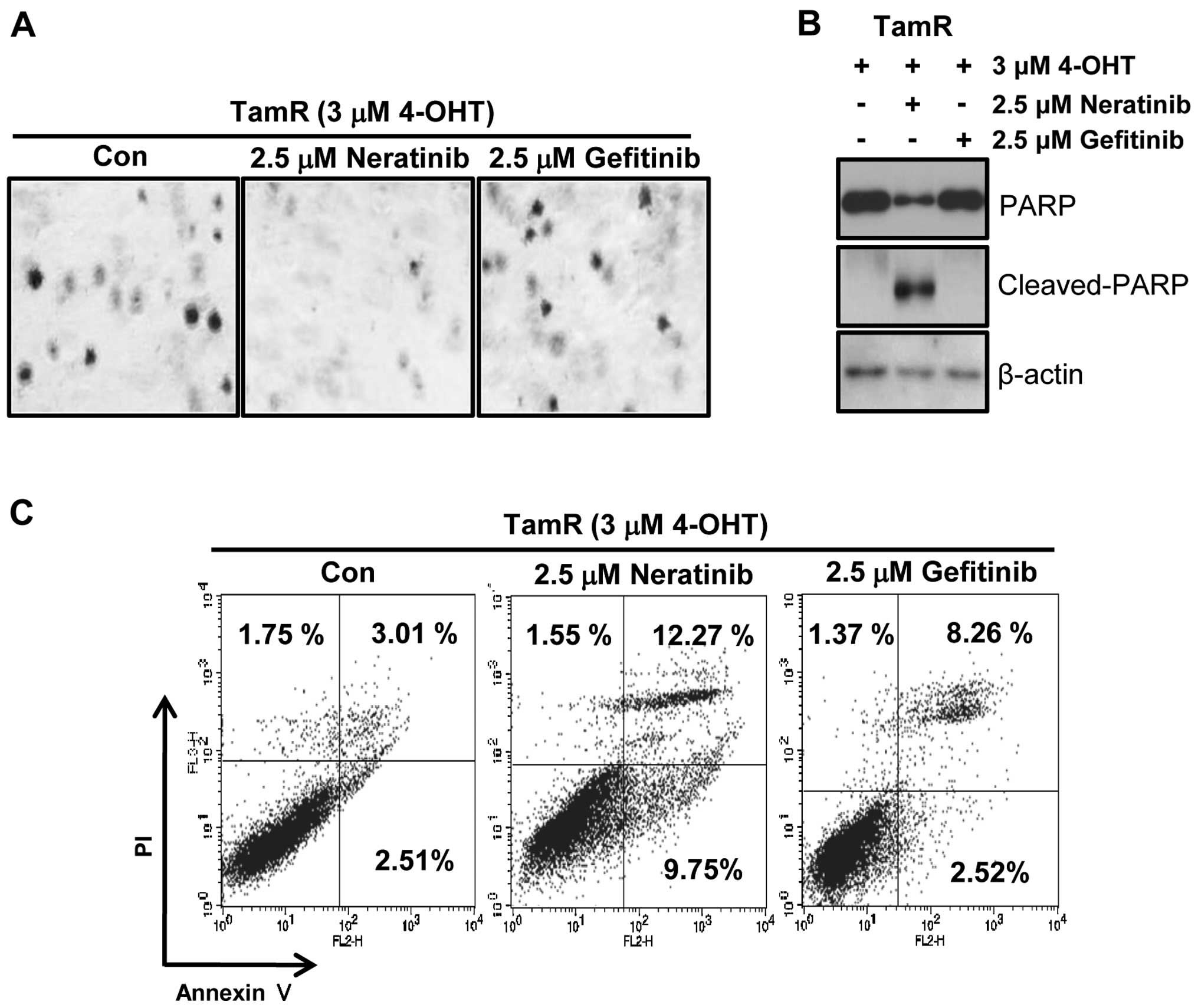
To evaluate the tumorigenicity of TamR cells by EGFR
inhibitors, we examined the effect of neratinib and gefitinib on
the ability of cells to form colonies using soft agar colony
formation assays. As shown in Fig.
5A, the colonies of TamR cells were completely decreased by 2.5
μM neratinib treatment while gefitinib-treated TamR cells
still maintained colonies. Furthermore, one of apoptosis marker
proteins, cleaved PARP-1 expression was also dramatically increased
by neratinib treatment (Fig. 5B).
Finally, we also measured the apoptotic cell death of TamR cells by
neratinib and gefitinib. Cells were treated with 2.5 μM
neratinib and gefitinib, respectively for 24 h. The apoptotic cell
population of TamR cells by neratinib was significantly increased
by 4-fold of control level (Fig.
5C). However, the apoptotic cell population by gefitinib was
slightly increased (Fig. 5C).
Therefore, we suggest that neratinib may be a new therapeutic drug
for treatment of TamR breast cancer.
Acknowledgments
This research was supported by a grant of the Korea
Health Technology R&D Project through the Korea Health Industry
Development Institute (KHIDI), funded by the Ministry of Health and
Welfare, Republic of Korea (HI14C3418) and by the Samsung
Biomedical Research Institute grant (SMX1131701).
References
|
1
|
Jemal A, Siegel R, Xu J and Ward E: Cancer
statistics, 2010. CA Cancer J Clin. 60:277–300. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Osborne CK, Zhao H and Fuqua SA: Selective
estrogen receptor modulators: structure, function, and clinical
use. J Clin Oncol. 18:3172–3186. 2000.PubMed/NCBI
|
|
3
|
Harvey JM, Clark GM, Osborne CK and Allred
DC: Estrogen receptor status by immunohistochemistry is superior to
the ligand-binding assay for predicting response to adjuvant
endocrine therapy in breast cancer. J Clin Oncol. 17:1474–1481.
1999.PubMed/NCBI
|
|
4
|
Herynk MH and Fuqua SA: Estrogen receptors
in resistance to hormone therapy. Adv Exp Med Biol. 608:130–143.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ali S and Coombes RC: Endocrine-responsive
breast cancer and strategies for combating resistance. Nat Rev
Cancer. 2:101–112. 2002. View
Article : Google Scholar
|
|
6
|
Kurebayashi J: Endocrine-resistant breast
cancer: underlying mechanisms and strategies for overcoming
resistance. Breast Cancer. 10:112–119. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Schiff R, Massarweh S, Shou J and Osborne
CK: Breast cancer endocrine resistance: how growth factor signaling
and estrogen receptor coregulators modulate response. Clin Cancer
Res. 9:447S–454S. 2003.PubMed/NCBI
|
|
8
|
Knowlden JM, Hutcheson IR, Jones HE,
Madden T, Gee JM, Harper ME, Barrow D, Wakeling AE and Nicholson
RI: Elevated levels of epidermal growth factor receptor/c-erbB2
heterodimers mediate an autocrine growth regulatory pathway in
tamoxifen-resistant MCF-7 cells. Endocrinology. 144:1032–1044.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kim S, Lee J, Lee SK, Bae SY, Kim J, Kim
M, Kil WH, Kim SW, Lee JE and Nam SJ: Protein kinase C-α
downregulates estrogen receptor-α by suppressing c-Jun
phosphorylation in estrogen receptor-positive breast cancer cells.
Oncol Rep. 31:1423–1428. 2014.
|
|
10
|
Benz CC, Scott GK, Sarup JC, Johnson RM,
Tripathy D, Coronado E, Shepard HM and Osborne CK:
Estrogen-dependent, tamoxifen-resistant tumorigenic growth of MCF-7
cells transfected with HER2/neu. Breast Cancer Res Treat. 24:85–95.
1992. View Article : Google Scholar
|
|
11
|
Li Z, Wang N, Fang J, Huang J, Tian F, Li
C and Xie F: Role of PKC-ERK signaling in tamoxifen-induced
apoptosis and tamoxifen resistance in human breast cancer cells.
Oncol Rep. 27:1879–1886. 2012.PubMed/NCBI
|
|
12
|
Martin MB, Franke TF, Stoica GE, Chambon
P, Katzenellenbogen BS, Stoica BA, McLemore MS, Olivo SE and Stoica
A: A role for Akt in mediating the estrogenic functions of
epidermal growth factor and insulin-like growth factor I.
Endocrinology. 141:4503–4511. 2000.PubMed/NCBI
|
|
13
|
deFazio A, Chiew YE, McEvoy M, Watts CK
and Sutherland RL: Antisense estrogen receptor RNA expression
increases epidermal growth factor receptor gene expression in
breast cancer cells. Cell Growth Differ. 8:903–911. 1997.PubMed/NCBI
|
|
14
|
Newby JC, Johnston SR, Smith IE and
Dowsett M: Expression of epidermal growth factor receptor and
c-erbB2 during the development of tamoxifen resistance in human
breast cancer. Clin Cancer Res. 3:1643–1651. 1997.
|
|
15
|
Kenny PA, Lee GY, Myers CA, Neve RM,
Semeiks JR, Spellman PT, Lorenz K, Lee EH, Barcellos-Hoff MH,
Petersen OW, et al: The morphologies of breast cancer cell lines in
three-dimensional assays correlate with their profiles of gene
expression. Mol Oncol. 1:84–96. 2007. View Article : Google Scholar
|
|
16
|
Zhou C, Zhong Q, Rhodes LV, Townley I,
Bratton MR, Zhang Q, Martin EC, Elliott S, Collins-Burow BM, Burow
ME, et al: Proteomic analysis of acquired tamoxifen resistance in
MCF-7 cells reveals expression signatures associated with enhanced
migration. Breast Cancer Res. 14:R452012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Borley AC, Hiscox S, Gee J, Smith C, Shaw
V, Barrett-Lee P and Nicholson RI: Anti-oestrogens but not
oestrogen deprivation promote cellular invasion in intercellular
adhesion-deficient breast cancer cells. Breast Cancer Res.
10:R1032008. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hiscox S, Jiang WG, Obermeier K, Taylor K,
Morgan L, Burmi R, Barrow D and Nicholson RI: Tamoxifen resistance
in MCF7 cells promotes EMT-like behaviour and involves modulation
of beta-catenin phosphorylation. Int J Cancer. 118:290–301. 2006.
View Article : Google Scholar
|
|
19
|
Hutcheson IR, Knowlden JM, Madden TA,
Barrow D, Gee JM, Wakeling AE and Nicholson RI: Oestrogen
receptor-mediated modulation of the EGFR/MAPK pathway in
tamoxifen-resistant MCF-7 cells. Breast Cancer Res Treat. 81:81–93.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kurokawa H, Lenferink AE, Simpson JF,
Pisacane PI, Sliwkowski MX, Forbes JT and Arteaga CL: Inhibition of
HER2/neu (erbB-2) and mitogen-activated protein kinases enhances
tamoxifen action against HER2-overexpressing, tamoxifen-resistant
breast cancer cells. Cancer Res. 60:5887–5894. 2000.PubMed/NCBI
|
|
21
|
Jiang Y, Zhao X, Xiao Q, Liu Q, Ding K, Yu
F, Zhang R, Zhu T and Ge G: Snail and Slug mediate tamoxifen
resistance in breast cancer cells through activation of EGFR-ERK
independent of epithelial-mesenchymal transition. J Mol Cell Biol.
6:352–354. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rabindran SK, Discafani CM, Rosfjord EC,
Baxter M, Floyd MB, Golas J, Hallett WA, Johnson BD, Nilakantan R,
Overbeek E, et al: Antitumor activity of HKI-272, an orally active,
irreversible inhibitor of the HER-2 tyrosine kinase. Cancer Res.
64:3958–3965. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sordella R, Bell DW, Haber DA and
Settleman J: Gefitinib-sensitizing EGFR mutations in lung cancer
activate anti-apoptotic pathways. Science. 305:1163–1167. 2004.
View Article : Google Scholar : PubMed/NCBI
|