Introduction
Quinazoline derivatives have drawn significant
attention ever since the discovery of gefitinib, a front line drug
for lung cancer. Several such as vandetanib, erlotinib and icotinib
are important antitumor agents containing a quinazoline core.
Gefitinib, also known as Iressa, is a synthetic anilinoquinazoline
compound that acts as a receptor tyrosine kinase inhibitor (TKI)
for its antitumor effect (1).
Gefitinib selectively binds to the epidermal growth factor receptor
(EGFR) tyrosine kinase domain through hydrogen bond formation of
the N1 atom of the quinazoline core with the Met769
residue of the kinase domain, preventing ATP from binding and
blocking subsequent signal transduction for their antitumor effect
(1–3).
EGFR is a receptor tyrosine kinase that is
overexpressed and occasionally mutated in cancers of epithelial
origin, including non-small cell lung cancer (NSCLC), breast,
colorectal and pancreatic cancer (4–6). Over
80% of lung squamous cell carcinomas and approximately half of all
lung adenocarcinomas overexpress EGFR (7,8). EGFR
expression is also associated with a poor response to therapy,
development of cytotoxic drug resistance, disease progression and
poor survival since the receptor tends to be hyperactivated
(9,10). Therefore, specific/selective
inhibition of EGFR tyrosine kinase activity has demonstrated
efficacy against certain types of tumors (11,12).
One of the important signaling mediators downstream
of EGFR signaling is signal transducer and activator of
transcription-3 (STAT3). STAT3 belongs to a protein family of
transcription factors which are essential for cellular functions
(13,14). In addition to EGFR, STAT3 can be
activated by other receptors and non-receptor tyrosine kinases,
such as the IL-6/gp130 receptor family, JAKs and Src kinases. STAT3
is persistently activated in a wide variety of tumors and has been
detected in over 50% of NSCLC primary tumors and cell lines
(15). STAT3 promotes tumor cell
cycle progression, cell proliferation and tumor metastasis through
differential gene regulation. In addition, aberrant STAT3
activation has been linked to gefitinib resistance (15,16).
Therefore, inhibitors of the JAK/STAT3 pathway have been developed
as promising antitumor agents.
We have developed methods to prepare novel
quinazolines (17,18), including ultrasound-promoted
synthesis (19), and KOtBu-mediated
stereoselective addition of quinazolines to alkynes (20). These methods have allowed the
preparation of quinazolines with 2-position substitutions,
including aryl groups and straight-, branched-chain, and
cyclo-alkyl groups with moderate to good yields (20). These compounds have not been fully
investigated for their biological activities. In view of the unique
structural feature of 2-substitution and the rapid progress in
preparation of 2-substituted quinazolines (21), we studied the antitumor activity of
2-alky substituted quinazolines (2-ASQs) and found that these novel
compounds possessed anticancer activity against lung carcinoma
cells. 2-ASQs showed no activity against EGFR activation, possibly
due to the steric effect caused by a bulky group near the
N1 atom. Instead, the compounds promoted apoptotic cell
death through inhibition of STAT3 phosphorylation. In the present
study, we report the antitumor activity of these newly synthesized
quinazolines.
Materials and methods
Chemicals and reagents
Primary antibodies were commercially purchased from
Cell Signaling Technology or from Santa Cruz Biotechnology.
HRP-conjugated secondary antibodies were purchased from
Sigma-Aldrich, and Alexa Fluor-conjugated secondary antibodies were
purchased from Life Technologies. Gefitinib, AG490 and S3I-201 were
purchased from Selleck and
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
was obtained from Sigma-Aldrich. The preparation and identification
of the quinozalines tested in the present study were described as
previously reported (20).
Cell culture
A549, H460, H1299, 293T and Vero cells were
purchased from the Cell Bank of the Chinese Academy of Sciences
(Shanghai, China). A549, H460, 293T and Vero cells were cultured in
high glucose Dulbecco's modified Eagle's medium (DMEM) supplemented
with non-essential amino acids, 10% heat-inactivated fetal bovine
serum (FBS), 2 mM L-glutamine and sodium pyruvate (Life
Technologies, Carlsbad, CA, USA). H1299 cells were cultured in
RPMI-1640 supplemented with non-essential amino acids, 10%
heat-inactivated FBS, sodium pyruvate and 2 mM L-glutamine. The
cells were maintained at 37°C in a humidified atmosphere with 5%
CO2.
Cell viability assay
For cell culture studies, the compounds were
dissolved in dimethylsulfoxide (DMSO) as a 1,000× stock and were
diluted with culture medium before testing. The cytotoxic effect
against Vero cells and IC50 values for tumor cells were
measured by cell viability using the MTT assay. Briefly, the cells
were seeded in 96-well plates at 5×103 cells/well in
complete medium. The cells were treated without or with (DMSO at
0.1% as a control) a compound at different concentrations for 72 h.
At the end of the experiment, MTT was added to each well to a final
concentration of 0.5 mg/ml for the measurement of formazan
formation. The absorbance was measured at 570 nm in a VersaMax
Microtiter Plate Reader (Molecular Devices, Sunnyvale, CA, USA). To
obtain an IC50 value, cells in 96-well plates were
treated with a test compound at concentrations of 0.3–30 µM
for 72 h. Cell viability was then determined using MTT assay and
the data were used to extrapolate IC50 values.
Western blot analysis
Cells were lysed in extraction buffer (300 mM NaCl,
1% Nonidet P-40 in 20 mM Tris-HCl, pH 7.9) supplemented with
protease and phosphatase inhibitors. The cell lysates were cleaned
by centrifugation at 10,000 × g and soluble proteins were separated
by SDS-PAGE, transferred to PVDF membranes and immunoblotted with
primary antibodies followed by HRP-conjugated secondary antibodies
and developed using an ECL Plus kit (GE Healthcare, Pittsburgh, PA,
USA). The images were collected using Alpha Innotech FluorChem FC2
imaging system (Alpha Innotech, San Leandro, CA, USA).
Flow cytometric assay
The percentage of apoptotic cells was detected by
FACS assay using an Annexin V-FITC/PI double labeling kit (Life
Technologies). Briefly, the cells at a density of 3×105
cells/ml were plated at 1.5 ml/well in 6-well plates and allowed to
attach overnight. The cells were treated without or with a test
compound at 1, 3 and 10 µM for 24 h. The cells then were
harvested and analyzed on FACSCalibur (BD Biosciences) following
the manufacturer's instructions.
Immunofluorescence analysis
To determine pSTAT3 distribution, H1299 cells were
pre-treated without or with 10 µM compound #7 and then
stimulated with IL-6 (20 ng/ml) for 15 min at 37°C. The cells were
then fixed with 4% paraformaldehyde, followed by permeabilization
with 0.2% Triton X-100 in phosphate-buffered saline (PBS). The
cells were incubated with anti-p-STAT3, followed by Alexa Fluor
568-conjugated secondary antibody (Life Technologies). Nuclei were
stained with 10 µg/ml of DAPI for 15 min. The images were
captured on an Olympus FluoView FV10i confocal microscope.
Transfections
siRNAs against human STAT3 were purchased from
GenePharma (Shanghai, China). Sequences of the oligos were as
follows: sense, 5′-CCACTTTGGTGTTTCATAA-3′; siRNA and STAT3C were
transfected using Lipofectamine 2000 (Life Technologies) following
the manufacturer's instructions. Overexpression of STAT3C and STAT3
siRNA were verified after transfected for 24 h by western
blotting.
STAT3 transcriptional activity
293T cells were plated into a 12-well plate with
4×105 cells/well. The cells were transiently transfected
with the p-STAT3-TA-luc plasmid (Beyotime, Nantong, China) using
pRL as an internal control. Twenty four hours after transfection,
the cells were then treated with or without #7 for 2 h before
treatment with IL-6 for another 12 h. The cells were lysed and used
for luciferase analysis using a Dual-Luciferase Reporter Assay
System and GloMax Luminometer (Promega Madison, WI, USA). The ratio
of firefly luciferase activity over that of Renilla
luciferase was used as a measurement of reporter gene activity.
Tumor xenograft study
Six-week-old female BALB/c nude mice were obtained
from the Model Animal Research Center of Nanjing University
(Nanjing, China). Animal care and experimental procedures were
conducted in accordance with the guidelines of the Institutional
Animal Care and Use Committee of the Medical School of Nanjing
University. To initiate the xenografts, 2×106 of A549
cells in 0.15 ml PBS were mixed with Matrigel at a 4:1 ratio (BD
Biosciences) and were subcutaneously injected into the flank area
of the nude mice. When palpable tumors were developed after 2
weeks, the mice were randomly divided into 4 groups, 6 in each
group and treated by gavage with vehicle, with compound #7 at 15
and 30 mg/kg, or with genfitinb at 30 mg/kg. The animals were
treated 3 times/week for 4 weeks. The body weight of the mice and
the size of the tumor mass were measured weekly. The tumor volume
was calculated as (half of the length times the square of the
width, in mm3). At the end of the experiment, the mice
were sacrificed, and the tumors were excised and weighed. A portion
of each tumor was fixed in buffered formalin and then embedded in
paraffin for immunohistochemical (IHC) staining or for hematoxylin
and eosin (H&E) staining. The remaining tissue was stored at
−70°C for further analysis.
For Ki-67 and cleaved caspase-3 staining, tissue
sections were de-paraffinized, fixed and stained with appropriate
antibodies. At least three randomly selected fields were examined
and photographed. The unfixed tumor tissues were homogenized and
the proteins extracted were subjected to western blot analysis.
Results
The 2-substituted quinazolines exert
antitumor effects by induction of cell apoptosis
The antitumor activity of the newly synthesized
2-ASQs and 2-aryl substituted quinazolines was first evaluated
against 3 different cell lines with wild-type (wt) EGFR. The
structures of the compounds tested are listed in Fig. 1A. Several 2-ASQs showed selective
cytotoxic effects on tumor cells, while none of these compounds
showed toxic effects on Vero cells after treatment for 72 h
(Fig. 1A). We then performed
experiments to quantitatively measure the cytotoxic effects of
compound 4 (#4) and 7 (#7) on tumor cell growth by determining the
half maximal inhibitory concentration (IC50) values.
A549, H460 and H1299 cells were cultured in the presence of varying
concentrations of a test compound or gefitinib as a positive
control. As shown in Fig. 1B, the
IC50 values for #4 and #7 against these tumor cells were
~3- to 5-fold lower than that of gefitinib. We used #7 as an
example to delineate the mechanism of its antitumor activity. The
more sensitive H460 and H1299 cells were incubated with #7 at
increased concentrations for 24 h. Annexin V/PI double staining was
used to assess apoptosis. As shown in Fig. 1C, treatment with #7 significantly
elevated the population of apoptotic cells. The apoptosis was also
validated with caspase activation determined by immunoblotting
studies. As shown in Fig. 1D, in
H460 and H1299 cells, treatment with #7 dose-dependently caused
caspase-9 and -3, and PARP cleavage, indicating that #7 exerted an
antitumor effect through induction of cell apoptosis.
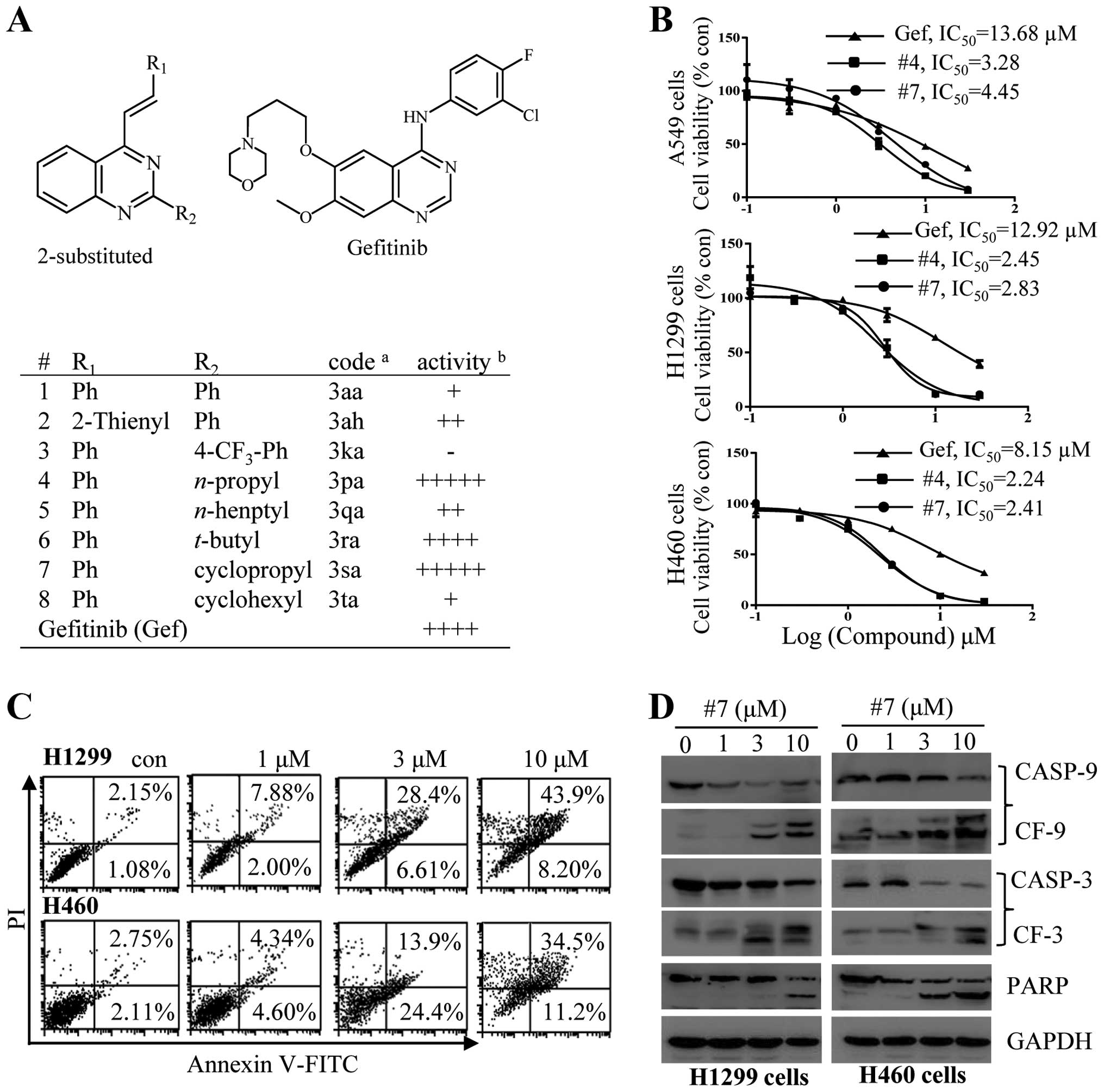 | Figure 12-Substituted quinazolines induce
apoptotic cell death. (A) The chemical structures of the
2-substituted quinazolines and gefitinib and summary of results
from an initial screening. For the initial screening, A549 lung
carcinoma cells were treated with a single dose of a test compound
at 30 µM, a concentration that showed no cytotoxic effect
against non-cancerous Vero cells. Cell viability was determined
using an MTT assay after 72 h. acode, names used in Zhao
et al (17);
bactivity, inhibition of cell viability: −, no activity;
+, ≤20%; ++, ≤40%; +++, ≤60%; ++++, ≤80%; and +++++, >80%. (B)
Determination of the IC50 values of compound #4 and #7
for A549, H1299 or H460 cells. Gef, gefitinib. (C and D) Compound
#7 promotes cell death through apoptosis. H1299 or H460 cells were
treated with #7 for 24 h at concentrations as indicated. (C) Cell
apoptosis was determined by Annexin V/PI staining. (D) Caspase-3
and -9, and PARP cleavage were detected by immunoblot analysis. |
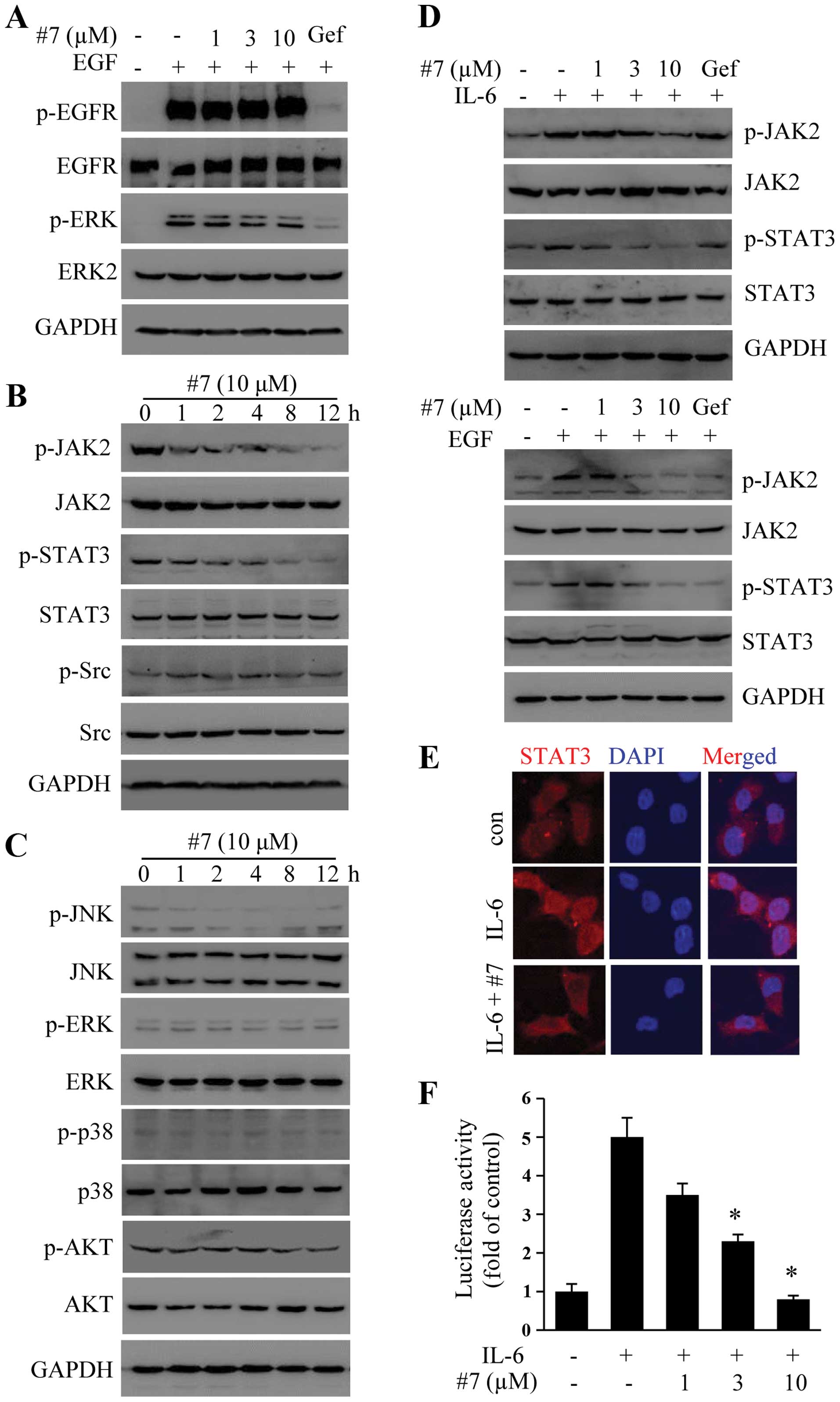
Compound #7 does not inhibit EGF-induced
EGFR phosphorylation
We sought to determine whether the antitumor
activity of #7 was associated with EGFR inhibition. As shown in
Fig. 2A, gefitinib at 10 µM
completely blocked EGF-induced EGFR phosphorylation and downstream
Erk activation. In contrast, pretreatment of H1299 cells with
varying concentrations of #7 showed no effect on EGFR activation
(Fig. 2A) nor ERK, suggesting that
the newly prepared 2-ASQs utilized a distinct mechanism to inhibit
tumor cell growth compared with that of gefitinib.
Compound #7 suppresses JAK2 and STAT3
phosphorylation and downstream signaling
STAT3 overexpression has been linked to
tumorigenesis of NSCLC (22,23).
We then examined whether #7 had the ability to inhibit STAT3
phosphorylation since both H460 and H1299 cells have
hyperphosphorylated STAT3. As shown in Fig. 2B, H1299 has hyperphosphorylated
STAT3 and JAK2. Treatment with 10 µM #7 for varying times
inhibited both JAK2 (Y1007) and STAT3 (Tyr705) phosphorylation. For
comparison, the treatment did not significantly affect Src (Y416)
phosphorylation. In addition, #7 showed no effect on Akt, p38, JNK
or ERK phosphorylation (Fig.
2C).
We also sought to determine whether #7 had an
inhibitory effect on IL-6 and EGF-induced STAT3 phosphorylation. As
shown in Fig. 2D, treatment with #7
blocked both IL-6 and EGF-induced STAT3 and JAK2 phosphorylation.
The effect of #7 on STAT3 activation was confirmed by staining for
IL-6-induced STAT3 nuclear translocation. As shown in Fig. 2E, treatment of H1299 cells with #7
effectively blocked STAT3 nuclear translocation (Fig. 2E). In addition, we also demonstrated
that #7 blocked STAT3-mediated transcription activity as was
determined using a reporter gene assay (Fig. 2F). These results together
demonstrated that #7 is a specific inhibitor of the STAT3
pathway.
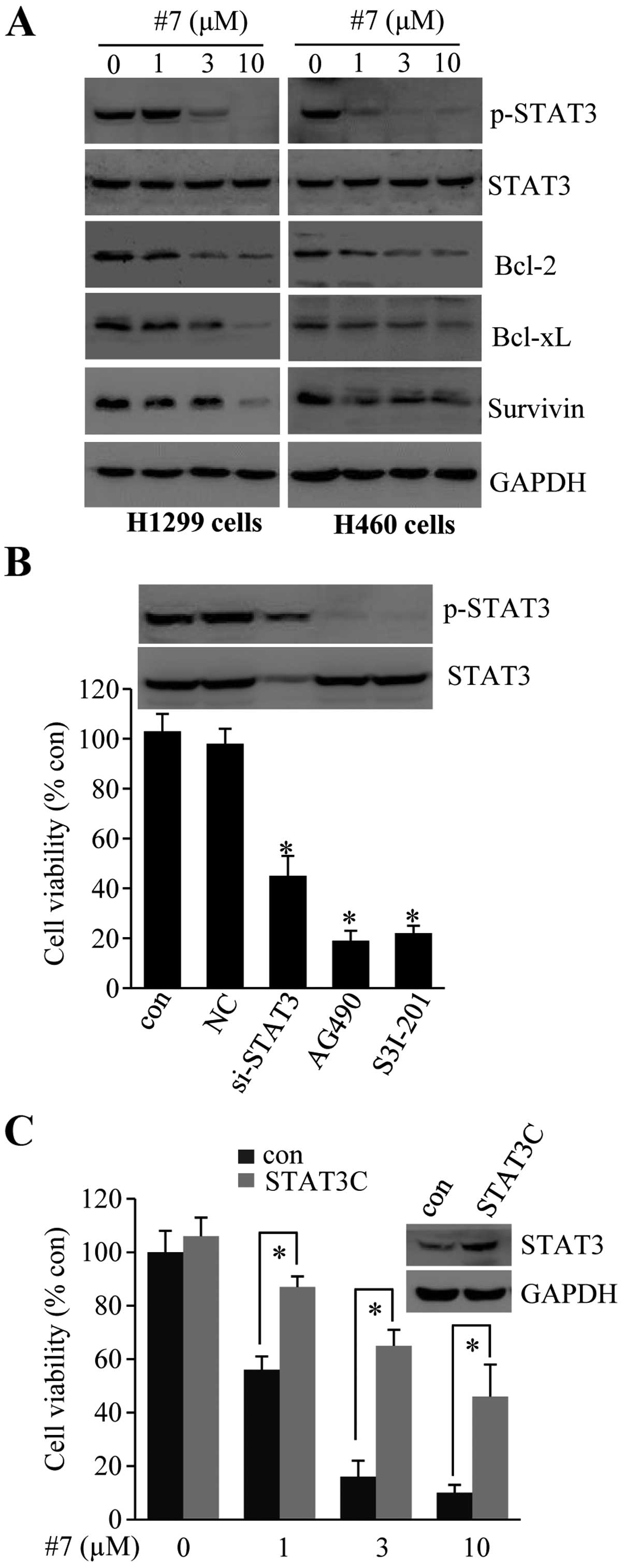
Compound #7 promotes apoptosis by
inhibition of STAT3 signaling
We determined whether compound #7-induced apoptosis
was dependent on STAT3 inhibition. First, we examined the
expression of anti-apoptosis protein expression in #7-treated
cells. As shown in Fig. 3A,
treatment with #7 resulted in reduced STAT3 phosphorylation in both
the H1299 and H460 cells. The treatment also suppressed Bcl-2,
Bcl-xL and survivin expression in these cells, indicating that #7
promoted tumor cell apoptosis through inhibition of STAT3
signaling.
We then performed two separate experiments to
substantiate the above conclusion. First, H1299 cells were
transfected with an siRNA to knockdown STAT3 expression or were
treated with specific inhibitors to block STAT3 phosphorylation. As
shown in Fig. 3B, knockdown of
STAT3 expression or inhibition of STAT3 phosphorylation resulted in
significant reduction in cell viability. Conversely, we found that
ectopic expression of a constitutively active mutant of STAT3
(STAT3C) was able to reverse the effect of #7 on cell viability
(Fig. 3C).
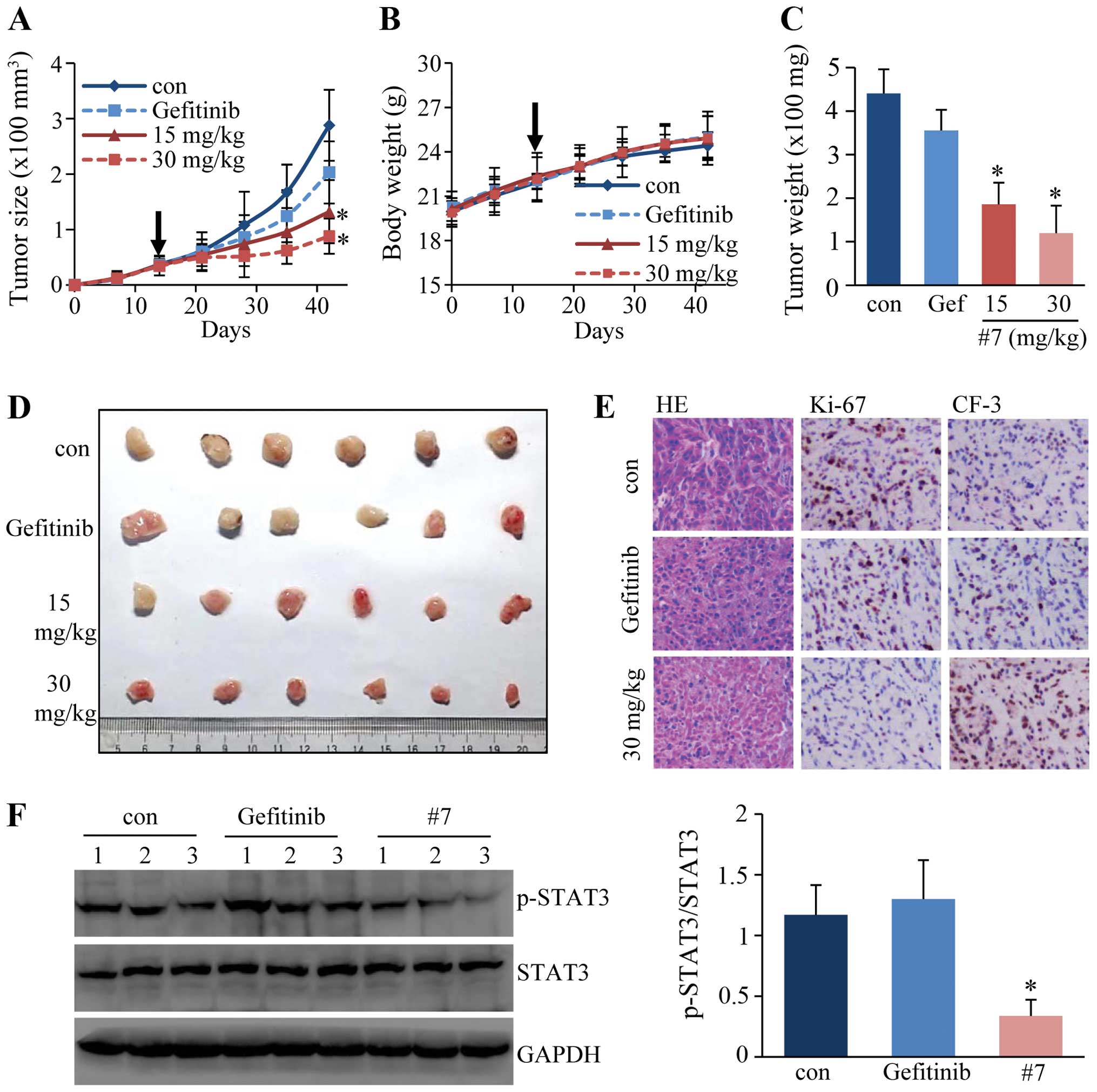
In vivo antitumor activity study in a
xenograft model
The antitumor activity of #7 was then evaluated in
nude mice bearing xenografts. We chose to use the A549 xenograft
since it is a well established animal for antitumor studies. BALB/c
athymic nude mice bearing A549 tumors were treated with #7 at 15
and 30 mg/kg, with gefitinib at 30 mg/kg, or with PBS by
intragastric gavage. After treatment for 4 weeks, the mice were
sacrificed and the size of the tumors was recorded. As shown in
Fig. 4D, tumors in mice treated
with #7 at 15 and 30 mg/kg were significantly smaller than tumors
in the control group. The dynamic change in tumor size also
demonstrated that #7 markedly attenuated A549 tumor growth in nude
mice without significantly affecting the body weight of the animals
(Fig. 4A-C). Examination after
H&E staining revealed that the tumor tissues from the 30 mg/kg
group were filled with fibrous tissues accompanied by reduced
numbers of cancer cells (Fig. 4E).
The level of Ki-67, a marker of cell proliferation detected by
immunohistochemical staining, was decreased in the treated groups
compared to that in the untreated controls. In contrast, the level
of cleaved caspase-3 was increased in the #7-treated tumors,
indicating that #7 promoted tumor cell apoptosis (Fig. 4E). We also examined the STAT3
phosphorylation status in the tumor tissues by western blot
analysis. Consistent with the results from the cell culture
studies, treatment with #7 also resulted in decreased
phosphorylation of STAT3 in these samples (Fig. 4F).
Taken together, these results demonstrated that
2-ASQ derivatives exert antitumor effects through STAT3
inhibition.
Discussion
Quinazoline derivatives have demonstrated distinct
therapeutic activities including anticancer, anti-inflammatory,
antibacterial, antiviral and antiobesity effects (21). We previously prepared a broad range
of quinazoline derivatives and characterized compounds #4 and #7 as
having potent antitumor activity through inhibition of JAK2/STAT3
activation. Several small organic molecules including ursolic acid
and curcumin have been identified as inhibitors of STAT3 pathway
signaling (24,25). Most of the JAK1/2 inhibitors that
are approved for clinical use or testing are heterocyclic compounds
including those containing a pyrimidine or pyrrolopyrimidine ring
(26,27). It is known that quinazolines block
EGFR activity by blocking ATP binding to the kinase domain.
Although we do not know the targets of 2-ASQ action, our data and
data published by other groups strongly suggest that the JAK/STAT3
pathway is a potential target of their action. In a recent docking
study, Yang et al (28)
predicted that quinazoline derivatives also could interact with the
active site of JAK2 and selectively block JAK2 activity. In
addition, 2-guanidino-substituted quinazolines have recently been
identified as new inhibitors of the STAT3 pathway (29). Further studies to optimize the
structures of the lead compounds and identify their molecular
targets are critical considering the importance of the role of the
JAK/STAT3 pathway in tumor development and antitumor resistance to
TKIs.
Nearly all patients develop resistance to TKIs and
relapse for a variety of reasons (30,31).
Given the fact that ~50% of tumors possess activated STAT3 and only
16% contain activating EGFR mutations that have increased
sensitivity to EGFR TKIs (10), it
is possible that further study of 2-ASQs may lead to potent
chemotherapeutics targeting STAT3 activation. We prepared 2-ASQs
with relatively high yields under mild conditions. 2-ASQ
derivatives with further modifications can also be readily prepared
with methods developed by us. STAT3 signaling is frequently
activated, often constitutively, in a variety of human
malignancies, including cancers of the head and neck, colorectum,
cervix, breast and esophagus. STAT3 regulates cancer progression
including cell proliferation, apoptosis resistance and
angiogenesis. As JAK/STAT signaling controls a variety of cellular
processes and affects the microenvironment of tumor growth and
antitumor immunity, therapeutics targeting the JAK/STAT3 pathway
may have a wide range of systemic ramifications for tumor treatment
and inflammatory diseases.
Acknowledgments
The authors would like to thank Dr Xuancheng Guo for
proofreading the manuscript. The present study was supported
financially by grants from NSFC of China (grant nos. 81121062 and
81371772). H.S.Q. was a recipient of a Predoctoral Fellowship from
Jiangsu Innovation Programs for Graduate Students.
References
|
1
|
Herbst RS, Fukuoka M and Baselga J:
Gefitinib - a novel targeted approach to treating cancer. Nat Rev
Cancer. 4:956–965. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wheeler DL, Dunn EF and Harari PM:
Understanding resistance to EGFR inhibitors - impact on future
treatment strategies. Nat Rev Clin Oncol. 7:493–507. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yun CH, Boggon TJ, Li Y, Woo MS, Greulich
H, Meyerson M and Eck MJ: Structures of lung cancer-derived EGFR
mutants and inhibitor complexes: Mechanism of activation and
insights into differential inhibitor sensitivity. Cancer Cell.
11:217–227. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Agus DB, Akita RW, Fox WD, Lewis GD,
Higgins B, Pisacane PI, Lofgren JA, Tindell C, Evans DP, Maiese K,
et al: Targeting ligand-activated ErbB2 signaling inhibits breast
and prostate tumor growth. Cancer Cell. 2:127–137. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Baselga J, Rischin D, Ranson M, Calvert H,
Raymond E, Kieback DG, Kaye SB, Gianni L, Harris A, Bjork T, et al:
Phase I safety, pharmacokinetic, and pharmacodynamic trial of
ZD1839, a selective oral epidermal growth factor receptor tyrosine
kinase inhibitor, in patients with five selected solid tumor types.
J Clin Oncol. 20:4292–4302. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Scagliotti GV, Selvaggi G, Novello S and
Hirsch FR: The biology of epidermal growth factor receptor in lung
cancer. Clin Cancer Res. 10:4227s–4232s. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Maurizi M, Almadori G, Ferrandina G,
Distefano M, Romanini ME, Cadoni G, Benedetti-Panici P, Paludetti
G, Scambia G and Mancuso S: Prognostic significance of epidermal
growth factor receptor in laryngeal squamous cell carcinoma. Br J
Cancer. 74:1253–1257. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tateishi M, Ishida T, Kohdono S, Hamatake
M, Fukuyama Y and Sugimachi K: Prognostic influence of the
co-expression of epidermal growth factor receptor and c-erbB-2
protein in human lung adenocarcinoma. Surg Oncol. 3:109–113. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Robertson SC, Tynan J and Donoghue DJ: RTK
mutations and human syndromes: When good receptors turn bad. Trends
Genet. 16:3682000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rosell R, Moran T, Queralt C, Porta R,
Cardenal F, Camps C, Majem M, Lopez-Vivanco G, Isla D, Provencio M,
et al Spanish Lung Cancer Group: Screening for epidermal growth
factor receptor mutations in lung cancer. N Engl J Med.
361:958–967. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Robinson KW and Sandler AB: EGFR tyrosine
kinase inhibitors: Difference in efficacy and resistance. Curr
Oncol Rep. 15:396–404. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhong D, Ru Y, Wang Q, Zhang J, Zhang J,
Wei J, Wu J, Yao L and Li X and Li X: Chimeric ubiquitin ligases
inhibit non-small cell lung cancer via negative modulation of EGFR
signaling. Cancer Lett. 359:57–64. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang L, Pan J, Dong Y, Tweardy DJ, Dong
Y, Garibotto G and Mitch WE: Stat3 activation links a C/EBPδ to
myostatin pathway to stimulate loss of muscle mass. Cell Metab.
18:368–379. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tierney MT, Aydogdu T, Sala D, Malecova B,
Gatto S, Puri PL, Latella L and Sacco A: STAT3 signaling controls
satellite cell expansion and skeletal muscle repair. Nat Med.
20:1182–1186. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gao SP, Mark KG, Leslie K, Pao W, Motoi N,
Gerald WL, Travis WD, Bornmann W, Veach D, Clarkson B, et al:
Mutations in the EGFR kinase domain mediate STAT3 activation via
IL-6 production in human lung adenocarcinomas. J Clin Invest.
117:3846–3856. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wu K, Chang Q, Lu Y, Qiu P, Chen B, Thakur
C, Sun J, Li L, Kowluru A and Chen F: Gefitinib resistance resulted
from STAT3-mediated Akt activation in lung cancer cells.
Oncotarget. 4:2430–2438. 2013.PubMed/NCBI
|
|
17
|
Zhao D, Zhu MX, Wang Y, Shen Q and Li JX:
Pd(0)-catalyzed benzylic arylation-oxidation of
4-methylquinazolines via sp3 C-H activation under air
conditions. Org Biomol Chem. 11:6246–6249. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhao D, Wang T, Shen Q and Li JX:
n-Bu4NI-catalyzed selective dual amination of
sp3 C-H bonds: Oxidative domino synthesis of
imidazo[1,5-c]quinazolines on a gram-scale. Chem Commun.
50:4302–4304. 2014. View Article : Google Scholar
|
|
19
|
Zhang L, Gao Z, Peng C, Bin ZY, Zhao D, Wu
J, Xu Q and Li JX: Ultrasound-promoted synthesis and
immunosuppressive activity of novel quinazoline derivatives. Mol
Divers. 16:579–590. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhao D, Shen Q, Zhou YR and Li JX:
KOtBu-mediated stereose-lective addition of quinazolines to alkynes
under mild conditions. Org Biomol Chem. 11:5908–5912. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang D and Gao F: Quinazoline derivatives:
Synthesis and bioactivities. Chem Cent J. 7:952013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yu H and Jove R: The STATs of cancer - new
molecular targets come of age. Nat Rev Cancer. 4:97–105. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Harada D, Takigawa N and Kiura K: The role
of STAT3 in non-small cell lung cancer. Cancers. 6:708–722. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pathak AK, Bhutani M, Nair AS, Ahn KS,
Chakraborty A, Kadara H, Guha S, Sethi G and Aggarwal BB: Ursolic
acid inhibits STAT3 activation pathway leading to suppression of
proliferation and chemosensitization of human multiple myeloma
cells. Mol Cancer Res. 5:943–955. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bharti AC, Donato N and Aggarwal BB:
Curcumin (diferuloylmethane) inhibits constitutive and
IL-6-inducible STAT3 phosphorylation in human multiple myeloma
cells. J Immunol. 171:3863–3871. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Baskin R, Majumder A and Sayeski PP: The
recent medicinal chemistry development of Jak2 tyrosine kinase
small molecule inhibitors. Curr Med Chem. 17:4551–4558. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Siveen KS, Sikka S, Surana R, Dai X, Zhang
J, Kumar AP, Tan BK, Sethi G and Bishayee A: Targeting the STAT3
signaling pathway in cancer: Role of synthetic and natural
inhibitors. Biochim Biophys Acta. 1845:136–154. 2014.PubMed/NCBI
|
|
28
|
Yang SH, Khadka DB, Cho SH, Ju HK, Lee KY,
Han HJ, Lee KT and Cho WJ: Virtual screening and synthesis of
quinazolines as novel JAK2 inhibitors. Bioorg Med Chem. 19:968–977.
2011. View Article : Google Scholar
|
|
29
|
LaPorte MG, da Paz Lima DJ, Zhang F, Sen
M, Grandis JR, Camarco D, Hua Y, Johnston PA, Lazo JS, Resnick LO,
et al: 2-Guanidinoquinazolines as new inhibitors of the STAT3
pathway. Bioorg Med Chem Lett. 24:5081–5085. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pao W, Miller VA, Politi KA, Riely GJ,
Somwar R, Zakowski MF, Kris MG and Varmus H: Acquired resistance of
lung adenocarcinomas to gefitinib or erlotinib is associated with a
second mutation in the EGFR kinase domain. PLoS Med. 2:e732005.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Engelman JA and Jänne PA: Mechanisms of
acquired resistance to epidermal growth factor receptor tyrosine
kinase inhibitors in non-small cell lung cancer. Clin Cancer Res.
14:2895–2899. 2008. View Article : Google Scholar : PubMed/NCBI
|