Introduction
Head and neck squamous cell carcinoma (HNSCC) is the
sixth most common cancer worldwide, with an annual incidence of
more than 500,000 cases (1).
Despite recent advances in treatment regimens including surgery,
radiotherapy, chemotherapy and introduction of novel therapeutic
agents, the 5-year survival rate of these patients has been
virtually unchanged in the past three decades, remaining at only
50% (2). Further improvements in
curative rates in HNSCC will require significant advances in the
development of new drugs and treatment strategies. A deeper
understanding of the precise molecular mechanisms responsible for
HNSCC tumorigenesis and progression will contribute significantly
toward the development of new and improved therapeutic agents.
It has been reported that many solid human cancers,
including HNSCC, are maintained by a small subpopulation of cells
called cancer stem cells (CSCs) (3–6). These
CSCs have the unique features of self-renewal, asymmetrical
differentiation into multiple lineages and enhanced
tumor-initiating capacity in xenograft models (7–9).
Accumulating evidence suggests that CSCs are responsible for tumor
metastasis and the acquisition of resistance to treatment with
conventional chemotherapeutic agents, which leads to tumor relapse
(10,11). Therefore, targeting CSCs with
conventional or other targeted therapies may be required to
effectively treat cancers (12–15).
Epigenetic regulations are required for normal
development and gene expression. Disruption of epigenetic
regulations often leads to aberrant gene expression and malignant
cellular transformation (16).
Epigenetic alterations commonly observed in malignant cells include
DNA methylation and changes in histone modification patterns as
well as expression profiles of chromatin-modifying enzymes
(16,17). Histone modification patterns are
dynamically regulated by histone acetyltransferases (HATs) and
histone deacetylases (HDACs), which add and remove specific
covalent modifications, respectively (18). An imbalance between HATs and HDACs
leads to aberrant gene expression and tumorigenesis (19).
Several epigenetic drugs that effectively reverse
aberrant DNA methylations and histone modifications have been
discovered. Valproic acid (VPA), a branched short-chain fatty acid,
inhibits HDACs causing increased chromatin acetylation (20). HDACs have been found to be
overexpressed or mutated in many types of human tumors (21,22). A
recent study demonstrated that HNSCCs are primarily hypoacetylated
which may account for the accumulation and maintenance of CSCs
(23). They also showed that
inhibition of HDACs using Trichostatin A reduced the number of
HNSCC CSCs and inhibited clonogenic sphere formation (23). In contrast, a separate report showed
that VPA promoted the expansion of breast CSCs through
reprogramming of differentiated cancer cells into stem-like cells.
The causes of this discrepancy might lie in the different cancer
cell systems as well as the different HDAC inhibitors used.
Unfortunately, previous reports regarding the effect
of VPA on HNSCC CSCs have been extremely scarce. The objectives of
the present study were to evaluate the effect of VPA on HNSCC CSCs
and to delineate the mechanisms by which VPA inhibits the
characteristics of CSCs derived from human primary HNSCC. Here, we
present evidence for the therapeutic value of VPA in combination
with conventional cisplatin, which suppressed the self-renewal and
proliferation and induced apoptosis of CSCs in HNSCC.
Materials and methods
Isolation and culture of HNSCC stem-like
cells
HNSCC stem-like cells (K3 and K5) were isolated and
characterized from the primary surgical specimens of HNSCC
patients, as previously described (8). The CSC phenotype of HNSCC CSCs was
confirmed by functional assays of sphere formation,
stemness-associated gene expression and xenograft tumor formation.
The cells were grown in serum-free media composed of DMEM
supplemented with B27 (Invitrogen), N2 supplement (Invitrogen),
basic fibroblast growth factor (bFGF; 20 ng/ml; R&D Systems,
Minneapolis, MN, USA) and epidermal growth factor (EGF; 20 ng/ml;
R&D Systems).
Sphere formation assays
To assess the self-renewal capacity of HNSCC CSCs
in vitro, the cells were dissociated into single cells,
seeded in a 24-well plate at a density of 200 cells/well, and
cultured in serum-free medium, with EGF and bFGF supplementation
every other day. Spheres with a diameter exceeding 10 µm
were counted after 14 days.
Western blot analysis
Western blot analysis of electrophoretically
separated proteins from cells was performed as previously described
(8). Specific antibodies against
Oct4, Sox2, Bcl-2, Bax and caspase 3 were purchased from Santa Cruz
Biotechnology (Santa Cruz, CA, USA). Secondary antibodies,
anti-rabbit IgG and anti-mouse IgG were purchased from Jackson
ImmunoResearch Laboratories (West Grove, PA, USA).
Detection of CD44 expression by flow
cytometry
HNSCC CSCs were dissociated into single cells and
washed twice in cold phosphate-buffered saline (PBS). Cells were
labeled with anti-CD44 and fluorescein isothiocyanate
(FITC)-labeled secondary antibodies, then subjected to flow
cytometry using a FACSCalibur machine (BD Biosciences). The
percentages of CD44+ cells in cultures were
determined.
Chemosensitivity assay
HNSCC CSCs were dissociated into single cells and
then plated in a 96-well plate at a density of 7×03
cells/well under serum-free culture conditions. Cells were treated
with cisplatin at the indicated concentrations and then cultured at
37°C under a humidified 5% CO2 atmosphere. Twenty-four
hours later, 20 µl of
3-(4,4-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
solution (5 mg/ml in PBS) was added to each well, and the plate was
placed at room temperature for 3 h. The absorbance at 570 nm was
measured using a SpectraMax 190 (Molecular Devices) instrument.
Quantitative reverse
transcription-polymerase chain reaction (qRT-PCR)
Total cellular RNA was reverse-transcribed using a
reverse transcriptase (RT) kit (Fermentas, Glen Burnie, MD, USA)
according to the manufacturer's instructions. For semi-quantitative
PCR, cDNA was added to a mixture of specific primers and 1 U of
Taq DNA polymerase (Roche Diagnostics, Indianapolis, IN,
USA), and amplified using an MJ Research MiniCycler (Bio-Rad
Laboratories, Waltham, MA, USA). PCR products were separated by
agarose gel electrophoresis (1.5% agarose gels) and detected under
ultraviolet light (Bio-Rad Laboratories, Hercules, CA, USA).
Real-time PCR was performed on an iCycler IQ real-time detection
system (Bio-Rad Laboratories), using IQ Supermix with SYBR-Green
(Bio-Rad Laboratories). The human sequence-specific primers used
are listed in Table I.
 | Table IPCR primer sequences (5′–3′). |
Table I
PCR primer sequences (5′–3′).
| Primer | Forward | Reverse |
|---|
| ABCB1 |
AGAGGGAGGACAAAGTCCCT |
CACTTCCTCAGACTGCTCCA |
| ABCC1 |
AGAGGGAGGACAAAGTCCCT |
CACTTCCTCAGACTGCTCCA |
| ABCC2 |
AGAGGGAGGACAAAGTCCCT |
CACTTCCTCAGACTGCTCCA |
| ABCC3 |
AGAGGGAGGACAAAGTCCCT |
CACTTCCTCAGACTGCTCCA |
| ABCC4 |
AGAGGGAGGACAAAGTCCCT |
CACTTCCTCAGACTGCTCCA |
| ABCC5 |
AGAGGGAGGACAAAGTCCCT |
CACTTCCTCAGACTGCTCCA |
| ABCC6 |
AGAGGGAGGACAAAGTCCCT |
AGAGGGAGGACAAAGTCCCT |
| ABCG2 |
AGAGGGAGGACAAAGTCCCT |
CACTTCCTCAGACTGCTCCA |
Xenograft tumor formation assay
All animal studies were approved by the
Institutional Animal Care and Use Committee of Konkuk University.
HNSCC CSCs were treated for 48 h with cisplatin (5 µM)
alone, cisplatin (5 µM) plus VPA (400 µM) or dimethyl
sulfoxide (DMSO; vehicle) in vitro. Following this,
105 or 106 cells were subcutaneously injected
into the flank of 8-week-old female BALB/c nude mice using a
22-gauge needle. Mice were visually inspected and palpated weekly
to monitor tumor formation. The mice were sacrificed 8 weeks after
transplantation, and subcutaneous tumor tissues were harvested for
estimating tumor size, weight and apoptosis.
Terminal deoxynucleotidyl transferase
dUTP nick-end labeling (TUNEL) assay
Tumor tissues collected from mice injected with
HNSCC CSCs treated with DMSO, cisplatin (5 µM), or cisplatin
(5 µM) and VPA (400 µM) were used for TUNEL assays.
Sections (4-µm) from formalin-fixed, paraffin-embedded
tumors were deparaffinized and rehydrated using xylene and ethanol,
respectively. The slides were rinsed twice with PBS and treated for
15 min at 37°C with proteinase K (15 µg/ml in 10 mM
Tris-HCl, pH 7.4–8.0). Endogenous peroxidases were blocked using 3%
hydrogen peroxide in methanol at room temperature for 10 min. The
tissue sections were then analyzed with an In Situ Cell Death
Detection kit-POD (Roche) following the manufacturer's
instructions.
Statistical analysis
The results are presented as mean ± SD. Statistical
analyses were performed with the SPSS 10.0 statistical software
(SPSS, Inc., Chicago, IL, USA). Statistical significance was
determined by one-way ANOVA followed by post hoc tests for multiple
comparisons. P<0.05 was considered to indicate a statistically
significant result.
Results
VPA suppresses the CSC properties in
HNSCC
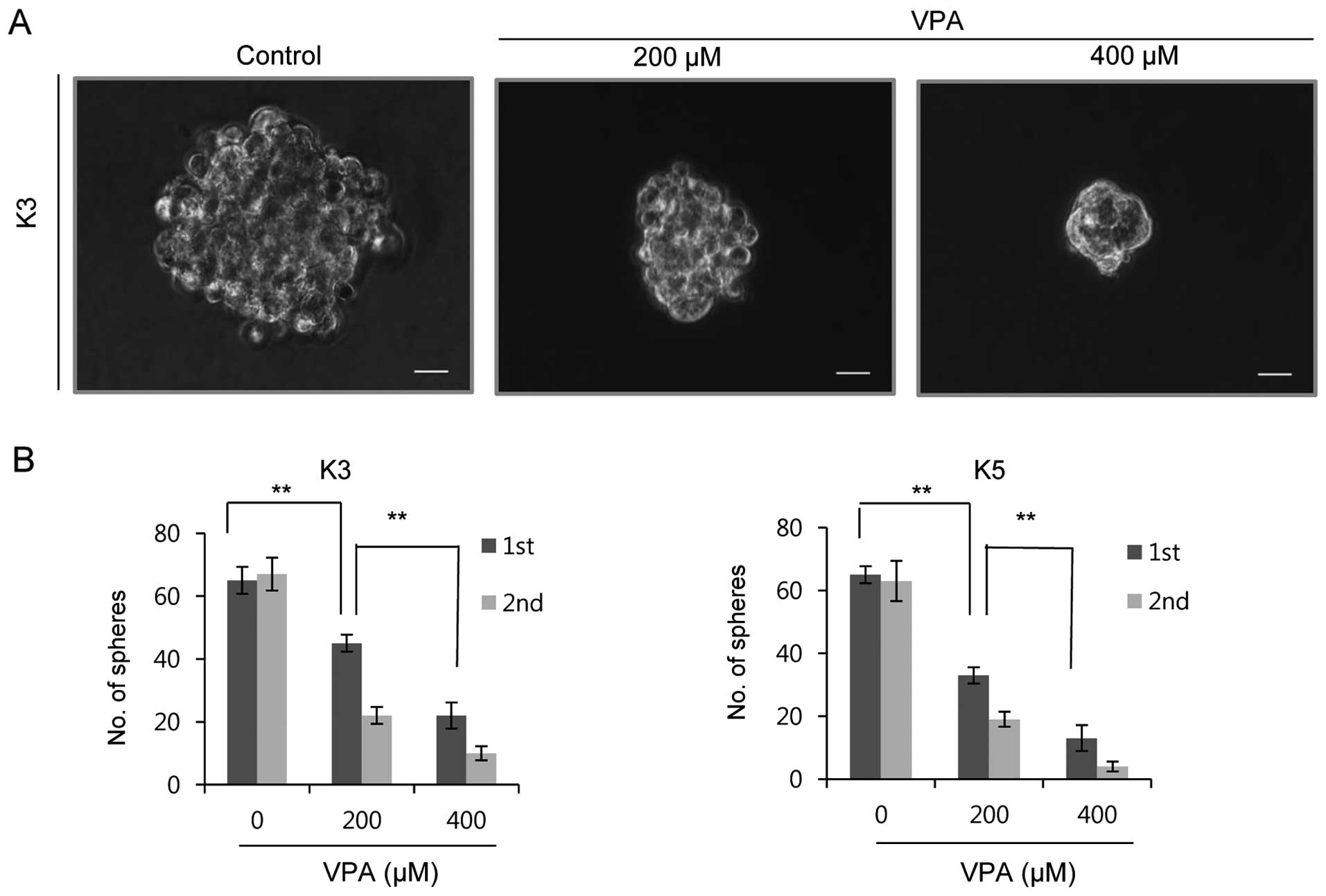
The sphere formation assay can be effectively used
to assess the self-renewal capacity of HNSCC CSCs, and has been
reported to correlate closely with tumorigenicity (8). We examined whether VPA had the ability
to inhibit the self-renewal capacity of HNSCC CSCs. VPA
significantly suppressed sphere formation in two HNSCC CSC cultures
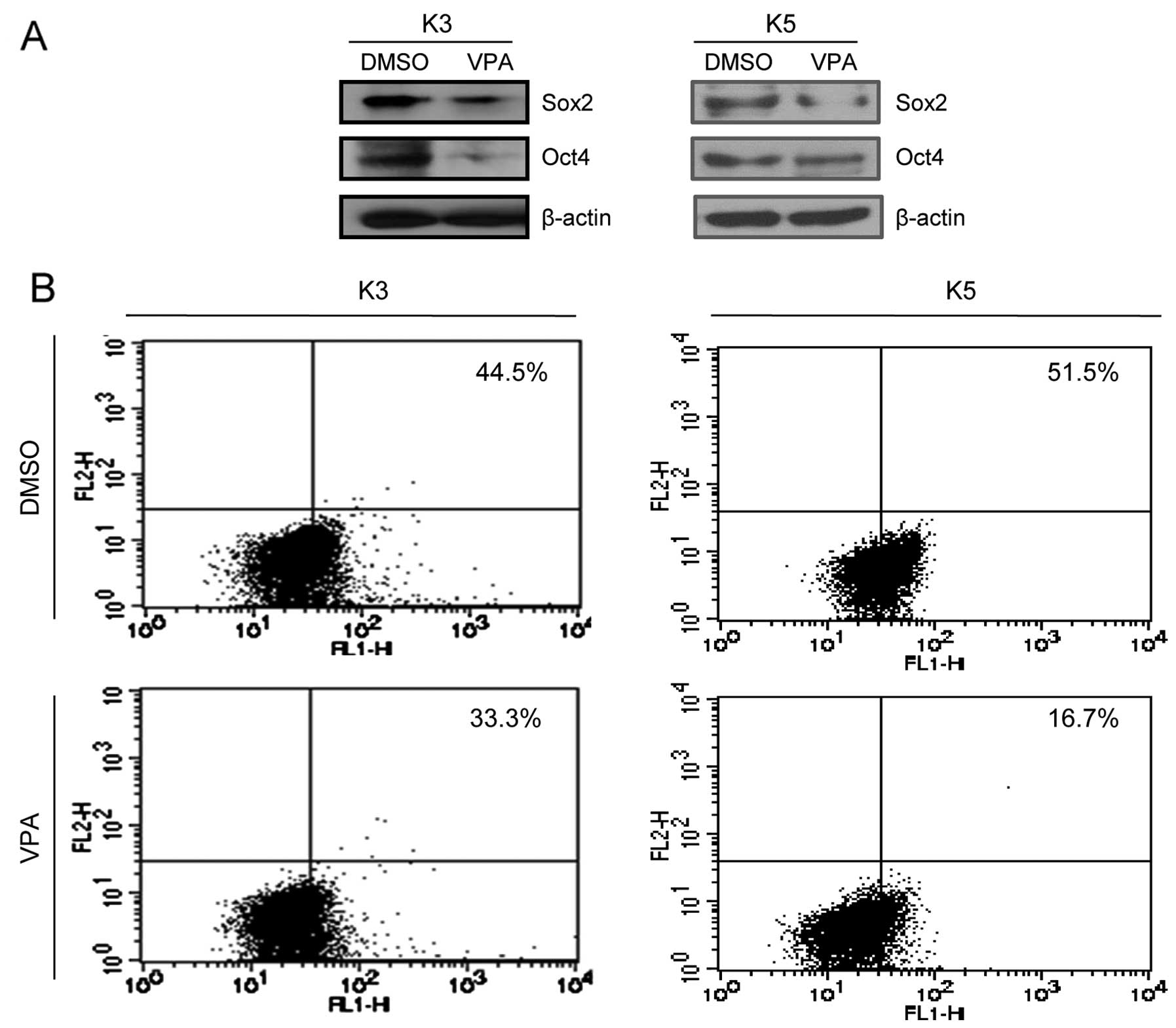
during two serial passages in a dose-dependent manner (Fig. 1). CSCs found in HNSCC express
typical stem cell markers, including Sox2, Oct4 and CD44.
Therefore, we examined whether VPA treatment interfered with the
expression of these markers. As evident from the results of the
western blot analysis, treatment of HNSCC CSCs with VPA
significantly suppressed Sox2 and Oct4 expression (Fig. 2A). Furthermore, VPA treatment
significantly reduced the number of CD44+ cells in the
HNSCC CSCs (Fig. 2B).
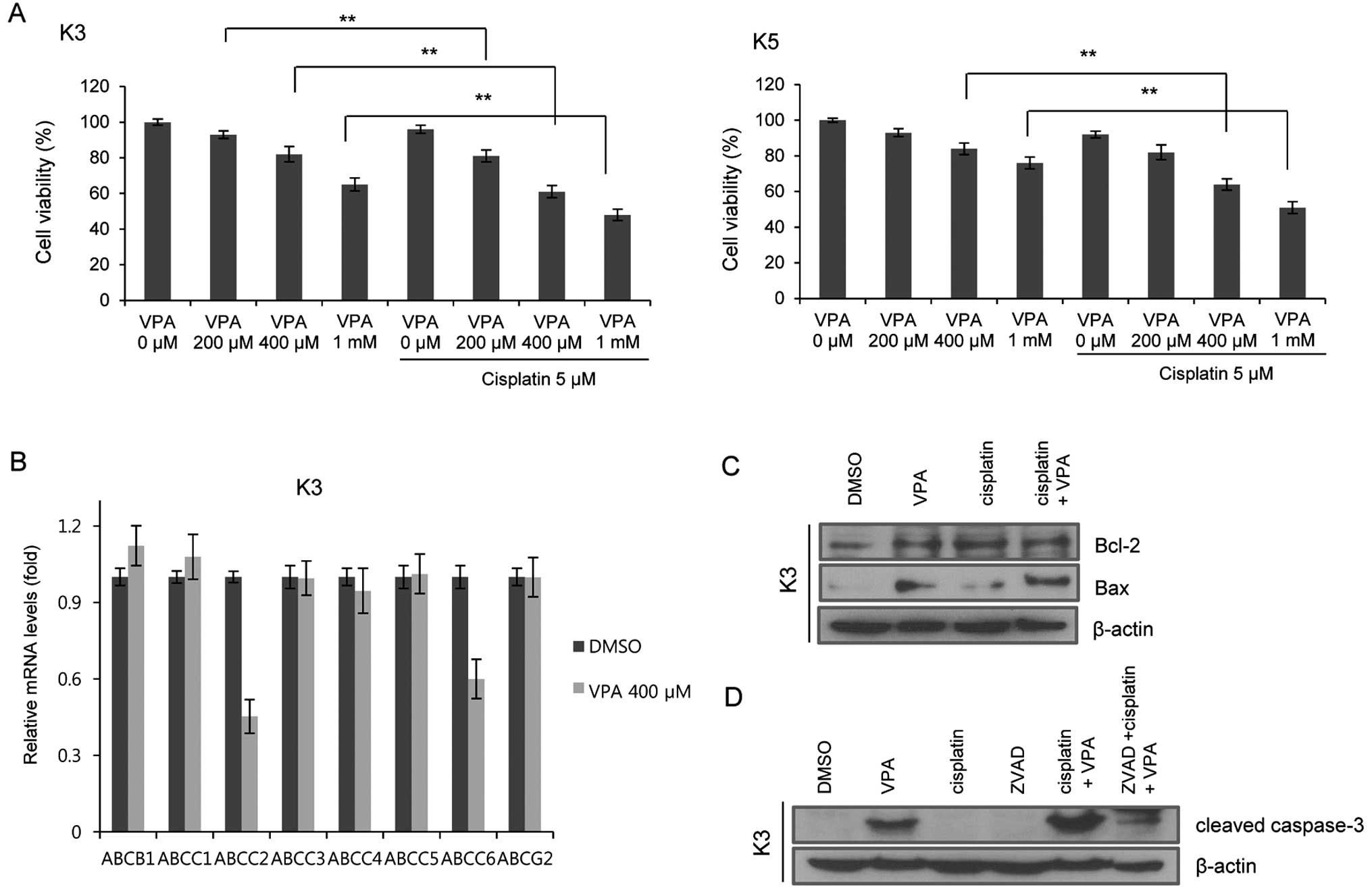
VPA enhances the chemosensitization of
cisplatin and apoptosis in HNSCC CSCs
Chemoresistance is one of the most important
characteristics of CSCs. We examined whether VPA increases the
susceptibility of HNSCC CSCs to cisplatin. A combination of VPA and
cisplatin increased the susceptibility of HNSCC CSCs to cisplatin
in a dose-dependent manner (Fig.
3A). In order to understand the mechanisms responsible for the
increased susceptibility of VPA-treated HNSCC CSCs to cisplatin, we
analyzed the changes in expression of ATP-binding cassette (ABC)
transporters following VPA treatment. Real-time qPCR analysis
revealed that VPA treatment (400 µM) reduced the transcript
levels of the ABCC2 and ABCC6 genes in the HNSCC CSCs (Fig. 3B).
To further determine whether the VPA-induced
suppression of CSC properties is due to increased apoptosis, we
investigated the expression of the apoptosis-related proteins Bax
and Bcl2. When compared to the cells treated with VPA (400
µM) or cisplatin (5 µM) alone, the expression of Bax
in the CSCs was increased following treatment with cisplatin (5
µM) and VPA (400 µM) (Fig. 3C). To test whether caspase activity
was required for VPA-mediated apoptosis, we examined the expression
of cleaved caspase-3 in the CSCs before and after VPA treatment.
Cleaved caspase-3 protein levels were significantly increased in
the cells treated with a combination of cisplatin and VPA. A known
caspase inhibitor, zVAD, abrogated the VPA-induced changes in
cleaved caspase-3 expression (Fig.
3D). These results suggested that VPA is a potent inducer of
apoptosis in HNSCC CSCs.
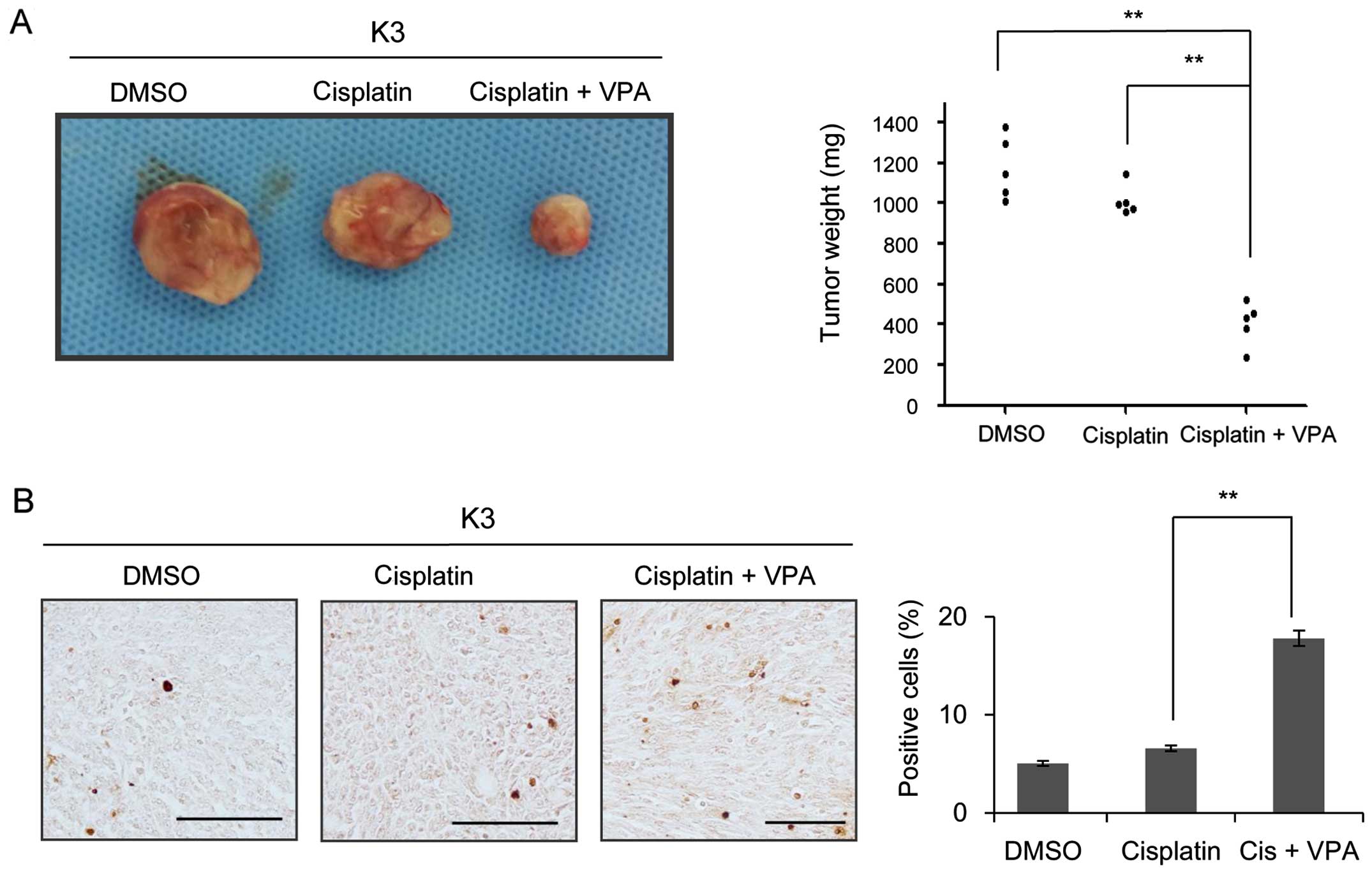
VPA inhibits the growth of HNSCC CSCs in
a xenograft model
To confirm the combined effect of VPA and cisplatin
on HNSCC CSCs, the inhibitory effect of VPA on the capacity of
HNSCC CSCs to initiate tumor formation in nude mice was examined.
Palpable tumor masses developed in 100% (6 of 6) of mice injected
with 105 DMSO-treated cells and 83.3% (5 of 6) of mice
injected with 105 cisplatin-treated cells; in contrast,
only 16.6% (1 of 6) cells with administration of VPA plus cisplatin
formed tumors when 105 cells were injected (Table II). In contrast to the large tumors
generated by HNSCC CSCs treated with cisplatin or DMSO alone, cells
treated with 400 µM VPA and 5 µM cisplatin generated
tumors that were small, and the average tumor weight in nude mice
(N=5) was significantly lower following treatment with cisplatin
and VPA (Fig. 4A). TUNEL assay
revealed a significant increase in the number of apoptotic cells in
tumors generated from CSCs treated with VPA and cisplatin compared
to those generated from cells treated with cisplatin or DMSO alone
(Fig. 4B). Taken together, these
data suggest that combination treatment of ciplatin and VPA induced
apoptosis and reduced the growth rate of CSCs in vivo.
| Table IITumor formation ability of injected
HNSCC CSCs in xenograft model. |
Table II
Tumor formation ability of injected
HNSCC CSCs in xenograft model.
| Injected cells | Control | Cisplatin | VPA +
cisplatin |
|---|
| 105 | 6/6 | 5/6 | 1/6 |
| 106 | 6/6 | 5/6 | 3/6 |
Discussion
Recent advances in the research of epigenetics have
shown that human cancer harbors global epigenetic alterations, in
addition to numerous genetic alterations (24). These genetic and epigenetic
alterations may have critical effects on all stages of cancer
development and progression. Unlike genetic alterations, epigenetic
alterations are potentially reversible. This reversibility of
epigenetic alterations has led to the possibility of developing a
new class of therapeutics that restores the normal epigenetic state
in malignant cell populations (16,25).
Thus, many drugs that target specific enzymes involved in the
epigenetic regulation of gene expression have been introduced, and
the utilization of these drugs is emerging as an effective and
valuable approach to combination treatment with conventional
chemotherapy (25). Of these, HDAC
inhibitors that help in restoring normal histone acetylation
patterns have been shown to induce growth arrest, apoptosis, and
differentiation by reactivating silenced tumor-suppressor genes
(26).
Valproic acid (VPA), a well-known anticonvulsive
agent, emerged in 1997 as an antineoplastic agent (27), and has been described to have
antiproliferative effects in a variety of human malignancies
(28,29). VPA modulates the biology of cancer
cells by inducing differentiation, inhibiting proliferation,
increasing apoptosis, and decreasing metastatic and angiogenetic
potential (30,31). A recent report showed that VPA
induced differentiation and apoptosis in ETO-positive leukemic
cells (32), and now this drug has
been tested in differentiation therapy of acute myeloid leukemia
(33). VPA also exerted inhibitory
effects on the migration and invasion of prostate cancer cells
(34). In HNSCC, VPA has been shown
to have acute and chronic growth inhibitory effects (35), and causes a 3- to 7-fold increase in
cisplatin cytotoxicity (28).
The recent interest in CSC research has emerged from
their expected role in the initiation and progression of cancer.
Moreover, CSCs are thought to be responsible for resistance to
current anticancer treatment and early recurrence in many cancers
(36). According to the CSC
hypothesis a different treatment strategy focusing mainly on CSCs
is required. However, only few attempts have been made to target
CSCs epigenetically. VPA, a known HDAC inhibitor, was found to
decrease proliferation potential and multilineage differentiation
capability of human mesenchymal stem cells (37). Therefore, we hypothesized that
inhibition of HDACs by VPA could suppress CSC activity in HNSCC. In
the present study, we demonstrated that VPA interfered with the
self-renewal of HNSCC CSCs. Accordingly, VPA effectively suppressed
the expression of stem cell markers, including Oct4, Sox2 and CD44.
A combination of VPA and cisplatin reduced HNSCC CSC
chemoresistance, likely by suppressing ABCC2 and ABCC6 expression
and increasing caspase-mediated apoptosis. Furthermore, this
combination treatment significantly inhibited the tumor growth and
induced apoptosis in a xenograft model.
Several studies have shown that these antitumor and
tumor cell differentiation effects of VPA are primarily mediated
through inhibition of HDACs (20,38).
Furthermore, inhibition of HDACs appears to interact with various
other signaling pathways through complex molecular mechanisms. HDAC
inhibition is linked to the modulation of
phosphatidylinositol-3-kinase/Akt signaling (39). The Akt signaling pathway has been
proved to interact with WNT/β-catenin signaling (40). HDAC inhibition was also found to be
increased in inhibition-associated phosphorylation of GSK3β
(41). Inactivation of the
E-cadherin gene was also demonstrated to be triggered by DNA
hypermethylation (42).
The present study further elucidated the VPA-induced
antitumor effects in HNSCC CSCs. VPA in combination with cisplatin
may disrupt the population of CSCs in HNSCC and thus be a potential
curative strategy for the management of HNSCC.
Acknowledgments
The present study was supported by the National
Research Foundation of Korea (NRF) grant funded by the Korea
government (MEST) (grant no. 2011-0014237 to S.H.L. and
2012R1A2A2A01046214 to Y.C.L.).
References
|
1
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: Estimates of worldwide burden of cancer in
2008: GLOBOCAN 2008. Int J Cancer. 127:2893–2917. 2010. View Article : Google Scholar
|
|
2
|
Posner MR, Hershock DM, Blajman CR,
Mickiewicz E, Winquist E, Gorbounova V, Tjulandin S, Shin DM,
Cullen K, Ervin TJ, et al TAX 324 Study Group: Cisplatin and
fluorouracil alone or with docetaxel in head and neck cancer. N
Engl J Med. 357:1705–1715. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Reya T, Morrison SJ, Clarke MF and
Weissman IL: Stem cells, cancer, and cancer stem cells. Nature.
414:105–111. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Al-Hajj M, Wicha MS, Benito-Hernandez A,
Morrison SJ and Clarke MF: Prospective identification of
tumorigenic breast cancer cells. Proc Natl Acad Sci USA.
100:3983–3988. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Collins AT, Berry PA, Hyde C, Stower MJ
and Maitland NJ: Prospective identification of tumorigenic prostate
cancer stem cells. Cancer Res. 65:10946–10951. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Prince ME, Sivanandan R, Kaczorowski A,
Wolf GT, Kaplan MJ, Dalerba P, Weissman IL, Clarke MF and Ailles
LE: Identification of a subpopulation of cells with cancer stem
cell properties in head and neck squamous cell carcinoma. Proc Natl
Acad Sci USA. 104:973–978. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jordan CT, Guzman ML and Noble M: Cancer
stem cells. N Engl J Med. 355:1253–1261. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lim YC, Oh SY, Cha YY, Kim SH, Jin X and
Kim H: Cancer stem cell traits in squamospheres derived from
primary head and neck squamous cell carcinomas. Oral Oncol.
47:83–91. 2011. View Article : Google Scholar
|
|
9
|
Oh SY, Kang HJ, Kim YS, Kim H and Lim YC:
CD44-negative cells in head and neck squamous carcinoma also have
stem-cell like traits. Eur J Cancer. 49:272–280. 2013. View Article : Google Scholar
|
|
10
|
Li F, Tiede B, Massagué J and Kang Y:
Beyond tumorigenesis: Cancer stem cells in metastasis. Cell Res.
17:3–14. 2007. View Article : Google Scholar
|
|
11
|
Dean M, Fojo T and Bates S: Tumour stem
cells and drug resistance. Nat Rev Cancer. 5:275–284. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gupta PB, Onder TT, Jiang G, Tao K,
Kuperwasser C, Weinberg RA and Lander ES: Identification of
selective inhibitors of cancer stem cells by high-throughput
screening. Cell. 138:645–659. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lim YC, Kang HJ, Kim YS and Choi EC:
All-trans-retinoic acid inhibits growth of head and neck cancer
stem cells by suppression of Wnt/β-catenin pathway. Eur J Cancer.
48:3310–3318. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lee SH, Nam HJ, Kang HJ, Kwon HW and Lim
YC: Epigallocatechin-3-gallate attenuates head and neck cancer stem
cell traits through suppression of Notch pathway. Eur J Cancer.
49:3210–3218. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang M, Atkinson RL and Rosen JM:
Selective targeting of radiation-resistant tumor-initiating cells.
Proc Natl Acad Sci USA. 107:3522–3527. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sharma S, Kelly TK and Jones PA:
Epigenetics in cancer. Carcinogenesis. 31:27–36. 2010. View Article : Google Scholar :
|
|
17
|
Portela A and Esteller M: Epigenetic
modifications and human disease. Nat Biotechnol. 28:1057–1068.
2010. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Haberland M, Montgomery RL and Olson EN:
The many roles of histone deacetylases in development and
physiology: Implications for disease and therapy. Nat Rev Genet.
10:32–42. 2009. View
Article : Google Scholar
|
|
19
|
Marks P, Rifkind RA, Richon VM, Breslow R,
Miller T and Kelly WK: Histone deacetylases and cancer: Causes and
therapies. Nat Rev Cancer. 1:194–202. 2001. View Article : Google Scholar
|
|
20
|
Göttlicher M, Minucci S, Zhu P, Krämer OH,
Schimpf A, Giavara S, Sleeman JP, Lo Coco F, Nervi C, Pelicci PG,
et al: Valproic acid defines a novel class of HDAC inhibitors
inducing differentiation of transformed cells. EMBO J.
20:6969–6978. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhu P, Martin E, Mengwasser J, Schlag P,
Janssen KP and Göttlicher M: Induction of HDAC2 expression upon
loss of APC in colorectal tumorigenesis. Cancer Cell. 5:455–463.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ropero S, Fraga MF, Ballestar E, Hamelin
R, Yamamoto H, Boix-Chornet M, Caballero R, Alaminos M, Setien F,
Paz MF, et al: A truncating mutation of HDAC2 in human cancers
confers resistance to histone deacetylase inhibition. Nat Genet.
38:566–569. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Giudice FS, Pinto DS Jr, Nör JE, Squarize
CH and Castilho RM: Inhibition of histone deacetylase impacts
cancer stem cells and induces epithelial-mesenchyme transition of
head and neck cancer. PLoS One. 8:e586722013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jones PA and Baylin SB: The epigenomics of
cancer. Cell. 128:683–692. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yoo CB and Jones PA: Epigenetic therapy of
cancer: Past, present and future. Nat Rev Drug Discov. 5:37–50.
2006. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Carew JS, Giles FJ and Nawrocki ST:
Histone deacetylase inhibitors: Mechanisms of cell death and
promise in combination cancer therapy. Cancer Lett. 269:7–17. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bolden JE, Peart MJ and Johnstone RW:
Anticancer activities of histone deacetylase inhibitors. Nat Rev
Drug Discov. 5:769–784. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Erlich RB, Rickwood D, Coman WB, Saunders
NA and Guminski A: Valproic acid as a therapeutic agent for head
and neck squamous cell carcinomas. Cancer Chemother Pharmacol.
63:381–389. 2009. View Article : Google Scholar
|
|
29
|
Starkova J, Madzo J, Cario G, Kalina T,
Ford A, Zaliova M, Hrusak O and Trka J: The identification of
(ETV6)/RUNX1-regulated genes in lymphopoiesis using histone
deacetylase inhibitors in ETV6/RUNX1-positive lymphoid leukemic
cells. Clin Cancer Res. 13:1726–1735. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cinatl J Jr, Cinatl J, Driever PH,
Kotchetkov R, Pouckova P, Kornhuber B and Schwabe D: Sodium
valproate inhibits in vivo growth of human neuroblastoma cells.
Anticancer Drugs. 8:958–963. 1997. View Article : Google Scholar
|
|
31
|
Blaheta RA, Michaelis M, Driever PH and
Cinatl J Jr: Evolving anticancer drug valproic acid: Insights into
the mechanism and clinical studies. Med Res Rev. 25:383–397. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zapotocky M, Mejstrikova E, Smetana K,
Stary J, Trka J and Starkova J: Valproic acid triggers
differentiation and apoptosis in AML1/ETO-positive leukemic cells
specifically. Cancer Lett. 319:144–153. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hrebackova J, Hrabeta J and Eckschlager T:
Valproic acid in the complex therapy of malignant tumors. Curr Drug
Targets. 11:361–379. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Witt D, Burfeind P, von Hardenberg S,
Opitz L, Salinas-Riester G, Bremmer F, Schweyer S, Thelen P, Neesen
J and Kaulfuss S: Valproic acid inhibits the proliferation of
cancer cells by re-expressing cyclin D2. Carcinogenesis.
34:1115–1124. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gan CP, Hamid S, Hor SY, Zain RB, Ismail
SM, Wan Mustafa WM, Teo SH, Saunders N and Cheong SC: Valproic
acid: Growth inhibition of head and neck cancer by induction of
terminal differentiation and senescence. Head Neck. 34:344–353.
2012. View Article : Google Scholar
|
|
36
|
Khalil MA, Hrabeta J, Cipro S, Stiborova
M, Vicha A and Eckschlager T: Neuroblastoma stem cells - mechanisms
of chemoresistance and histone deacetylase inhibitors. Neoplasma.
59:737–746. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lee S, Park JR, Seo MS, Roh KH, Park SB,
Hwang JW, Sun B, Seo K, Lee YS, Kang SK, et al: Histone deacetylase
inhibitors decrease proliferation potential and multilineage
differentiation capability of human mesenchymal stem cells. Cell
Prolif. 42:711–720. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gurvich N, Tsygankova OM, Meinkoth JL and
Klein PS: Histone deacetylase is a target of valproic acid-mediated
cellular differentiation. Cancer Res. 64:1079–1086. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chou CW, Wu MS, Huang WC and Chen CC: HDAC
inhibition decreases the expression of EGFR in colorectal cancer
cells. PLoS One. 6:e180872011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wang XH, Meng XW, Sun X, Liu BR, Han MZ,
Du YJ, Song YY and Xu W: Wnt/β-catenin signaling regulates MAPK and
Akt1 expression and growth of hepatocellular carcinoma cells.
Neoplasma. 58:239–244. 2011. View Article : Google Scholar
|
|
41
|
De Sarno P, Li X and Jope RS: Regulation
of Akt and glycogen synthase kinase-3 beta phosphorylation by
sodium valproate and lithium. Neuropharmacology. 43:1158–1164.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Graff JR, Herman JG, Lapidus RG, Chopra H,
Xu R, Jarrard DF, Isaacs WB, Pitha PM, Davidson NE and Baylin SB:
E-cadherin expression is silenced by DNA hypermethylation in human
breast and prostate carcinomas. Cancer Res. 55:5195–5199.
1995.PubMed/NCBI
|