Introduction
Nasopharyngeal carcinoma (NPC) is a common
epithelial squamous-cell head and neck carcinoma, which originates
from the nasopharyngeal mucosa and is associated with Epstein-Barr
virus (EBV) infection (1). This
type of cancer shows a distinct racial and geographical
distribution and is prevalent among Southern Chinese, as well as
natives in Southeast Asia including Malaysia (2,3). In
the early stages, NPC may be asymptomatic or present with
apparently trivial symptoms and is therefore likely to be ignored
resulting in late diagnosis (4,5).
NPC is usually treated with radiotherapy and/or
chemotherapy (cisplatin and 5-fluorouracil) (6). Concurrent chemo-radiotherapy is the
main modality of treatment for advanced-stage NPC (7). However, advanced-stage NPC is
associated with poor prognosis and therapeutic failure (7). Recurrence, distant metastases,
treatment resistance and adverse effects of treatment remain the
major challenges in the treatment of NPC (8–10).
p53, a tumor suppressor gene, is a transcription
factor which controls genes involved in DNA repair, cell cycle
arrest and apoptosis (11). Μurine
double minute (Mdm2) is an important negative regulator of p53
(12). It is an E3 ubiquitin ligase
which promotes degradation of p53 via the ubiquitin-proteosome
pathway (13,14) and functions in an autoregulatory
feedback loop with p53. Inhibition of the p53-Mdm2 interaction
prevents this degradation, thus resulting in increased p53
levels.
Nutlins are cis-imidazoline analogs (14) which compete with Mdm2 for binding to
p53. Nutlin-3 has been reported to be effective in killing cancer
cells retaining functional wt p53 (15). Nutlin-3 exerts anticancer effects on
acute myeloid (16) and chronic
lymphocytic leukemia (17),
multiple myeloma (18), Kaposi
sarcoma (19), liposarcoma
(20), rhabdomyosarcoma (21), Ewing's sarcoma (22), colon (23) and testicular cancer (24), osteosarcoma and other types of
cancer (25). Nutlin-3 suppressed
not only tumor growth, but also distant metastasis in a xenograft
model of wt p53 neuroblastoma (26). Moreover, Nutlin-3 selectively
enhanced apoptosis in wt p53 cancer cells by activating the p53
pathway (16,26). Currently, drugs which reactivate the
p53 pathway are undergoing clinical trials (27). Although Nutlin-3 has been reported
to be effective against a wide variety of tumors bearing wt p53,
the effects of Nutlin-3 on NPC cells have yet to be reported.
p53 mutations have been reported to be rare in NPC
(28,29), even in recurrent radioresistant NPC
(30), thus making this cancer a
potential candidate for treatment with p53-Mdm2 inhibitors, such as
Nutlin-3. However, increased staining of p53 protein by
immunohistochemistry has been reported, suggesting the deregulation
of the p53 pathway in NPC (29,31,32).
In addition, latent genes of EBV, such as LMP1 which is known to be
expressed in NPC, has been reported to inhibit the p53 pathway
(33). Interestingly, it has been
suggested that induced overexpression of p53 using an adenoviral
vector was found to be effective against NPC cells (34,35),
indicating that further elevation of the p53 levels using the p53
activator, Nutlin-3, may be effective to further improve NPC
treatment.
In the present study, we sought to investigate the
effects of Nutlin-3 alone or in combination with cisplatin on an
EBV-positive, wt p53, NPC cell line, C666-1. We also tested whether
extended treatment with Nutlin-3 would result in the emergence of
p53 mutations in NPC cells. The findings of the present study
provide further insights for the potential use of Nutlin-3 as an
NPC therapeutic agent.
Materials and methods
In vitro cell culture
C666-1 cells were cultured in Roswell Park Memorial
Institute (RPMI)-1640 medium (Gibco Life Technologies, Carlsbad,
CA, USA) supplemented with 15% heat-inactivated fetal calf serum
(FCS) (Gibco Life Technologies). EBV-negative NPC HK1 cells were
cultured in 10% FCS RPMI-1640 medium. HCT116, a colorectal
carcinoma cell line, and MDA-MB-231, a metastatic breast
adenocarcinoma cell line were cultured in Dulbecco's modified
Eagle's medium (DMEM) (Gibco Life Technologies) supplemented with
10% heat-inactivated FCS. Nasopharyngeal epithelial (NPE) cell
line, NP69, was cultured in keratinocyte serum-free medium (K-SFM)
(Gibco Life Technologies) supplemented with bovine pituitary
extract and 0.16 ng/ml recombinant epidermal growth factor (Gibco
Life Technologies), while NPE NP460 cells were maintained in a 1:1
ratio of defined K-SFM medium supplemented with growth factor
(Gibco Life Technologies) and EpiLife® Defined Growth
Supplement (Cascade Biologics, Life Technologies, Carlsbad, CA,
USA). All of the cell lines were maintained in an exponential
growth phase in the presence of 10 U/ml of penicillin (Gibco Life
Technologies) and 10 µg/ml streptomycin (Gibco Life
Technologies) at 37°C in a 5% CO2 humidified atmosphere.
Mycoplasma-free status of the cells was verified monthly with the
e-myco™ Mycoplasma PCR Detection kit (Intron Biotechnology, Inc.,
Seongnam, Korea).
PCR and DNA sequencing
Genomic DNA was extracted from cultured cells using
QIAamp DNA Mini kit (Qiagen, Valencia, CA, USA) following the
manufacturer's instructions. p53 gene amplification spanning from
exons 2 to 11 was amplified in separate PCR reactions using
specific designed oligonucleotide forward (F) and reverse (R)
primers synthesized by First BASE Laboratories, Malaysia. Primer
sequences used for PCR were: exon 2 (F), 5′-AGC TGT CTC AGA CAC TGG
CA-3′ and (R), 5′-GAG CAG AAA GTC AGT CCC ATG-3′; exon 3+4 (F),
5′-AGA CCT ATG GAA ACT GTG AGT GGA-3′ and (R), 5′-GAA GCC TAA GGG
TGA AGA GGA-3′; exon 5 (F), 5′-CGG AAT TCT TAT CTG TTC ACT TGT GCC
C-3′ and (R), 5′-CGG GAT CCA CCC TGG GCA ACC AGC CCT G-3′; exon 6
(F), 5′-CGG AAT TCG GTC CCC AGG CCT CTG ATT CCT -3′ and (R), 5′-CGG
GAT CCA CCC GGA GGG CCA CTG ACA AC-3′; exon 7 (F), 5′-CTG CTT GCC
ACA GGT CTC-3′ and (R), 5′-TGG ATG GGT AGT AGT ATG GAA G-3′; exon 8
(F), 5′-CGG AAT TCT TGG GAG TAG ATG GAG CCT-3′ and (R), 5′-CGG GAT
CCC TCC TCC ACC GCT TCT TGT CCT-3′; exon 9 (F), 5′-AGC AAG CAG GAC
AAG AAG CG-3′ and (R), 5′-CCA GGA GCC ATT GTC TTT GA-3′; exon 10
(F), 5′-CTC AGG TAC TGT GTA TAT ACT TAC-3′ and (R), 5′-ATA CTA CGT
GGA GGC AAG AAT-3′; exon 11 (F), 5′-TCC CGT TGT CCC AGC GTT-3′ and
(R), 5′-TAA CCC TTA ACT GCA AGA ACAT-3′. The quality and purity of
the resulting PCR products were evaluated on an ethidium bromide
stained-agarose gel (2%). The products were further purified using
Qiagen QIA quick PCR Purification kit followed by Sanger
sequencing. The data were aligned and compared against the
published human p53 sequences in NCBI GenBank (X54156). The
nucleotide variants were analyzed using Mutation Surveyor V4.0.7
(SoftGenetics, LLC., State College, PA, USA).
Western blot analysis
Cells were lysed using cold lysis buffer containing
2 mM DL-dithiothreitol, 0.2 mM phenylmethylsulfonylfluoride
(Bio-Rad, Hercules, CA, USA) and protease inhibitor cocktail (Roche
Diagnostics GmbH, Mannheim, Germany). Total protein lysate (10
µg) was separated on 10–15% sodium dodecyl
sulphate-polyacrylamide gel electrophoresis, followed by
immunoblotting with monoclonal antibodies for p53 (Promega
Corporation, Madison, WI, USA), Mdm2 (Santa Cruz Biotechnology,
Inc., Santa Cruz, CA, USA), p21Waf1/Cip1, PUMA, BAX, Bcl2 and PARP
(Cell Signaling Technology, Inc., Beverly, MA, USA). β-actin (Santa
Cruz Biotechnology) was used as a loading control and secondary
antibody reactions were performed with anti-mouse or anti-rabbit
(Promega Corporation) horseradish peroxidase-conjugated IgG.
Chemiluminescent imaging was conducted using ImageQuant™ LAS 500
(GE Healthcare Life Sciences, Buckinghamshire, UK). Densitometric
analysis was performed on an Alpha Imager HP System with AlphaView
software (Alpha Innotech Corporation, San Leandro, CA, USA).
MTS cell viability assay
Ready-to-use 1 mg/ml stock cisplatin
[cis-PtCl2(NH3)2]
(Kemoplat, Fresenius Kabi, Maharashtra, India) and Nutlin-3 (Alexis
Biochemicals, San Diego, CA, USA) were utilized for the in
vitro treatment. Briefly, cells were seeded at densities of
1.5×104 cells/ml for NP69 and NP460; and
3×104 cells/ml for C666-1 cells in 96-well plates (TPP
Techno Plastic Products AG, Trasadingen Switzerland). The cells
were cultured for 24 h before treatment. The dose-response curves
and half maximal inhibitory concentration (IC50) values
were determined by 96® AQueous One Solution Cell
Proliferation MTS solution (Promega Corporation) followed by
measurement using an EnVision multilabel plate reader (PerkinElmer,
Waltham, MA, USA).
Soft agar colony formation assay
The C666-1 cells were seeded at a density of
5×104 cells/ml in 96-well plates (TPP), followed by
cisplatin treatment with or without Nutlin-3. The cells were then
plated into a two-layer soft agar made from DNA grade Seakem
agarose (Lonza, Rockland, ME, USA) culture system (comprised of a
layer of 0.3% agarose in complete media; and with 0.6% agar as a
base layer) in 6-well plates (TPP). Anchorage-independent growth
was measured by counting the numbers of viable colonies using an
Olympus stereomicroscope model SZX7 (Olympus, Tokyo, Japan). The
colonies were scored by using Image Pro Plus AMS version 6.3 (Media
Cybernetics, Inc. Rockville, MD, USA). Colonies with a minimum
diameter of 60 µm, area 2,800 µm2 and
roundness score ranging from 0.25 to 0.50 (roundness
=4A/πD2; A is the area; D is the maximum diameter; with
1.0 indicating a perfect circle) were counted in order to exclude
abortive colonies.
p53 knockdown with small-hairpin RNA
(shRNA)
p53 knockdown was performed using four
lentiviral-based shRNA constructs (Sigma-Aldrich, St. Louis, MO,
USA). The shRNA p53-target sequences were as follows: p53si-2 (D3),
5′-CAC CATCCACTACAACTACAT-3′; p53si-3 (C12), 5′-CGGCGC
ACAGAGGAAGAGAAT-3′; p53si-4 (E1), 5′-GAGGGATGT TTGGGAGATGTA-3′;
p53si-5 (E2), 5′-GTCCAGATGAAG CTCCCAGAA-3′ and non-specific (NS),
5′-CAACAAGAT GAAGAGCACCAA-3′. Lentiviral stocks were generated by
co-transfecting the HEK-293T cells (ATCC® CRL-3216™;
American Type Culture Collection, Manassas, VA, USA) with the
plasmid vector, the psPAX2 packaging plasmids (Addgene plasmid
12260) and pMD2G envelope plasmid (Addgene plasmid 12259) (Addgene,
Inc., Cambridge, MA, USA) using Lipofectamine 2000 (Invitrogen,
Life Technologies, Carlsbad, CA, USA) according to the
manufacturer's recommendations. The knockdown was verified by
western blot analysis.
High content analysis of apoptosis
Briefly, C666-1 cells were seeded at a density of
3×104 cells/ml in View Plate-96 Black 96-well plates
(PerkinElmer) and were allowed to grow for 24 h prior to cisplatin
and/or Nutlin-3 treatments for 48 and 72 h. The cells were cultured
with 0.1% DMSO (Sigma-Aldrich) or basal media, which served as
vehicle controls for Nutlin-3 and cisplatin treatments,
respectively. The cells were stained with AnnexinV-FITC, propidium
iodide (PI) and Hoechst 33342 (BD Biosciences, San Jose, CA, USA)
according to the manufacturer's instructions. Well to well imaging
with three filter channels (DAPI, FITC and TRITC) was performed
using a Metamorph screening acquisition module, on a Nikon
Ti-ECLIPSE inverted fluorescence microscope (Nikon Corporation,
Tokyo, Japan), at a magnification of ×20. Nine fields were imaged
and scored for each well using Metamorph software version 7.7.0.0
(Molecular Devices, Downingtown, PA, USA). The percentages of
apoptotic cells were calculated from triplicate wells.
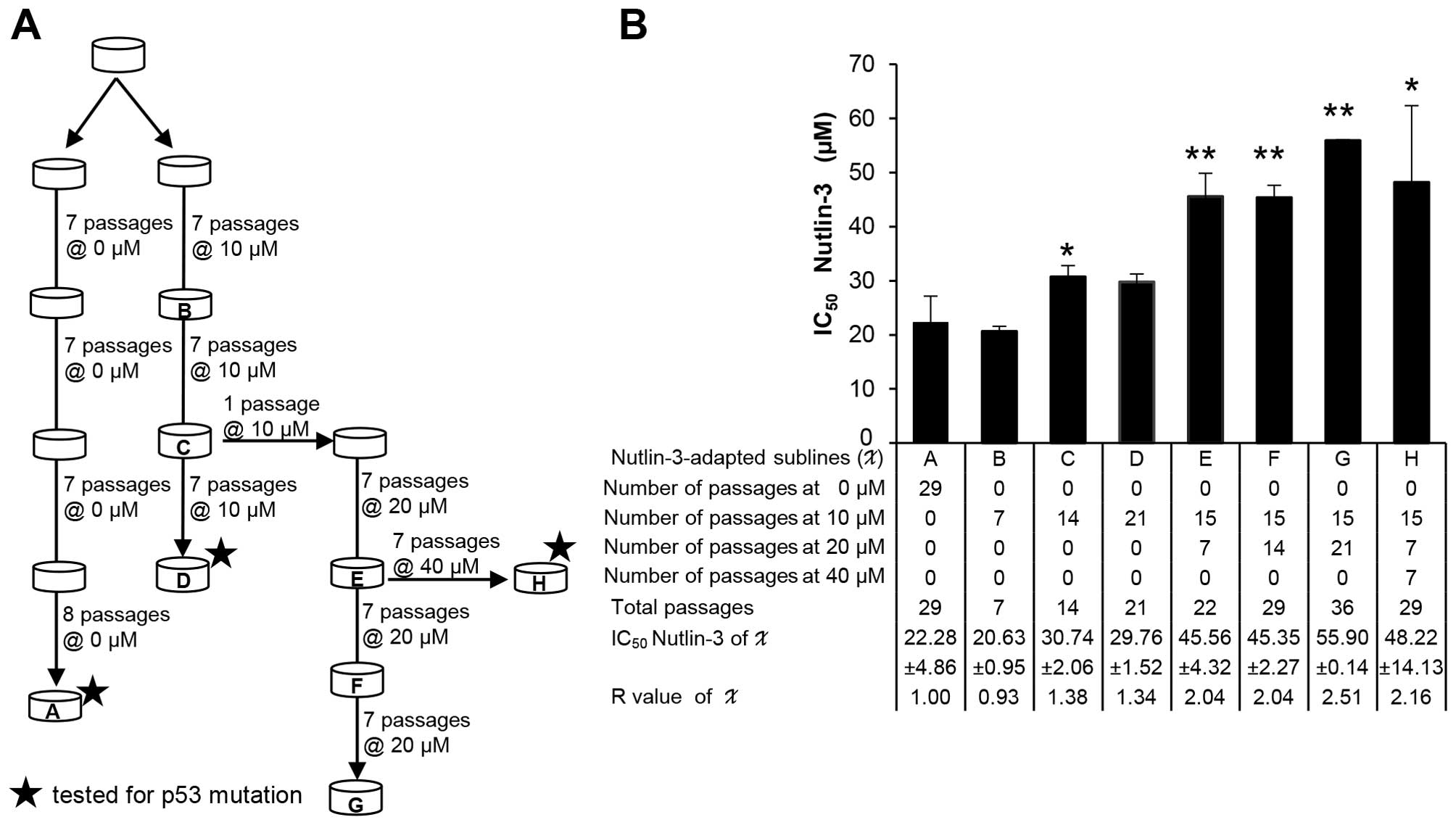
Establishment of Nutlin-3-resistant NPC
C666-1 cells
Nutlin-3-resistant cells were generated by
propagation of parental C666-1 cells in stepwise ascending
concentrations (10, 20 and 40 µM) of Nutlin-3 for a varying
total number of passages (7–36) over
a period of up to six months. Cell viability of the
Nutlin-3-resistant sublines relative to the control parental C666-1
cells was determined by MTS viability assay after a 72-h treatment
with Nutlin-3. The resistance index (R) (R= IC50
resistant cells/IC50 sensitive cells) was calculated to
determine the degree of acquired resistance to its relative control
parental C666-1 cells.
Statistical analysis
Data were analyzed by Microsoft Excel and/or
GraphPad Prism version 5 (GraphPad Software Inc., San Diego, CA,
USA). Statistical significance was measured using the Student's
paired t-test and P-values <0.05 were considered to be
statistically significant.
Results
p53 mutation status of NPC and NPE
cells
In the present study, p53 sequences in NPC (C666-1;
HK1) and NPE (NP69; NP460) cell lines were first examined by
sequencing exons 2 through 11 of the p53 gene. The p53 sequences in
C666-1 cells had a single G>C base substitution detected at
position 12139 at codon 72 of exon 4, while no alteration was
detected in other exons (data not shown). An identical alteration
was also detected in the NP69 and NP460 cell lines. However, a
homozygous C>G base substitution at codon 130 of exon 5 was
detected in the HK1 cells. Overall, these results suggest that the
p53 mutation was only detected in the HK1 cells, while C666-1, NP69
and NP460 cells retained the wt p53 status while expressing a
polymorphism at codon 72, common in the wt p53 gene.
Effects of single drug treatments of
cisplatin and Nutlin-3 on NPC and NPE cells
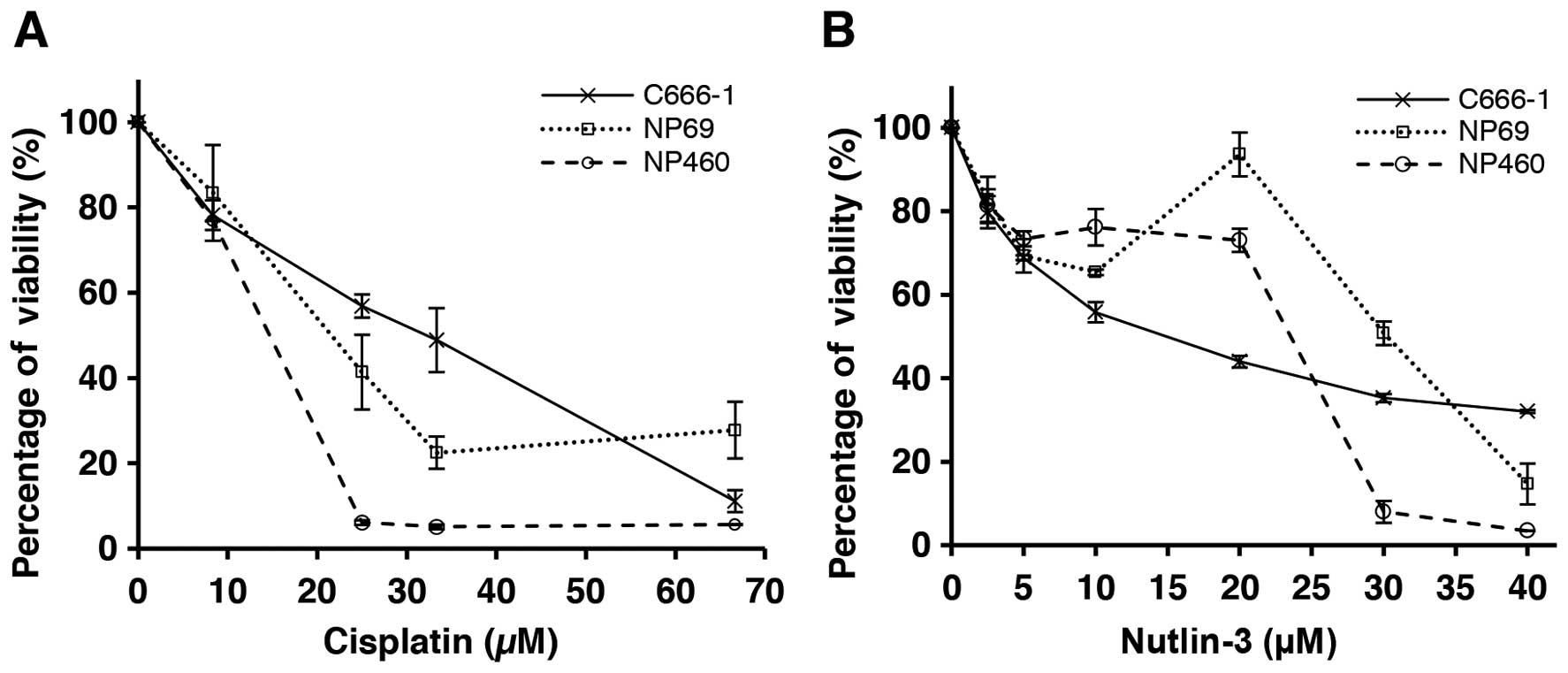
The effects of single drug treatment, cisplatin
(Fig. 1A) or Nutlin-3 (Fig. 1B), on the growth and viability of
the NPC C666-1 and NPE cell lines were evaluated. The results
indicated that both NPC and NPE cells were sensitive to cisplatin
and showed clear dose dependence, whereas the NPE cells were less
sensitive than the NPC cells to Nutlin-3. The IC50
values showed that cisplatin was more cytotoxic against the NPE
cell lines compared to the NPC cells (Table I). In contrast, Nutlin-3 showed
stronger cytotoxic effects against the NPC cells compared to the
effects in the NPE cells, suggesting that Nutlin-3 may be more
selective towards cancer cells.
 | Table ISensitivity of the NPC and NPE cell
lines to cisplatin and Nutlin-3 as indicated by IC50 ±
SD values. |
Table I
Sensitivity of the NPC and NPE cell
lines to cisplatin and Nutlin-3 as indicated by IC50 ±
SD values.
| Cell lines | IC50
values (µM)
|
|---|
| Cisplatin | Nutlin-3 |
|---|
| NP69 | 19.31±4.75 | 31.69±2.54 |
| NP460 | 14.18±2.97 | 22.85±1.18 |
| C666-1 | 32.07±4.18 | 19.95±8.93 |
Combination treatment of cisplatin and
Nutlin-3 on NPC cells
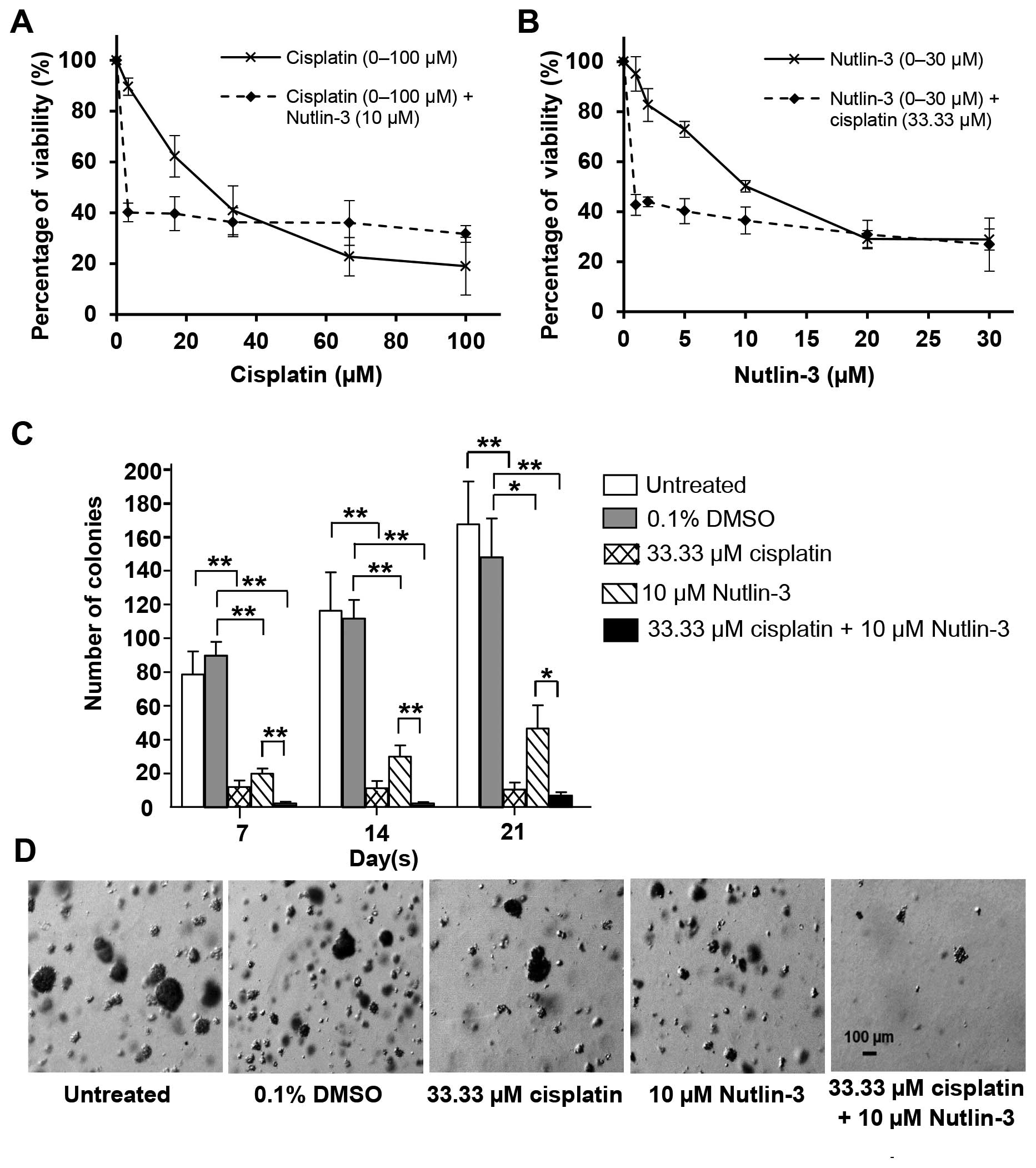
The effects of Nutlin-3 in combination with
cisplatin on the C666-1 cells were investigated (Fig. 2). Cisplatin alone inhibited the
growth of the C666-1 cells in a dose-dependent manner with an
IC50 value of 26.55±6.15 µM. When C666-1 cells
were treated with a combination of cisplatin (0–100 µM) and
a fixed concentration of 10 µM Nutlin-3, the IC50
value was markedly reduced to 3.67±0.88 µM (Fig. 2A). Similarly, when the cells were
treated with a combination of Nutlin-3 (0–30 µM) and a fixed
concentration of 33.33 µM cisplatin, the IC50
value of Nutlin-3 was markedly decreased from 10.23±0.87 µM
to 0.86±0.13 µM (Fig. 2B),
suggesting that the cisplatin combined with Nutlin-3 was more
effective than each agent when used separately. These findings
provide an early indication that Nutlin-3 sensitizes C666-1 cells
to the cytotoxic effects of cisplatin.
Effects of Nutlin-3 on the tumorigenicity
of NPC cells in soft agar
The effects of Nutlin-3 in combination with
cisplatin on the tumorigenicity of C666-1 cells were assessed by
determining the efficiency of anchorage-independent colony
formation using soft agar colony formation assay. Cisplatin (33.33
µM) and Nutlin-3 (10 µM) significantly reduced the
number of colonies formed when administered alone as compared to
the untreated and DMSO-treated controls. When the two treatments
were combined, the numbers of colonies formed were markedly reduced
(Fig. 2C) and the sizes of colonies
were also significantly smaller (Fig.
2D). This further supports the earlier observation that
Nutlin-3 sensitizes C666-1 cells to the cytotoxic effect of
cisplatin and suppresses colony formation.
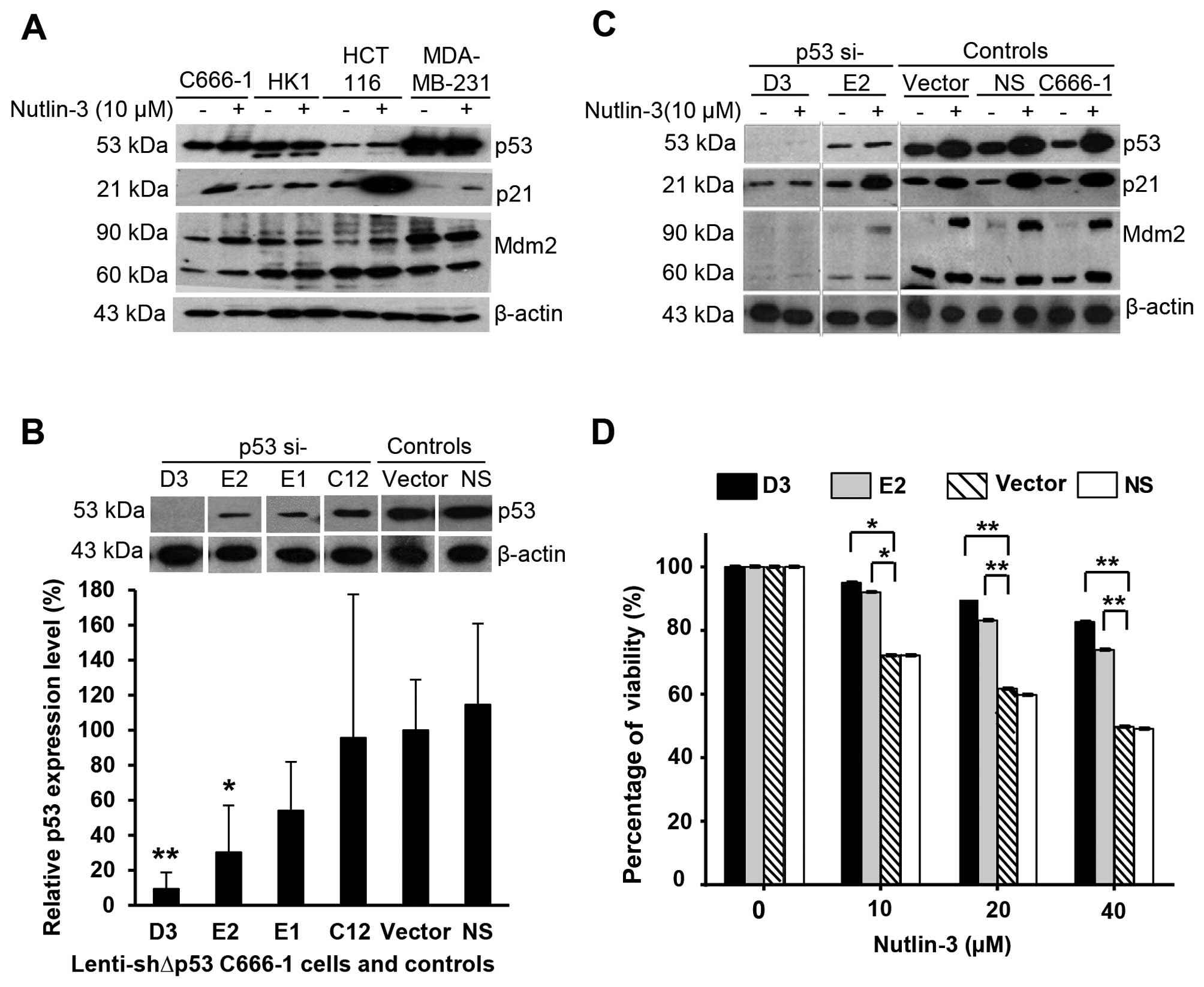
Nutlin-3 activates the p53 pathway in wt
p53 NPC cells
The effects of Nutlin-3 on the p53 pathway were
investigated on NPC C666-1 and HK1 cells harboring wt and mutant
p53, respectively. Investigation was also performed in parallel
with colorectal (HCT116) and breast (MDA-MB-231) cancer cells
harboring wt and mutant p53, respectively. The expression levels of
cellular proteins p53, p21Waf1/Cip1 and Mdm2 are shown in Fig. 3A. Upon treatment of C666-1 or HCT116
cells with Nutlin-3, p53 was activated and this significantly
induced the expression of p21 protein and to a lesser extent, Mdm2
protein. The effects of Nutlin-3 were stronger in cells bearing wt
p53 compared to cells with mutated p53 (HK1 and MDA-MB-231 cells).
These findings suggest that Nutlin-3 activates the p53 pathway and
induces upregulation of p53, p21 and Mdm2 in cells bearing wt
p53.
Nutlin-3 activates the p53 pathway and
exerts its cytotoxic effects on NPC cells in a p53-dependent
manner
To verify that the cytotoxic effects of Nutlin-3 are
mediated through the p53 pathway, we knocked down the p53 gene in
the C666-1 cells using four different lentiviral-based shRNA
constructs (p53si-E1, -E2, -C12 or -D3). The efficiency of
knockdown was verified by examining the p53 protein expression
level as shown in Fig. 3B;
Vector-pLKO and NS were used as controls. Cells transduced with
p53si-E2 and p53si-D3 constructs had 70 (P<0.05) and 90%
(P<0.005) knockdown of p53 protein expression, respectively;
whereas, the control Vector-pLKO and NS had unaltered p53 protein
expression. Next, the effects of Nutlin-3 on the expression of
p53-related proteins following the knockdown of p53 protein in the
lenti-shΔp53-transduced cells were studied. When the parental
C666-1 cells were exposed to 10 µM Nutlin-3, increased
expression levels of p53, p21 and Mdm2 were observed in comparison
to the untreated-parental C666-1 cells and control Vector-pLKO and
NS (Fig. 3C). However, these
effects were markedly inhibited in the p53-knockdown C666-1 cells
transduced with the p53si-D3 or p53si-E2 construct. The percentages
of viability relative to control Vector-pLKO and NS following
treatment with Nutlin-3 are shown in Fig. 3D. Cells with p53 knockdown were
found to be less sensitive to Nutlin-3 in comparison to the
controls. Collectively, these findings suggest that the effect of
Nutlin-3 on C666-1 cell viability is p53-dependent.
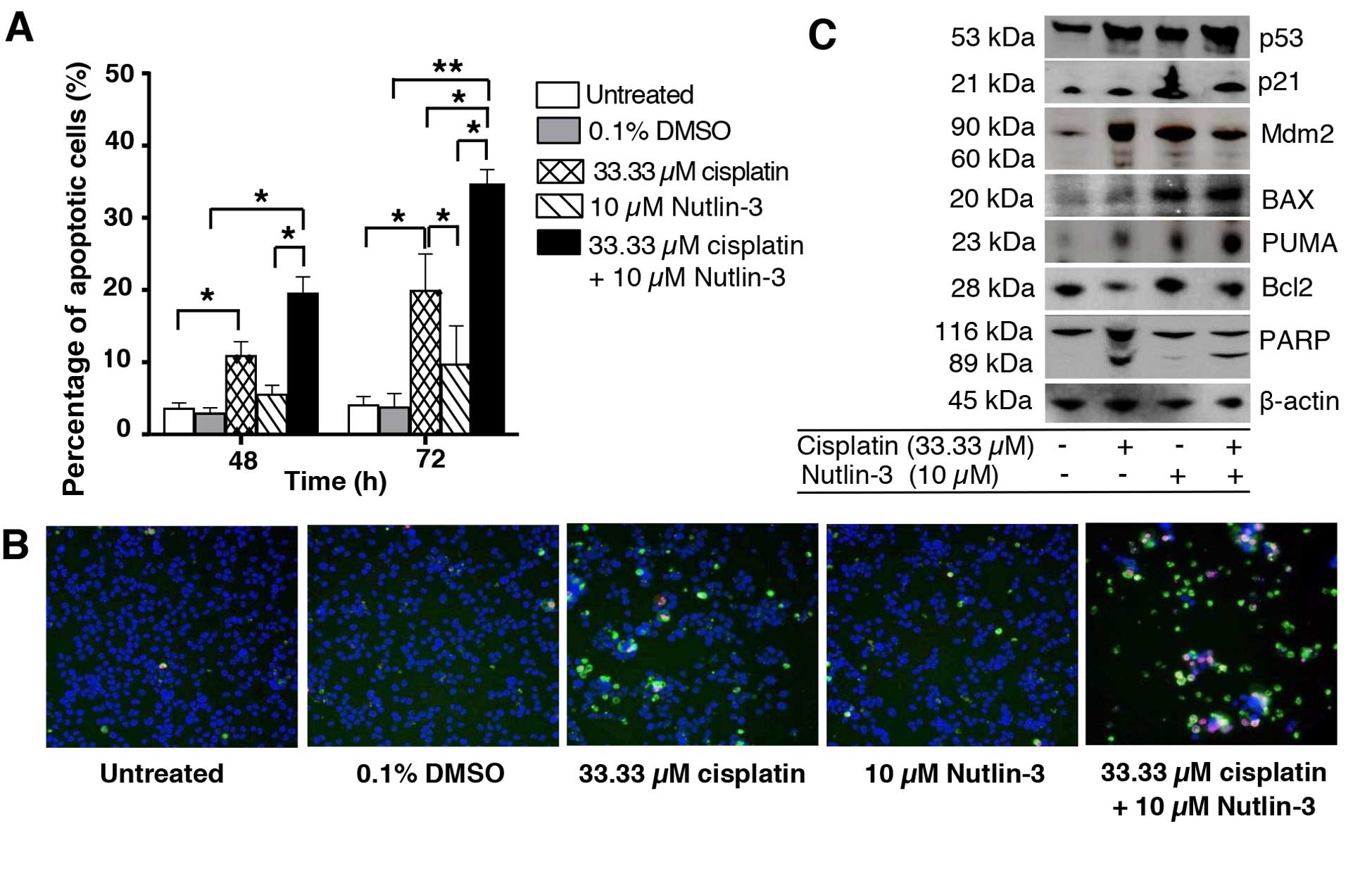
Nutlin-3 sensitizes NPC cells to
cisplatin-induced apoptosis
As shown in Fig. 2,
the combination of cisplatin and Nutlin-3 effectively impaired cell
viability and markedly suppressed the tumorigenicity of C666-1
cells. We used high content imaging of Annexin V/PI-stained cells
to determine the effects of the drug combination on induction of
apoptosis (Fig. 4A and B).
Treatment of C666-1 cells with cisplatin resulted in apoptosis.
Apoptosis increased significantly in the cells treated with both
cisplatin and Nutlin-3. These observations indicated that Nutlin-3
sensitized C666-1 cells to cisplatin and enhanced apoptotic cell
death. Next, we investigated whether Nutlin-3 activates other
p53-mediated pro-apoptotic genes (Fig.
4C). Treatment with Nutlin-3 alone significantly upregulated
BAX and PUMA protein expression. Similarly, the expression levels
of p53, BAX and PUMA were further enhanced by the combination of
cisplatin and Nutlin-3. Furthermore, the cleaved PARP level
detected in the cells treated with the combination drugs was
consistent with apoptosis. Collectively, these results suggest that
Nutlin-3 sensitized the C666-1 cells to cisplatin-induced apoptosis
by modulating pro-apoptotic targets PUMA and BAX.
Extended treatment with Nutlin-3 results
in reduced sensitivity without emergence of p53 mutation
p53 mutations are known to result in resistance to
Nutlin-3 (36). To investigate
whether Nutlin-3 induces the emergence of p53 mutant cells, C666-1
cells were treated with various concentrations of Nutlin-3 for
extended periods (Fig. 5A). The
Nutlin-3-treated NPC sublines showed a doubling in the
IC50 value to Nutlin-3 when compared to the parental
C666-1 cells, suggesting that the C666-1 cells were developing
relative resistance to Nutlin-3 (Fig.
5B). Next, the Nutlin-3-resistant sublines were investigated
for p53 status. The p53 gene sequence (spanning exon 2 to 11) of
the investigated sublines was found to be wt compared to the
parental C666-1 cells. These results indicate that, while extended
treatment of NPC cells with Nutlin-3 resulted in decreased
sensitivity to Nutlin-3, it was not associated with emergence of
p53 mutations at the investigated doses and durations.
Discussion
Targeting p53-Mdm2 interaction to reactivate p53
tumor-suppressing function is a promising cancer therapeutic
strategy for tumors retaining wt p53 (37). The disruption of p53-Mdm2
interaction to reactivate the p53 pathway by small-molecule
inhibitor Nutlin-3 is restricted to tumors with wt p53 (14). Hence, in the present study, a prior
verification of the p53 status of our NPC and NPE cell lines was
performed. A base change of G>C at codon 72 of the 4th exon of
the p53 gene, known as codon 72 polymorphism which encodes for
variant amino acids: arginine (CGC) or proline (CCC) was previously
reported (38). In the present
study, we found the identical codon 72 polymorphism in the p53 gene
of our NP69, NP460 and C666-1 cell lines. This polymorphism is
common in the wt p53 gene and has been detected in lung cancer
(39), teratoma (40) and esophageal squamous cell carcinoma
(41). As expected, our HK1 cell
line had a homozygous point mutation C>G at codon 130 of the 5th
exon of p53 gene, substituting leucine (CTC) with valine (GTC),
similar to a previous report (42).
The present study demonstrated that Nutlin-3
sensitized NPC cells to cisplatin-induced cytotoxicity in a
p53-dependent manner. Nutlin-3 reduced the viability and colony
formation of wt p53 NPC cells. Furthermore, Nutlin-3 significantly
reduced the viability of NPC, but not NPE cells at a concentration
of 10–20 µM, consistent with recent studies that showed that
Nutlin-3 was not toxic to normal cells at the concentrations of
10–20 µM (18,43).
Nutlin-3 activated p53 and upregulated p21 and Mdm2
in the C666-1 and HCT116 cancer cells retaining wt p53, but not in
the HK1 and MDA-MB-231 cancer cells lacking wt p53. These findings
are in concordance with previous reports that Nutlin-3 induced
apoptosis in p53 wt, but not p53 mutant cancer cells (14,16,26,44).
However, Nutlin-3 has also been reported to act via p53-independent
pathways (45). Nevertheless,
suppression of endogenous p53 by lentiviral-based shRNA inhibited
the effects of Nutlin-3 on C666-1 cells, thus indicating that the
effects of Nutlin-3 on C666-1 cells were p53-dependent.
The anti-proliferative activity of cisplatin on NPC
cells was found to be enhanced when combined with Nutlin-3. Our
data indicate that Nutlin-3 sensitized the NPC cells to
cisplatin-induced apoptosis. Similarly, these synergistic
activities have been reported in neuroblastoma (46), gastric cancer (47) and ovarian cancer cells (48) and testicular germ cell tumors
(49). Collectively, the
enhancement of cisplatin-induced apoptotic cell death of NPC cells
by Nutlin-3 was evidenced by the upregulation of p53, p21, Mdm2,
BAX and PUMA expressions. These findings are in agreement with the
notion that p53 promotes apoptosis by activating p53 pro-apoptotic
targets.
Long-term treatment of cancer cells with increasing
concentrations of Nutlin-3 could result in the emergence of
Nutlin-3-resistant p53-mutated cells (36). In the present study, we found that
although long-term exposure of wt p53 C666-1 cells to 10–40
µM Nutlin-3 in stepwise dose increments resulted in reduced
sensitivity to Nutlin-3, the treatment did not result in the
emergence of p53-mutated cells. This finding suggests that the
acquisition of Nutlin-3 resistance in these cells could be due to
other mechanisms. The reduction in sensitivity to Nutlin-3 in
long-term treatment suggests that optimization in the clinical dose
could be important to enhance the efficacy of Nutlin-3. However,
such clinical implications have yet to be investigated fully.
In conclusion, we present evidence that Nutlin-3 is
an effective p53 activator in NPC cells. Nutlin-3 was more toxic to
NPC cells compared to non-malignant cells. The effects of Nutlin-3
on NPC cells were p53-dependent. In combination with cisplatin,
Nutlin-3 promoted apoptosis induction in the NPC cells expressing
wt p53. Collectively, the present study suggests that the potential
use of inhibitors of p53-Mdm2 interaction should be explored
further for the treatment of NPC.
Acknowledgments
The authors would like to thank the Director General
of Health Malaysia for his permission to publish this paper and the
Director of the Institute for Medical Research for her support. We
would like to thank Professor Kwok-Wai Lo (Chinese University of
Hong Kong) and Professor George Tsao (University of Hong Kong) for
kindly providing the C666-1 and HK1 cells, respectively. The
present study was supported and funded by the Ministry of Health
Malaysia.
References
|
1
|
Pathmanathan R, Prasad U, Chandrika G,
Sadler R, Flynn K and Raab-Traub N: Undifferentiated,
nonkeratinizing, and squamous cell carcinoma of the nasopharynx.
Variants of Epstein-Barr virus-infected neoplasia. Am J Pathol.
146:1355–1367. 1995.PubMed/NCBI
|
|
2
|
Devi BC, Pisani P, Tang TS and Parkin DM:
High incidence of nasopharyngeal carcinoma in native people of
Sarawak, Borneo Island. Cancer Epidemiol Biomarkers Prev.
13:482–486. 2004.PubMed/NCBI
|
|
3
|
Zeng MS and Zeng YX: Pathogenesis and
etiology of nasopharyngeal carcinoma. Nasopharyngeal Cancer Medical
Radiology. Springer Berlin; Heidelberg: pp. 9–25. 2010, View Article : Google Scholar
|
|
4
|
Khoo AS and Pua KC: Diagnosis and clinical
evaluation of nasopharyngeal carcinoma. Nasopharyngeal Carcinoma:
Keys for Translational Medicine and Biology. Landes Bioscience and
Springer Science:Business Media; New York, NY: pp. 1–9. 2013,
View Article : Google Scholar
|
|
5
|
Pua KC, Khoo AS, Yap YY, Subramaniam SK,
Ong CA, Gopala Krishnan G and Shahid H; The Malaysian
Nasopharyngeal Carcinoma Study Group: Nasopharyngeal Carcinoma
Database. Med J Malaysia. 63(Suppl C): 59–62. 2008.
|
|
6
|
Lee AW, Lin JC and Ng WT: Current
management of nasopharyngeal cancer. Semin Radiat Oncol.
22:233–244. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang L, Chen QY, Liu H, Tang LQ and Mai
HQ: Emerging treatment options for nasopharyngeal carcinoma. Drug
Des Devel Ther. 7:37–52. 2013.PubMed/NCBI
|
|
8
|
Chee Ee Phua V, Loo WH, Yusof MM, Wan
Ishak WZ, Tho LM and Ung NM: Treatment outcome for nasopharyngeal
carcinoma in University Malaya Medical Centre from 2004–2008. Asian
Pac J Cancer Prev. 14:4567–4570. 2013. View Article : Google Scholar
|
|
9
|
Razak AR, Siu LL, Liu FF, Ito E,
O'Sullivan B and Chan K: Nasopharyngeal carcinoma: The next
challenges. Eur J Cancer. 46:1967–1978. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tuan JK, Ha TC, Ong WS, Siow TR, Tham IW,
Yap SP, Tan TW, Chua ET, Fong KW and Wee JT: Late toxicities after
conventional radiation therapy alone for nasopharyngeal carcinoma.
Radiother Oncol. 104:305–311. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Brown CJ, Lain S, Verma CS, Fersht AR and
Lane DP: Awakening guardian angels: Drugging the p53 pathway. Nat
Rev Cancer. 9:862–873. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Michael D and Oren M: The p53-Mdm2 module
and the ubiquitin system. Semin Cancer Biol. 13:49–58. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fuchs SY, Adler V, Buschmann T, Wu X and
Ronai Z: Mdm2 association with p53 targets its ubiquitination.
Oncogene. 17:2543–2547. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Vassilev LT, Vu BT, Graves B, Carvajal D,
Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C and Klein C:
In vivo activation of the p53 pathway by small-molecule antagonists
of MDM2. Science. 303:844–848. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wiman KG: Strategies for therapeutic
targeting of the p53 pathway in cancer. Cell Death Differ.
13:921–926. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kojima K, Konopleva M, Samudio IJ, Shikami
M, Cabreira-Hansen M, McQueen T, Ruvolo V, Tsao T, Zeng Z, Vassilev
LT, et al: MDM2 antagonists induce p53-dependent apoptosis in AML:
Implications for leukemia therapy. Blood. 106:3150–3159. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Saddler C, Ouillette P, Kujawski L,
Shangary S, Talpaz M, Kaminski M, Erba H, Shedden K, Wang S and
Malek SN: Comprehensive biomarker and genomic analysis identifies
p53 status as the major determinant of response to MDM2 inhibitors
in chronic lymphocytic leukemia. Blood. 111:1584–1593. 2008.
View Article : Google Scholar
|
|
18
|
Stühmer T, Chatterjee M, Hildebrandt M,
Herrmann P, Gollasch H, Gerecke C, Theurich S, Cigliano L, Manz RA,
Daniel PT, et al: Nongenotoxic activation of the p53 pathway as a
therapeutic strategy for multiple myeloma. Blood. 106:3609–3617.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ye F, Lattif AA, Xie J, Weinberg A and Gao
S: Nutlin-3 induces apoptosis, disrupts viral latency and inhibits
expression of angiopoietin-2 in Kaposi sarcoma tumor cells. Cell
Cycle. 11:1393–1399. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Müller CR, Paulsen EB, Noordhuis P,
Pedeutour F, Saeter G and Myklebost O: Potential for treatment of
liposarcomas with the MDM2 antagonist Nutlin-3A. Int J Cancer.
121:199–205. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Miyachi M, Kakazu N, Yagyu S, Katsumi Y,
Tsubai-Shimizu S, Kikuchi K, Tsuchiya K, Iehara T and Hosoi H:
Restoration of p53 pathway by nutlin-3 induces cell cycle arrest
and apoptosis in human rhabdomyosarcoma cells. Clin Cancer Res.
15:4077–4084. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sonnemann J, Palani CD, Wittig S, Becker
S, Eichhorn F, Voigt A and Beck JF: Anticancer effects of the p53
activator nutlin-3 in Ewing's sarcoma cells. Eur J Cancer.
47:1432–1441. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hori T, Kondo T, Kanamori M, Tabuchi Y,
Ogawa R, Zhao QL, Ahmed K, Yasuda T, Seki S, Suzuki K, et al:
Nutlin-3 enhances tumor necrosis factor-related apoptosis-inducing
ligand (TRAIL)-induced apoptosis through up-regulation of death
receptor 5 (DR5) in human sarcoma HOS cells and human colon cancer
HCT116 cells. Cancer Lett. 287:98–108. 2010. View Article : Google Scholar
|
|
24
|
Koster R, Timmer-Bosscha H, Bischoff R,
Gietema JA and de Jong S: Disruption of the MDM2-p53 interaction
strongly potentiates p53-dependent apoptosis in cisplatin-resistant
human testicular carcinoma cells via the Fas/FasL pathway. Cell
Death Dis. 2:e1482011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tovar C, Rosinski J, Filipovic Z, Higgins
B, Kolinsky K, Hilton H, Zhao X, Vu BT, Qing W, Packman K, et al:
Small-molecule MDM2 antagonists reveal aberrant p53 signaling in
cancer: Implications for therapy. Proc Natl Acad Sci USA.
103:1888–1893. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Van Maerken T, Ferdinande L, Taildeman J,
Lambertz I, Yigit N, Vercruysse L, Rihani A, Michaelis M, Cinatl J
Jr, Cuvelier CA, et al: Antitumor activity of the selective MDM2
antagonist nutlin-3 against chemoresistant neuroblastoma with
wild-type p53. J Natl Cancer Inst. 101:1562–1574. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Khoo KH, Verma CS and Lane DP: Drugging
the p53 pathway: Understanding the route to clinical efficacy. Nat
Rev Drug Discov. 13:217–236. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Effert P, McCoy R, Abdel-Hamid M, Flynn K,
Zhang Q, Busson P, Tursz T, Liu E and Raab-Traub N: Alterations of
the p53 gene in nasopharyngeal carcinoma. J Virol. 66:3768–3775.
1992.PubMed/NCBI
|
|
29
|
Hoe SL, Lee ES, Khoo AS and Peh SC: p53
and nasopharyngeal carcinoma: A Malaysian study. Pathology.
41:561–565. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chang KP, Hao SP, Lin SY, Tsao KC, Kuo TT,
Tsai MH, Tseng CK and Tsang NM: A lack of association between p53
mutations and recurrent nasopharyngeal carcinomas refractory to
radiotherapy. Laryngoscope. 112:2015–2019. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hui AB, Lo KW, Leung SF, Teo P, Fung MK,
To KF, Wong N, Choi PH, Lee JC and Huang DP: Detection of recurrent
chromosomal gains and losses in primary nasopharyngeal carcinoma by
comparative genomic hybridisation. Int J Cancer. 82:498–503. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang ZC, Fu S, Wang F, Wang HY, Zeng YX
and Shao JY: Oncogene mutational profile in nasopharyngeal
carcinoma. Onco Targets Ther. 7:457–467. 2014.PubMed/NCBI
|
|
33
|
Liu MT, Chang YT, Chen SC, Chuang YC, Chen
YR, Lin CS and Chen JY: Epstein-Barr virus latent membrane protein
1 represses p53-mediated DNA repair and transcriptional activity.
Oncogene. 24:2635–2646. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pan JJ, Zhang SW, Chen CB, Xiao SW, Sun Y,
Liu CQ, Su X, Li DM, Xu G, Xu B, et al: Effect of recombinant
adenovirus-p53 combined with radiotherapy on long-term prognosis of
advanced nasopharyngeal carcinoma. J Clin Oncol. 27:799–804. 2009.
View Article : Google Scholar
|
|
35
|
Weinrib L, Li JH, Donovan J, Huang D and
Liu FF: Cisplatin chemotherapy plus adenoviral p53 gene therapy in
EBV-positive and -negative nasopharyngeal carcinoma. Cancer Gene
Ther. 8:352–360. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Michaelis M, Rothweiler F, Barth S, Cinatl
J, van Rikxoort M, Löschmann N, Voges Y, Breitling R, von Deimling
A, Rödel F, et al: Adaptation of cancer cells from different
entities to the MDM2 inhibitor nutlin-3 results in the emergence of
p53-mutated multi-drug-resistant cancer cells. Cell Death Dis.
2:e2432011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shangary S and Wang S: Small-molecule
inhibitors of the MDM2-p53 protein-protein interaction to
reactivate p53 function: A novel approach for cancer therapy. Annu
Rev Pharmacol Toxicol. 49:223–241. 2009. View Article : Google Scholar :
|
|
38
|
Matlashewski GJ, Tuck S, Pim D, Lamb P,
Schneider J and Crawford LV: Primary structure polymorphism at
amino acid residue 72 of human p53. Mol Cell Biol. 7:961–963.
1987.PubMed/NCBI
|
|
39
|
Fan R, Wu MT, Miller D, Wain JC, Kelsey
KT, Wiencke JK and Christiani DC: The p53 codon 72 polymorphism and
lung cancer risk. Cancer Epidemiol Biomarkers Prev. 9:1037–1042.
2000.PubMed/NCBI
|
|
40
|
Udin N, Ahmad KA, Ahmad F, Omar E, Aziz
ME, Kumar R and Abdullah JM: Molecular genetic analysis of a
suprasellar immature teratoma : Mutation of exon 4 p53 gene. Malays
J Med Sci. 15:43–46. 2008.PubMed/NCBI
|
|
41
|
Yang W, Zhang Y, Tian X, Ning T and Ke Y:
p53 Codon 72 polymorphism and the risk of esophageal squamous cell
carcinoma. Mol Carcinog. 47:100–104. 2008. View Article : Google Scholar
|
|
42
|
Spruck CH III, Tsai YC, Huang DP, Yang AS,
Rideout WM III, Gonzalez-Zulueta M, Choi P, Lo KW, Yu MC and Jones
PA: Absence of p53 gene mutations in primary nasopharyngeal
carcinomas. Cancer Res. 52:4787–4790. 1992.PubMed/NCBI
|
|
43
|
Jiang M, Pabla N, Murphy RF, Yang T, Yin
XM, Degenhardt K, White E and Dong Z: Nutlin-3 protects kidney
cells during cisplatin therapy by suppressing Bax/Bak activation. J
Biol Chem. 282:2636–2645. 2007. View Article : Google Scholar
|
|
44
|
Chang LJ and Eastman A: Differential
regulation of p21 (waf1) protein half-life by DNA damage and
Nutlin-3 in p53 wild-type tumors and its therapeutic implications.
Cancer Biol Ther. 13:1047–1057. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Valentine JM, Kumar S and Moumen A: A
p53-independent role for the MDM2 antagonist Nutlin-3 in DNA damage
response initiation. BMC Cancer. 11:792011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Barbieri E, Mehta P, Chen Z, Zhang L,
Slack A, Berg S and Shohet JM: MDM2 inhibition sensitizes
neuroblastoma to chemotherapy-induced apoptotic cell death. Mol
Cancer Ther. 5:2358–2365. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Endo S, Yamato K, Hirai S, Moriwaki T,
Fukuda K, Suzuki H, Abei M, Nakagawa I and Hyodo I: Potent in vitro
and in vivo antitumor effects of MDM2 inhibitor nutlin-3 in gastric
cancer cells. Cancer Sci. 102:605–613. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Mir R, Tortosa A, Martinez-Soler F, Vidal
A, Condom E, Pérez-Perarnau A, Ruiz-Larroya T, Gil J and
Giménez-Bonafé P: Mdm2 antagonists induce apoptosis and synergize
with cisplatin overcoming chemoresistance in TP53 wild-type ovarian
cancer cells. Int J Cancer. 132:1525–1536. 2013. View Article : Google Scholar
|
|
49
|
Bauer S, Mühlenberg T, Leahy M, Hoiczyk M,
Gauler T, Schuler M and Looijenga L: Therapeutic potential of Mdm2
inhibition in malignant germ cell tumours. Eur Urol. 57:679–687.
2010. View Article : Google Scholar
|