Introduction
The incidence of testicular cancer has been on the
increase over the last few decades due to increasing environmental
toxicants (1). Testicular germ cell
cancer is a curable cancer and cisplatin is the potent
chemotherapeutic agent used to combat this disease. However,
cisplatin also affects germ, Sertoli and Leydig cells during
chemotherapy, which results in a 30–50% infertility rate for
testicular cancer survivors (2,3).
Identification of a strategy to attenuate cisplatin-induced
testicular toxicity without affecting the killing of cancer cells
largely improves long-term quality of life for testicular cancer
patients.
Gap junction-mediated intercellular communication
(GJIC) is reported to have enhancing effects on the toxicity of
chemotherapeutic agents such as cisplatin and etoposide (4,5) to
cancer cells. Gap junction (GJ) is formed by the docking of two
hemichannels from neighbouring cells. Each hemichannel is composed
of six homogenous or heterogeneous connexins (Cxs). The connexin
family comprises 20 members in mouse and 21 in human, in which
connexin 43 (Cx43) is a predominant Cx member that expressed in
human testicular tissues and testicular cancer cells (6,7).
Gap junctions provide a direct pathway for the rapid
inter-cytoplasmic diffusion of hydrophilic metabolites and
signaling molecules between adjacent cells. Accumulated evidence
suggests that various death signals triggered by anti-tumor agents
propagated through gap junctions from target cells to neighbouring
cells, thereby promoting antineoplastic efficacy in cancer cells
(4,8). This 'bystander' effect is not limited
to death signals. Previous findings have demonstrated that the
protective signal may spread through gap junctions between normal
cells in response to oxidative stress and ischemic injury (9–11).
Hong et al reported that gap junctions between testicular
cancer cells communicated predominantly toxic effects and
increasing cisplatin toxicity, while gap junctions between normal
testicular cells communicated predominantly protective effects,
decreasing cisplatin toxicity to Sertoli and Leydig cells (12). This finding suggests that an
increase in GJIC enhances cisplatin-induced toxic effect on cancer
cells while protecting normal testicular cells against
cisplatin-induced injury.
Statins are competitive inhibitors of
3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA) reductase. They
reduce plasma cholesterol levels and are widely used for
cardiovascular disease resulting from hypercholesterolemia. There
is considerable interest concerning the therapeutic utilization of
statins in antineoplastic and antioxidation treatment (13,14).
Our previous results showed that simvastatin increased GJ function
in Leydig tumor cells and increased etoposide cytotoxicity to these
cells (15). Nevertheless, to the
best of our knowledge, there is no evidence to demonstrate the
effect of simvastatin on GJIC between normal testicular cells and
whether this interaction is involved in the effect of simvastatin
on the response of testicular normal cells to chemotherapeutic
compounds.
In the present study, we confirmed that GJ protected
TM4 Sertoli cells against cisplatin toxicity. Simvastatin
attenuated cisplatin toxicity, which was decreased when GJIC was
inhibited by a chemical inhibitor or Cx43-siRNA. Moreover, we
revealed that simvastatin induced enhancing effects on the gap
junction through the downregulation of phosphorylated Cx43. Our
results indicated that the inhibitory effect of simva statin on
cisplatin toxicity in TM4 cells may be attributed to an increase of
GJIC by decreasing Cx43 phosphorylation. Simvastatin, a sensitizer
of testicular tumor cells to the antineoplastic agent demonstrated
in our previous study, protects normal testicular cells from
cisplatin toxicity.
Materials and methods
Materials
Cisplatin was purchased from Sigma-Aldrich, St.
Louis, MO, USA, and was dissolved in phosphate-buffered saline
(PBS) for a stock concentration. Cell culture reagents were all
obtained from Invitrogen Life Technologies (Carlsbad, CA, USA).
Simvastatin was purchased from Sigma-Aldrich. Simvastatin (20 mg),
which is a lactone prodrug, was diluted in 0.5 ml 100% ethanol,
followed by the addition of 0.75 ml 0.1 M NaOH and heating at 50°C
for 2 h. The solution was neutralized with 0.1 M HCl to pH 7.2 and
adjusted with PBS to a final concentration of 5 mM, sterilized by
filtration and aliquots were stored at −20°C.
Cell lines and cell culture
The mouse Sertoli cell line (TM4) was obtained from
the American Type Culture Collection (ATCC; Manassas, VA, USA). TM4
cells were cultured in Dulbecco's modified Eagle's medium
(DMEM)/F-12 supplemented with 5% horse serum and 2.5% fetal bovine
serum, 100 U/ml penicillin and 100 g/ml streptomycin. The cells
were grown at 37°C in an atmosphere of 5% CO2 in
air.
RNA interference
RNA interference of Cx43 expression was performed by
stable cell lines endogenously expressing shRNA. pSilencer 2.1-U6
neo plasmids expressing shRNA targeting Cx43 (target sequence,
5′-GCTCACGTGTTCTATGTGA-3′) or pSilencer 2.1-U6 negative control
plasmids (both from Ambion Europe, UK) were transfected into cells,
and stable cell lines were selected by 0.1 mg/ml G418 and
identified by western blotting and by parachute dye transfer assay
(12).
Standard colony-forming assay
Cell survival was assayed by a standard
colony-forming assay, adapted for use at high and low cell density,
corresponding to conditions in which junctional channel formation
was permitted or not, respectively (8). In the high-density condition, the
cells were seeded at 3×104 cells/cm2 thus
that cultures were 70–100% confluent at the time of cisplatin
exposure. The cells were treated with cisplatin for 1 h in the
dark, then washed with PBS, harvested by trypsinization, counted,
diluted and seeded in 6-well dishes (100 cells/well). Colony
formation was assessed 5–7 days later by fixation and staining with
crystal violet. Colonies containing ≥50 cells were scored. In the
low-density condition, the cells were directly seeded at a density
of 500 cells/cm2 in 6-well plates and treated with
cisplatin for 1 h after attachment. The plates were rinsed and
assessed for colony formation as previously described. Colony
formation was normalized to the colony-forming efficiency of
non-cisplatin-treated cells. There was no significant difference in
the plating efficiency between the low- and high-density cultures
in the untreated samples (data not shown). Simvastatin was added to
the cultural medium 3 h prior to cisplatin treatment.
Evaluation of the GJIC capacity
To evaluate GJIC capacity, we performed 'Parachute'
dye-coupling assay as described by Goldberg et al and Koreen
et al (16). Donor and receiver TM4 cells were grown to
80–90% confluency. The donor cells were double-labeled with 10
μM CM-DiI, a membrane dye that cannot spread to coupled
cells, and 10 μM calcein-AM (both from Invitrogen Life
Technologies), which is converted inter-cellularly into the gap
junction-permeable dye calcein. The donor cells were washed,
trypsinized and then resuspended with serum-free medium and seeded
onto the receiver cells at a 1:150 donor/receiver ratio.
Simvastatin was added in the donor cell suspension at the same
time. The donor cells were allowed to attach for 4 h at 37°C to
form gap junctions with receiver cells, followed by examination
with a fluorescence microscope. For each experimental condition,
the average number of receiver cells containing dye calcein around
one donor cell was determined and normalized to that of the
control.
Western blotting
Protein extractions were carried out by direct
dissolution of cells in protein lysis buffer containing protease
inhibitors (Sigma-Aldrich). Protein concentrations were determined
using Bio-Rad Dc protein assay reagent (Bio-Rad, Hercules, CA,
USA). Approximately 20 μg of lysate protein was separated on
the 10% SDS-PAGE gel, then electrically transferred to
polyvinylidene difluoride (PVDF) membranes (Bio-Rad) and blocked
with 5% skim milk TBST. Individual membranes were probed with
monoclonal anti-Cx43 antibody produced in mouse (1:4,000;
Sigma-Aldrich) and monoclonal antibodies against p-Cx43 (ser368)
(Cell Signaling Technology, Beverly, MA, USA). The membranes were
incubated with horseradish peroxidase (HRP)-conjugated secondary
antibodies (GE Healthcare, Pittsburgh, PA, USA). The immunoreactive
bands were visualized by ECL plus western blotting detection system
(GE Healthcare). Blots were re-probed with an anti β-tubulin
antibody (Sigma-Aldrich) and developed in an identical manner for
assessing β-tubulin protein levels. The intensities were quantified
by Quantity One software on GS-800 densitometer (Bio-Rad).
Immunofluorescent staining
TM4 cells were plated on glass coverslips in 24-well
plates and treated with 10 μM simvastatin when 80%
confluency was reached. The cells were fixed in 4% paraformaldehyde
at room temperature, washed and blocked with 5% BSA for 30 min at
room temperature. This process was followed by incubation with
monoclonal anti-mouse Cx43 antibody (1:1,500; Sigma-Aldrich)
dissolved in 5% BSA. The cells were incubated with the goat
anti-mouse secondary antibody conjugated with fluorescein
isothiocyanate (1/400; Invitrogen Life Technologies), washed and
mounted with a solution containing P-phenylenediamine + glycerol +
PBS at the proportions 29:9:1. Fluorescence was viewed and images
were captured with a fluorescence microscope.
PKC activity assay
The total protein kinase C activity of TM4 cells was
determined using the PepTag non-radioactive PKC assay kit (Promega,
Madison, WI, USA), according to the manufacturer's instructions.
The PepTag assay uses fluorescent peptide substrate which is highly
specific for PKC. Phosphorylation of this substrate by PKC alters
the peptides net charge from +1 to −1. This change in the net
charge of the substrate allows the phosphorylated and
non-phosphorylated versions of the substrate to be rapidly
separated on an agarose gel (0.8%). The phosphorylated species
migrates towards the positive electrode, while the
non-phosphorylated substrate migrates towards the negative
electrode. The bands were then visualized under UV light and
excised from the gel, and heated at 95°C until the gel was
sectioned. The bands were melted and mixed with Gel solubilization
solution, glacial acetic acid and distilled water. Absorbency was
read at 570 nm using a spectrophotometer in a 96-well plate.
Statistical analysis
Data were statistically analyzed using the unpaired
Student's t-test at a significance level of P<0.05. Data are
presented as means ± SEM unless otherwise indicated using SigmaPlot
(Jandel Scientific, San Rafael, CA, USA).
Results
Gap junctions in TM4 cells attenuate
cisplatin cytotoxicity
TM4 cells expressing Cx43 were cultured under
conditions where gap junction formation was not possible
(low-density; 100 cells/cm2; cells not in direct contact
with each other) and where gap junction formation was possible
(high-density; 3×104 cells/cm2). The cells
were treated with cisplatin for 1 h and then cell survival was
assessed as described in Materials and methods. As shown in
Fig. 1A, treatment with cisplatin
reduced the clonogenic survival of TM4 cell at low and high density
in a concentration-dependent manner. The toxic effect of cisplatin
was much less at a high cell density than at a low cell
density.
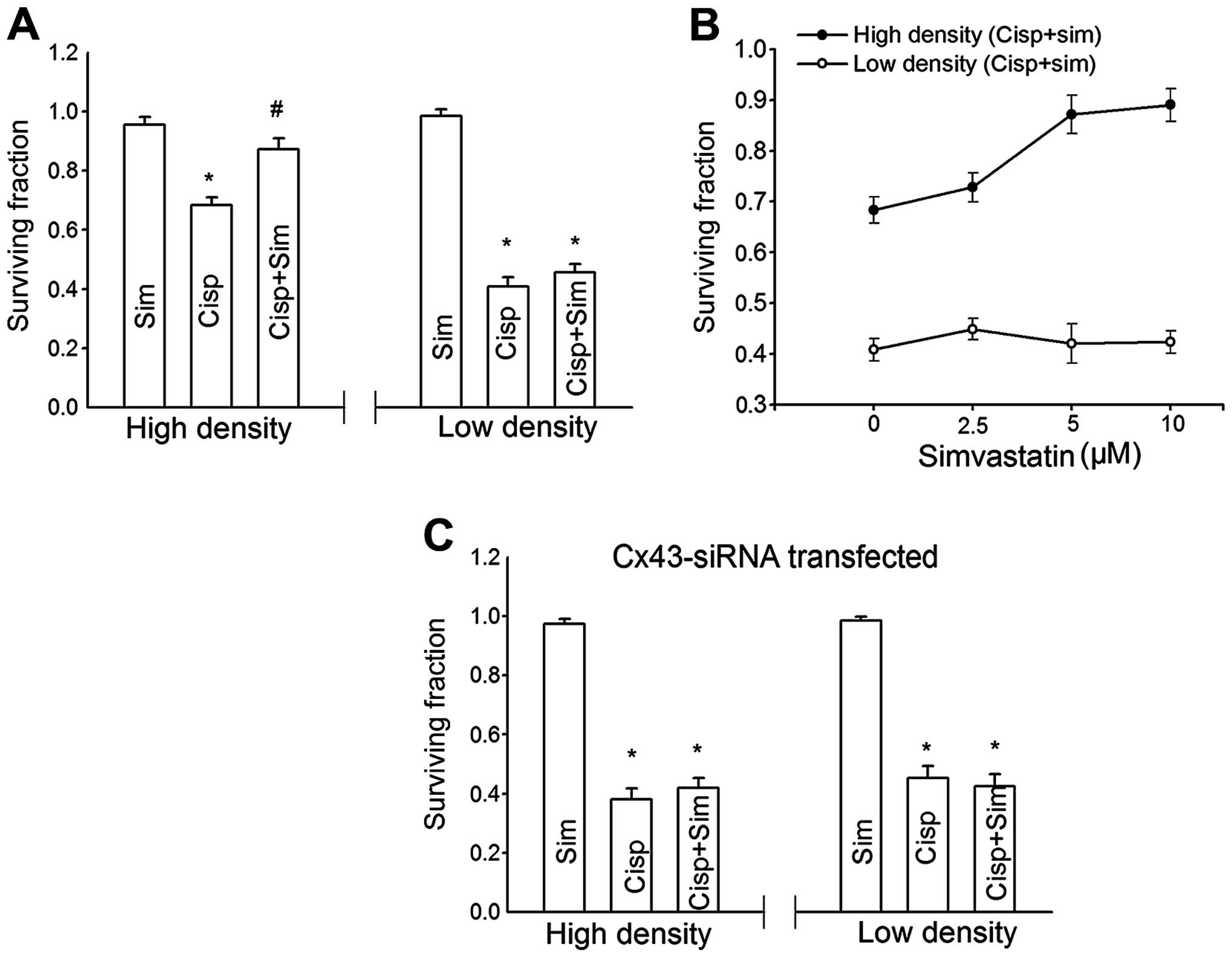
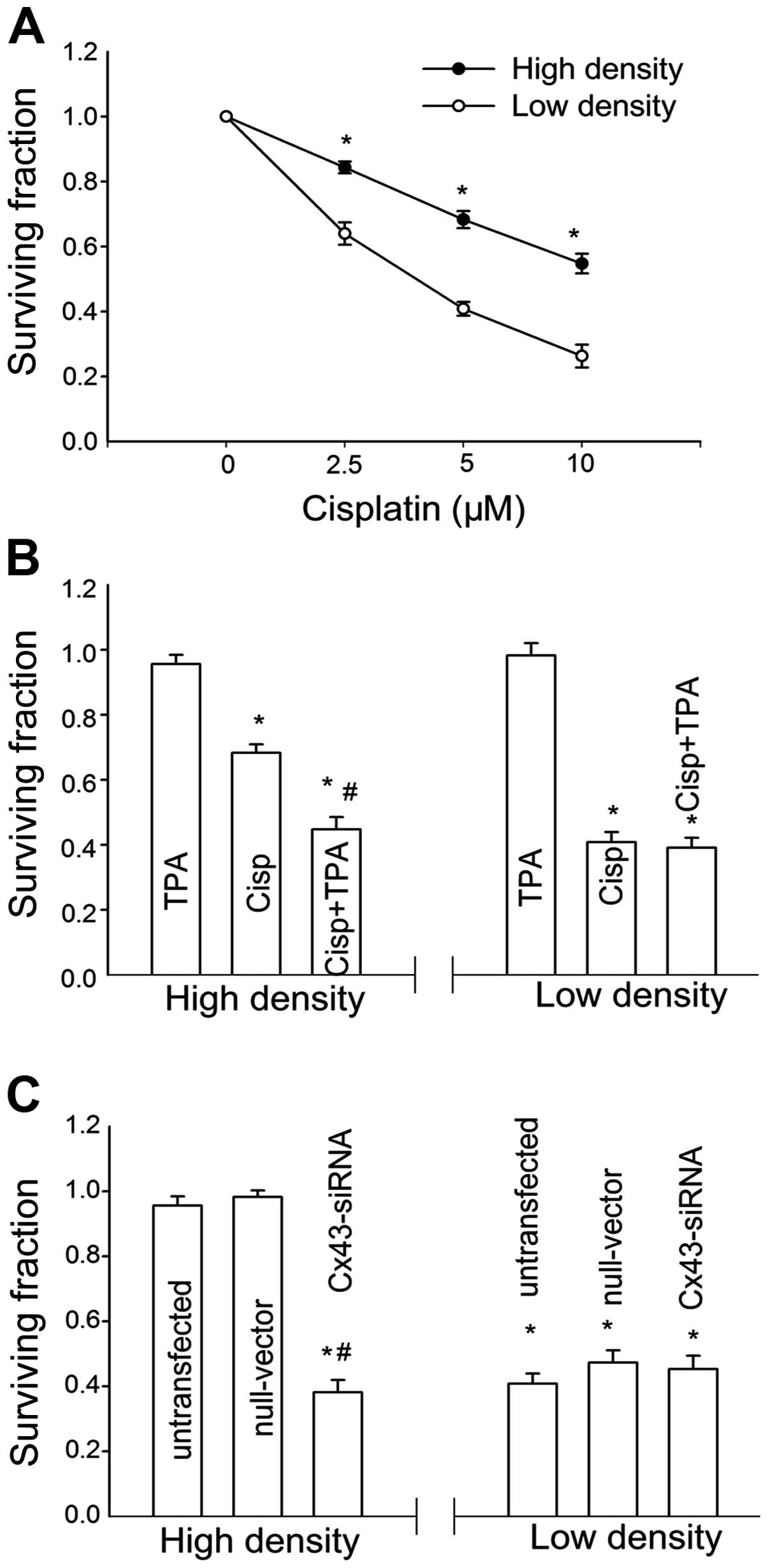 | Figure 1The effect of cell density on
cisplatin cytotoxicity in TM4 cells is mediated by gap junction.
(A) Clonogenic survival of cells treated with a range
concentrations of cisplatin at a low cell density (-○-, 100
cells/ml) and high cell density (-•-, 3×104 cells/ml).
Points, mean for five to seven experiments; bars, SEM. *,
Significantly different from low cell density group, P<0.05. (B)
Clonogenic survival of cells treated with 5 μM cisplatin
with or without 50 nM TPA at low and high cell density. (C)
Clonogenic survival of siRNA-transfected and untransfected cells
treated with 5 μM cisplatin at low and high cell density.
Columns, mean for five experiments; bars, SEM. *, Significantly
different from control, P<0.05; #, significantly
different from the cisplatin bar in (B) and from the untransfected
cells bar in (C), P<0.05. |
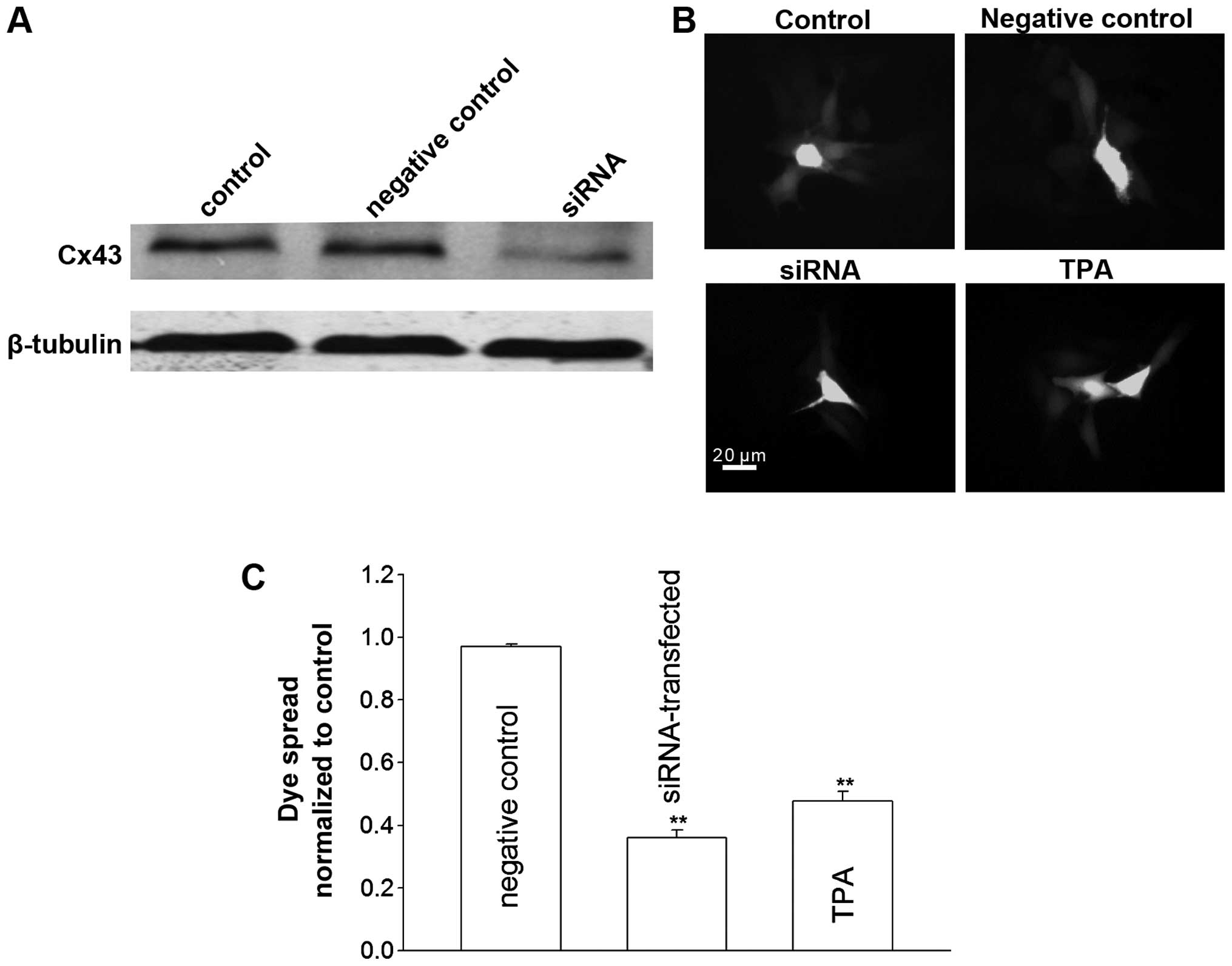
To investigate the role of GJIC in the
density-dependence of cisplatin response in TM4 cells, the gap
junction function was regulated by two methods: pharmacological
inhibition on junctional channels by TPA and downregulation of Cx43
expression by siRNA. TPA is a well-recognized chemical inhibitor
for Cx43 channels. Fig. 2 shows
that, TPA significantly suppressed the dye spread through
junctional channels in TM4 cells. At high cell density,
pretreatment of TM4 cells with TPA increased cisplatin toxicity,
and manifested a reduction in survival fraction from 68 to 43%. By
contrast, TPA had no effect on cisplatin cytotoxicity when the
cells were cultured at a low cell density (Fig. 1B). Cx43 protein expression and
junctional coupling were downregulated in TM4 cells stably
transfected with Cx43 siRNA (Fig.
2). Consistent with the TPA results, cisplatin toxicity in
siRNA-transfected cells was greater than that of the untransfected
cells at high cell density. At a low cell density, cisplatin
responses showed no differences between siRNA-transfected and
-untransfected cells (Fig. 1C).
These results suggested that inhibition of GJIC substantially
increased cisplatin cytotoxicity only at high density, suggesting
the density-dependence of cisplatin cytotoxity in TM4 cells is
mediated by gap junctions.
Simvastatin reduces cisplatin
cytotoxicity in TM4 cells
We examined the effect of simvastatin on cisplatin
cytotoxicity in TM4 cells. TM4 cells were pretreated with 10
μM simvastatin for 3 h and exposed to simvastatin and
cisplatin for another 1 h. As shown in Fig. 3A, simvastatin itself had no toxic
effect on TM4 cells at high and low density. At high density, the
inhibition of clonogenic survival by cisplatin was attenuated by
simvastatin. However, simvastatin exerted no effect on cisplatin
toxicity at low density. As shown in Fig. 3B the simvastatin-induced suppression
of cisplatin toxicity in TM4 cells was concentration-dependent. The
cytotoxicity of cisplatin was reduced with increasing
concentrations of simvastatin.
Simvastatin-induced suppression of
cisplatin cytotoxicity is reversed by GJIC reduction
To investigate whether the density-dependent
simvastatin effects were mediated by GJIC, Cx43-siRNA stably
transfected cells were used. In Fig.
3C, simvastatin-induced reduction in cisplatin toxicity was
markedly attenuated in Cx43-siRNA stably transfected cells at a
high density. At low cell density where gap junction rarely formed,
there was no difference between the control and
Cx43-siRNA-transfected cells. These results indicated that the
simvastatin-induced decrease of cisplatin cytotoxicity was reversed
by GJIC reduction.
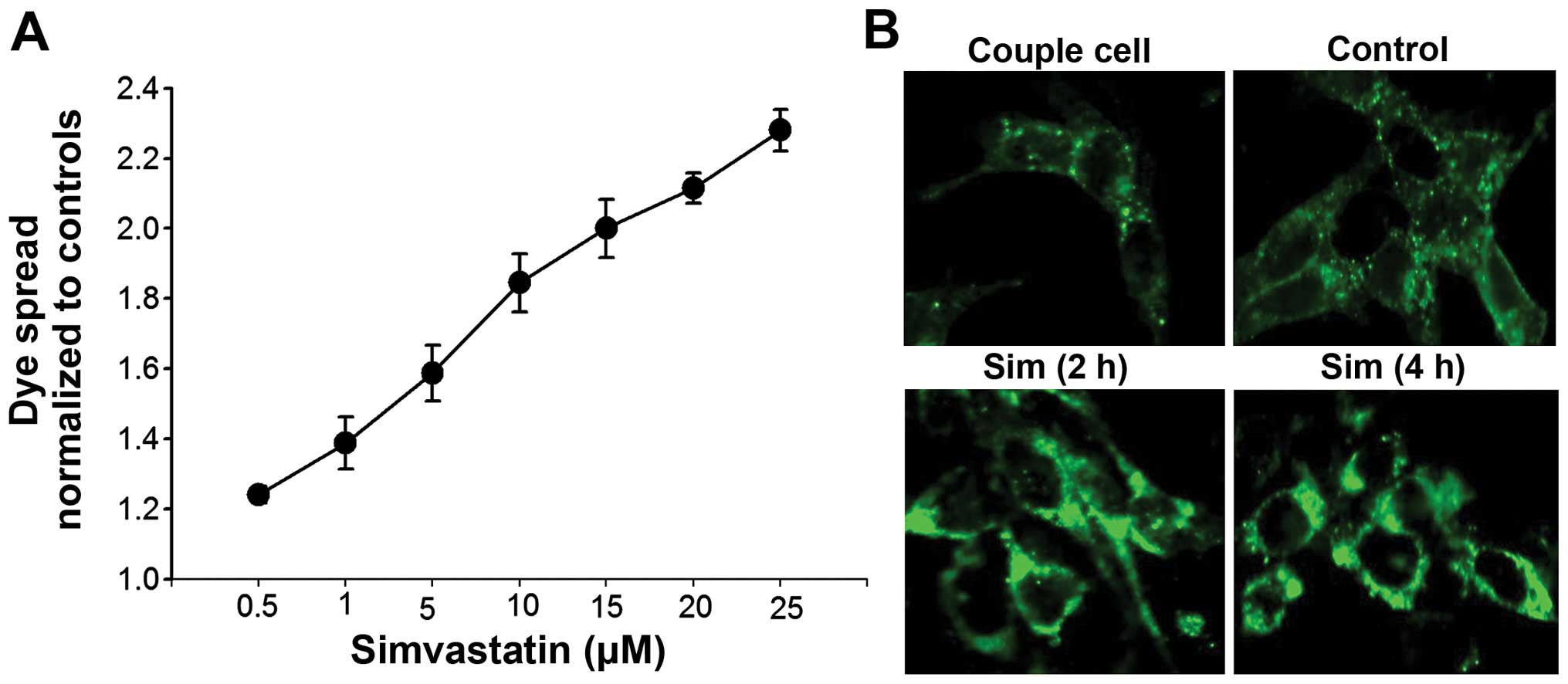
Effect of simvastatin on GJIC and Cx43
membrane localization in TM4 cells
The results described above support that the
inhibitory effect of simvastatin on cisplatin cytotoxicity requires
functional gap junctions, indicating that simvastatin may regulate
gap junction activity. At high cell density when gap junction is
formed, simvastatin may enhance GJIC, which mediated the transfer
of possible protection signals among normal tesicular cells thereby
reducing cisplatin toxicity. To confirm this hypothesis, we
examined the effect of simvastatin on junctional coupling between
TM4 cells. Fig. 4A shows that the
cells that have been treated with simvastatin for 4 h have a marked
increase in calcein dye diffusion through gap junctions. The dye
coupling was gradually enhanced with increasing concentrations of
simvastatin from 0.5 to 25 μM.
Immunofluorescent staining was used to examine
whether simvastatin-enhanced GJIC occurred due to an increase in
functional gap junction proteins on plasma membrane. In Fig. 4B, Cx43 was observed at appositional
plasma membranes between coupled TM4 cells. Treatment with
simvastatin to TM4 cells for 30 min or up to 4 h induced a great
increase in Cx43 immunostaining density in the plasma membrane
regions. These results suggested that simvastatin enhanced GJIC in
TM4 cells by increasing the distribution of constituent Cx43 on the
plasma membrane.
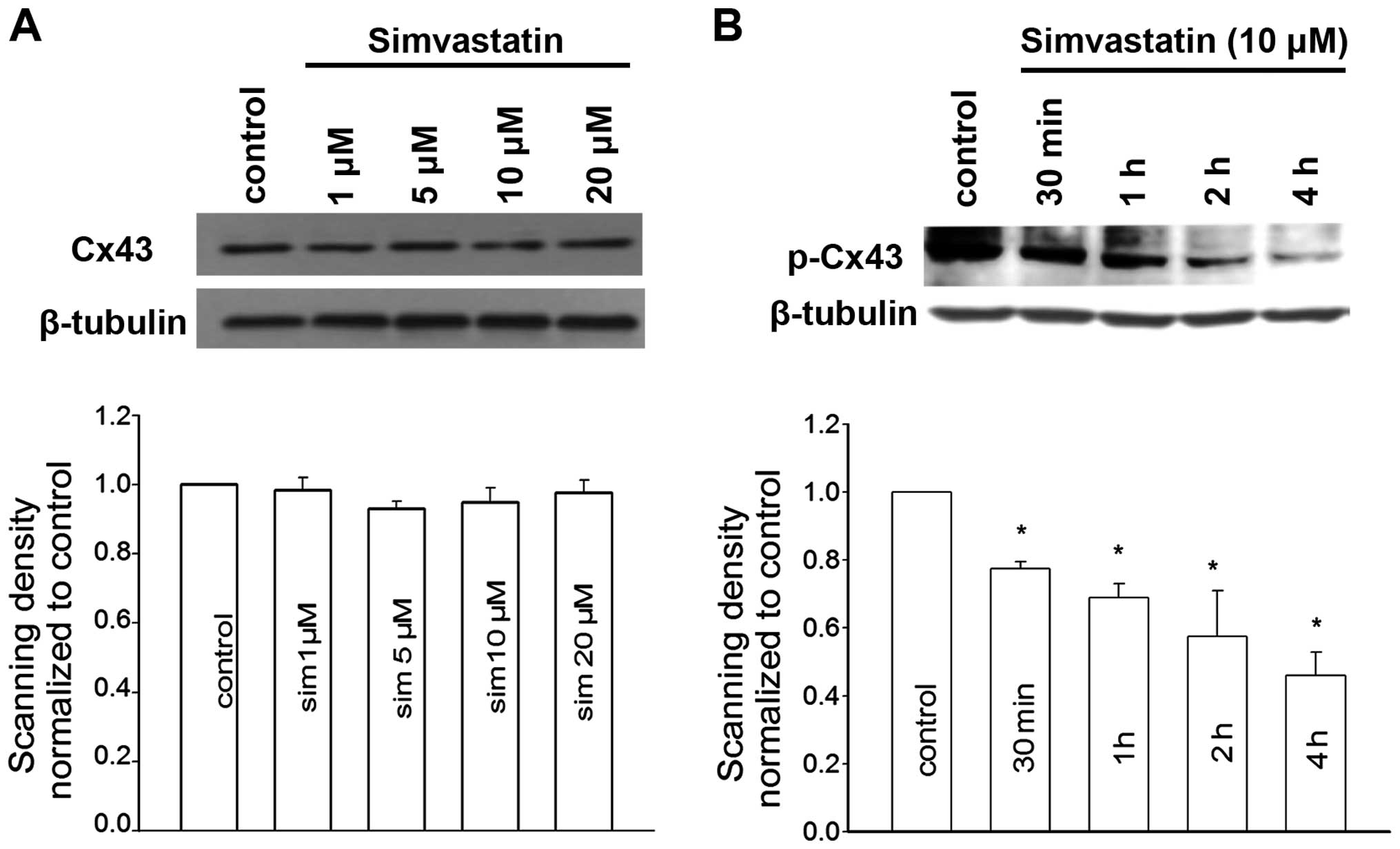
Simvastatin inhibits PKC-mediated Cx43
phosphorylation
In order to investigate the mechanism by which
simvastatin enhanced GJIC, western blotting was performed to
examine the expression of constituent Cx43 in simvastatin-treated
cells. As shown in Fig. 5A,
simvastatin did not alter the level of total Cx43.
Cx43 phosphorylation is known to play an important
role in gap junction disassembly and internalization from the cell
surface (17). Our previous study
on Leydig tumor cells raises the possibility of targeting PKC as a
regulatory signal for simvastin to induce Cx43 dephosphorylation
(15). We then investigated the
effect of simvastatin on the phosphorylation status of Cx43. In
Fig. 5B, the amount of
phosphorylated Cx43 at serine 368 (ser368) which was identified to
be a specific phosphorylation site for PKC was markedly reduced in
cells pretreated with simvastatin. This reduction was enhanced with
increasing simvastatin concentrations. The expression of
phosphorylated Cx43 in Sertoli cells was almost 50% of control
after simvastatin treatment for 4 h.
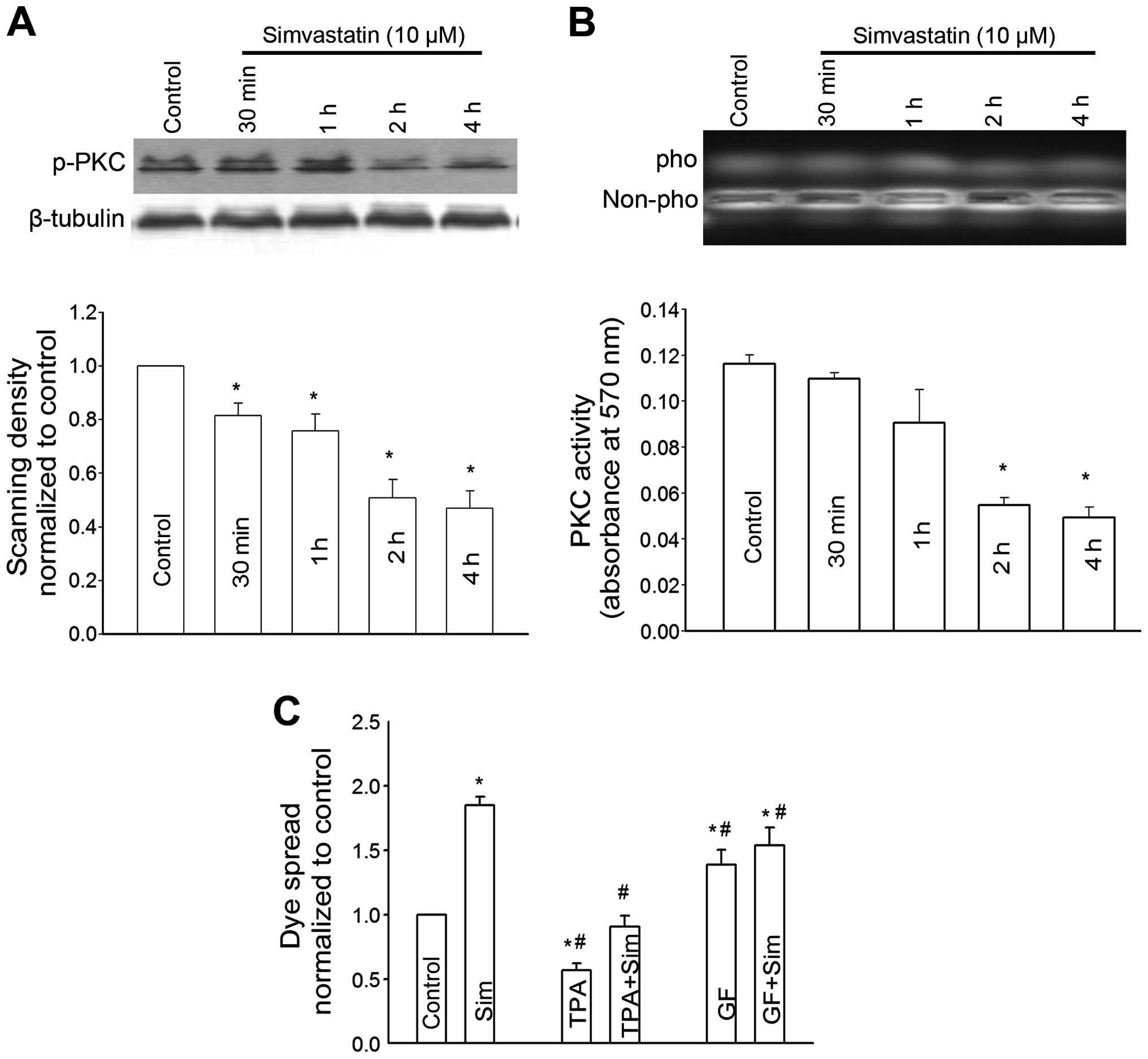
As shown in Fig. 6,
simvastatin markedly reduced PKC expression. The decrease of PKC
expression occurred at 30 min and peaked in 4 h. Consistent with
the alteration of PKC expression, PKC activity was also reduced in
a time-dependent manner in simvastatin-treated cells (Fig. 6B). To investigate that PKC-mediated
connexin phosphorylation was associated with the
simvastatin-induced enhancement of GJIC, dye spread of Sertoli
cells was then assessed in the cells treated with simvastatin and
GF109203X, a specific PKC inhibitor or TPA, a PKC activator.
GF109203X increased the dye spread of Sertoli cells through GJ,
while TPA markedly decreased the dye spread. The
simvastatin-induced increase of GJIC in Sertoli cells was slightly
affected by simultaneous pretreatment with GF109203X. TPA exerted a
significant suppression effect on the simvastatin-induced
enhancement of dye spread through GJ.
Discussion
Infertility is one of the most serious late adverse
consequences after cisplatin-based chemotherapy treatment (2,18).
Cisplatin induces toxicity in germ cells and in Sertoli and Leydig
cells, and eventually results in long-lasting azoospermia and
testicular atrophy in male adults (19). Disruption of the cell junction has
been identified between Sertoli cells in rats after intraperitonal
injection with cisplatin (20).
Investigation into cultured cells has also demonstrated that
cisplatin reduced the production of transferrin, androgen-binding
protein, lactate and estradiol in Sertoli cells (21). In concordance with this observation,
we found that cisplatin induced cytotoxicity in Sertoli cells, with
clonogenic survivals decreasing 32% in controls after incubation
with cisplatin for 1 h.
The concentration of cisplatin used in the present
study was lower relative to some in vitro models, but was
approximately the peak plasma concentration during chemotherapy
(22). Our paradigm allowed the
GJIC-mechanism observed in the present study to be applied. Thus, a
much higher concentration than that used in our text led to
extensive cell death and no GJIC-mediated effects were identified.
Investigators have shown that the inter-Sertoli cell junctions
constituting the blood-testis barrier become leaky even at
low-cisplatin doses (3).
Clinically, normal cells are less sensitive to anti-tumor agents
compared with cancer cells. Thus, long-lasting continuous cisplatin
exposure in testicular tumor patients produces toxicity on Sertoli
cells.
Cx43 is the predominately expressed Cx in testis. It
is localized between Sertoli and Germ cells in seminiferous
epithelium as well as in Leydig cells (23). Cx43 is not associated with the
physiological functions of Leydig cells. However, Cx43 is critical
for the formation of junctions and coordination of junctional
communication between Sertoli and Germ cells and plays a vital role
in the processes of Sertoli proliferation as well as Germ cell
proliferation and differentiation (23). Cx43 in Sertoli cells is considered
important in spermatogenesis (7).
It has been revealed that Cx43 expression was significantly reduced
in testes of infertile patients with secretory azoospermia. In
Sertoli cell-only syndrome rat, Cx43 expression in Sertoli cells
was undetected and the GJIC between them was impaired. Sridharan
et al and Günther et al have confirmed that lack of
Cx43 solely in Sertoli cells was sufficient to induce the arrest of
spermatogenesis (24,25). By contrast, enviroment chemicals
have been found to affect Sertoli cell interactions through
junctional proteins, and specifically Cx43 exerted deleterious
effects on spermatogenesis (26).
In light of those findings, an extensive exploitation of drugs or
biologic approach to restore Cx43 expression and increase GJIC in
Sertoli cells is of considerable value for improving reproduction.
Our studies therefore suggest that future investigations in the
putative therapeutic roles of simvastatin in decreasing
reproduction toxicity among male patients should be performed.
Statins, classical cholesterol-lowering drugs, exert
pleiotropic effects on osteoporosis, inflammation and other
diseases generated by different issues (27,28).
Cx43 also plays essential roles in coordinating activities in the
majority of organs. To the best of our knowledge, GJIC, which we
demonstrated to be enhanced by simvastatin in the present study,
seems to provide a probable mechanism as evidence for pleiotropic
actions. An increase of GJ conductance protects heart from
life-threatening arrhythmia (29).
The antiarrhythmic ability of simvastatin thus seems due to its
action on GJ (30). Furthermore,
whether simvastatin-induced effects on GJIC ameliorate
cisplatin-induced cardiovascular toxicity, which was also shown to
be associated with Cx43 merit future investigation.
Phosphorylation is critically involved in Cx
disassembly, degradation and internalization. It therefore altered
the amounts of constituent Cxs localized on membrane and GJ
function. Cx43 is the most easily phosphorylated Cx subtype,
containing 12 or more serine or tyrosine residues at carboxyl
terminal that can be extensively phosphorylated by a multiplicity
of phosphorylation protein kinases (17,31).
Simvastatin is a lipophilic statin that permeates through plasma
membrane by passive diffusion. It has been observed that
simvastatin inhibits the activation of different kinases such as
PKC or MAPKs in various cell culture (15,32).
Li et al focused on Sertoli cells, showing that PKC played a
crucial role in regulating tight junctions during spermatogenesis
(33). Our findings confirm that
simvastatin induced a significant decrease in PKC activity and the
simvastatin-induced increase of junctional dye transfer was
abolished by TPA (a PKC activator) and imitated by GF109203X (a PKC
inhibitor). Therefore, our results suggest that PKC is a regulatory
signal for simvastin to induce Cx43 dephosphorylation in Sertoli
cells.
The GJIC-mediated protection effect in normal cells
has also been confirmed by other authors showing that neuronal
vulnerability to oxidative stress and ischemia was significantly
increased by the inhibition of astrocytic gap junctions (11,34),
and that Cx43 exerted a protective effect against oxidative
stress-induced cell death in human retinal pigment epithelial cells
and in cultured primary osteocytes (10,35).
Cx43 itself has been suggested to contribute to the activation of a
major cytoprotective signaling pathway in cardiomyocytes (36). The cells exposed to cisplatin
usually produce apoptosis by the formation of crosslinks, including
intra- and inter-DNA crosslinks. It has also been demonstrated that
cisplatin toxicity in rat liver epithelial cells was enhanced by
GJIC (8). However, in normal
testicular cells, cisplatin-induced DNA crosslinks were increased
when GJIC was blocked by 18-GA or siRNA. GJIC is believed to
communicate predominantly protective signals in normal testicular
cells through gap junction in response to cisplatin (12). Nevertheless, which and how
protective signals were triggered after cisplatin exposure have not
been identified and whether it directly or indirectly induces the
decrease of DNA crosslinks remains to be investigated. Glutathione
is speculated to be a likely candidate. Glutathione (GSH) is a
tripeptide with a molecular weight of 307 Da and is permeable to
gap junctions. As reported by Nakamura et al (37), metabolic coupling of GSH between
mouse and quail myocytes through gap junctions played an essential
role in resistance of mouse myocytes to oxidative stress.
Hepatotoxicity induced by acetaminophen was more higher in Cx32-KO
mice and this was associated with a lower level of cellular GSH
concentration. That study identified the protective effect may be
due to GSH transmission between neighboring cells through GJIC
(38).
To the best of our knowledge, the present study is
the first to demonstrate that the enhan cement effect of
simvastatin on GJ has a protective effect against cisplatin-induced
toxicity on Sertoli cells. The beneficial role of GJ in
cisplatin-based chemotherapy therefore is bidirectional. An
increase of GJIC by simvastatin has synergistically toxic effects
on tumor cells, while the increase of GJIC on Sertoli cells
alleviated their sensitivity to antineoplastic agents and therefore
improved reproductive potency in male adults. The present study
provides an update on the pharmacologic intervention of GJ or
connexins and highlights the importance of basic cell biology in
decreasing reproduction toxicity caused by exposure to
chemotherapy.
Acknowledgments
The present study was supported by a grant from the
National Natural Science Foundation of China (nos. 81401017,
81373439 and U1303221).
Abbreviations:
|
GJIC
|
gap junctional intercellular
communication
|
|
TPA
|
12-O-tetradecanoylphorbol
13-acetate
|
References
|
1
|
Elzinga-Tinke JE, Dohle GR and Looijenga
LH: Etiology and early pathogenesis of malignant testicular germ
cell tumors: Towards possibilities for preinvasive diagnosis. Asian
J Androl. 17:381–393. 2015.PubMed/NCBI
|
|
2
|
Brydøy M1, Fosså SD, Klepp O, Bremnes RM,
Wist EA and Wentzel-Larsen T: Paternity and testicular function
among testicular cancer survivors treated with two to four cycles
of cisplatin-based chemotherapy. Eur Urol. 58:134–140. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Efstathiou E and Logothetis CJ: Review of
late complications of treatment and late relapse in testicular
cancer. J Natl Compr Canc Netw. 4:1059–1070. 2006.PubMed/NCBI
|
|
4
|
He B, Tong X, Wang L, Wang Q, Ye H, Liu B,
Hong X, Tao L and Harris AL: Tramadol and flurbiprofen depress the
cytotoxicity of cisplatin via their effects on gap junctions. Clin
Cancer Res. 15:5803–5810. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shishido SN and Nguyen TA: Gap junction
enhancer increases efficacy of cisplatin to attenuate mammary tumor
growth. PLoS One. 7:e449632012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Saez JC, Berthoud VM, Branes MC, Martinez
AD and Beyer EC: Plasma membrane channels formed by connexins:
Their regulation and functions. Physiol Rev. 83:1359–1400. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Weider K, Bergmann M and Brehm R: Connexin
43: Its regulatory role in testicular junction dynamics and
spermatogenesis. Histol Histopathol. 26:1343–1352. 2011.PubMed/NCBI
|
|
8
|
Jensen R and Glazer PM:
Cell-interdependent cisplatin killing by Ku/DNA-dependent protein
kinase signaling transduced through gap junctions. Proc Natl Acad
Sci USA. 101:6134–6139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Contreras JE, Sánchez HA, Véliz LP,
Bukauskas FF, Bennett MV and Sáez JC: Role of connexin-based gap
junction channels and hemichannels in ischemia-induced cell death
in nervous tissue. Brain Res Brain Res Rev. 47:290–303. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kar R, Riquelme MA, Werner S and Jiang JX:
Connexin 43 channels protect osteocytes against oxidative
stress-induced cell death. J Bone Miner Res. 28:1611–1621. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nakase T, Fushiki S and Naus CC:
Astrocytic gap junctions composed of connexin 43 reduce apoptotic
neuronal damage in cerebral ischemia. Stroke. 34:1987–1993. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hong X, Wang Q, Yang Y, Zheng S, Tong X,
Zhang S, Tao L and Harris A: Gap junctions propagate opposite
effects in normal and tumor testicular cells in response to
cisplatin. Cancer Lett. 317:165–171. 2012. View Article : Google Scholar
|
|
13
|
Subramanian S, Emami H, Vucic E, Singh P,
Vijayakumar J, Fifer KM, Alon A, Shankar SS, Farkouh M, Rudd JH, et
al: High-dose atorvastatin reduces periodontal inflammation: A
novel pleiotropic effect of statins. J Am Coll Cardiol.
62:2382–2391. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hwang KE, Kwon SJ, Kim YS, Park DS, Kim
BR, Yoon KH, Jeong ET and Kim HR: Effect of simvastatin on the
resistance to EGFR tyrosine kinase inhibitors in a non-small cell
lung cancer with the T790M mutation of EGFR. Exp Cell Res.
323:288–296. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang L, Fu Y, Peng J, Wu D, Yu M, Xu C,
Wang Q and Tao L: Simvastatin-induced up-regulation of gap
junctions composed of connexin 43 sensitize Leydig tumor cells to
etoposide: An involvement of PKC pathway. Toxicology. 312:149–157.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Koreen IV, Elsayed WA, Liu YJ and Harris
AL: Tetracycline-regulated expression enables purification and
functional analysis of recombinant connexin channels from mammalian
cells. Biochem J. 383:111–119. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Laird DW: Connexin phosphorylation as a
regulatory event linked to gap junction internalization and
degradation. Biochim Biophys Acta. 1711:172–182. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Boekelheide K: Mechanisms of toxic damage
to spermatogenesis. J Natl Cancer Inst Monogr. 34:6–8. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Harman JG and Richburg JH:
Cisplatin-induced alterations in the functional spermatogonial stem
cell pool and niche in C57/BL/6J mice following a clinically
relevant multi-cycle exposure. Toxicol Lett. 227:99–112. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pogach LM, Lee Y, Gould S, Giglio W,
Meyenhofer M and Huang HF: Characterization of cis-platinum-induced
Sertoli cell dysfunction in rodents. Toxicol Appl Pharmacol.
98:350–361. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nambu A, Kumamoto Y and Mikuma N: Effects
of anti-cancer agents on cultured rat Sertoli cells. Nihon
Hinyokika Gakkai Zasshi. 86:1132–1136. 1995.In Japanese. PubMed/NCBI
|
|
22
|
Erdlenbruch B, Nier M, Kern W, Hiddemann
W, Pekrun A and Lakomek M: Pharmacokinetics of cisplatin and
relation to nephrotoxicity in paediatric patients. Eur J Clin
Pharmacol. 57:393–402. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Carette D, Weider K, Gilleron J, Giese S,
Dompierre J, Bergmann M, Brehm R, Denizot JP, Segretain D and
Pointis G: Major involvement of connexin 43 in seminiferous
epithelial junction dynamics and male fertility. Dev Biol.
346:54–67. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sridharan S, Simon L, Meling DD, Cyr DG,
Gutstein DE, Fishman GI, Guillou F and Cooke PS: Proliferation of
adult Sertoli cells following conditional knockout of the Gap
junctional protein GJA1 (connexin 43) in mice. Biol Reprod.
76:804–812. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Günther S, Fietz D, Weider K, Bergmann M
and Brehm R: Effects of a murine germ cell-specific knockout of
connexin 43 on connexin expression in testis and fertility.
Transgenic Res. 22:631–641. 2013. View Article : Google Scholar
|
|
26
|
Qiu L, Zhang X, Zhang X, Zhang Y, Gu J,
Chen M, Zhang Z, Wang X and Wang SL: Sertoli cell is a potential
target for perfluorooctane sulfonate-induced reproductive
dysfunction in male mice. Toxicol Sci. 135:229–240. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
van der Meij E, Koning GG, Vriens PW,
Peeters MF, Meijer CA, Kortekaas KE, Dalman RL, van Bockel JH,
Hanemaaijer R, Kooistra T, et al: A clinical evaluation of statin
pleiotropy: Statins selectively and dose-dependently reduce
vascular inflammation. PLoS One. 8:e538822013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Uzzan B, Cohen R, Nicolas P, Cucherat M
and Perret GY: Effects of statins on bone mineral density: A
meta-analysis of clinical studies. Bone. 40:1581–1587. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Greener ID, Sasano T, Wan X, Igarashi T,
Strom M, Rosenbaum DS and Donahue JK: Connexin43 gene transfer
reduces ventricular tachycardia susceptibility after myocardial
infarction. J Am Coll Cardiol. 60:1103–1110. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Naji F, Suran D, Kanic V, Vokac D and
Sabovic M: Comparison of atorvastatin and simvastatin in prevention
of atrial fibrillation after successful cardioversion. Int Heart J.
50:153–160. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Aravindakshan J and Cyr DG: Nonylphenol
alters connexin 43 levels and connexin 43 phosphorylation via an
inhibition of the p38-mitogen-activated protein kinase pathway.
Biol Reprod. 72:1232–2140. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lee YM, Chen WF, Chou DS, Jayakumar T, Hou
SY, Lee JJ, Hsiao G and Sheu JR: Cyclic nucleotides and
mitogen-activated protein kinases: Regulation of simvastatin in
platelet activation. J Biomed Sci. 17:452010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li JC, Mruk D and Cheng CY: The
inter-Sertoli tight junction permeability barrier is regulated by
the interplay of protein phosphatases and kinases: An in vitro
study. J Androl. 22:847–856. 2001.PubMed/NCBI
|
|
34
|
Le HT, Sin WC, Lozinsky S, Bechberger J,
Vega JL, Guo XQ, Sáez JC and Naus CC: Gap junction intercellular
communication mediated by connexin43 in astrocytes is essential for
their resistance to oxidative stress. J Biol Chem. 289:1345. 2014.
View Article : Google Scholar :
|
|
35
|
Pocrnich CE, Shao Q, Liu H, Feng MM,
Harasym S, Savage M, Khimdas S, Laird DW and Hutnik CM: The effect
of connexin43 on the level of vascular endothelial growth factor in
human retinal pigment epithelial cells. Graefes Arch Clin Exp
Ophthalmol. 250:515–522. 2012. View Article : Google Scholar
|
|
36
|
Rottlaender D, Boengler K, Wolny M,
Michels G, Endres-Becker J, Motloch LJ, Schwaiger A, Buechert A,
Schulz R, Heusch G, et al: Connexin 43 acts as a cytoprotective
mediator of signal transduction by stimulating mitochondrial
KATP channels in mouse cardiomyocytes. J Clin Invest.
120:1441–1453. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Nakamura TY, Yamamoto I, Kanno Y, Shiba Y
and Goshima K: Metabolic coupling of glutathione between mouse and
quail cardiac myocytes and its protective role against oxidative
stress. Circ Res. 74:806–816. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Igarashi I, Maejima T, Kai K, Arakawa S,
Teranishi M and Sanbuissho A: Role of connexin 32 in acetaminophen
toxicity in a knockout mice model. Exp Toxicol Pathol. 66:103–110.
2014. View Article : Google Scholar
|