1. Introduction
Inactivation of tumor suppressor genes (TSGs), whose
structural abnormalities and functional disorders lead to changes
in their regulatory mechanism, plays an important role in the
development and progression of human tumors. The role of TSGs
involved in normal differentiation of tissues and inhibition of
carcinoma has gained increasing attention (1,2).
Numerous studies have shown that the mutations and/or deletions of
IKKα frequently occur in human epithelial-derived
carcinogenesis, while IKKα stable expression appears in
corresponding normal tissues (3–6).
2. Structure and traditional functions of
IKKα
IKKα is a 85-kDa polypeptide containing an
amino-terminal serine-threonine kinase catalytic domain and
carboxyl- proximal helix-loop-helix (HLH) and leucine zipper-like
(LZip) amphipathic α-helical domains (7,8).
IKKα and IKKβ, which share similar substrate
specificities as catalytic subunits, form a specific IκB kinase
complex with the regulatory subunit IKKγ/NEMO, thus
contributing to kinase activity regulation in the cytoplasm
(9).
A series of surrounding signals, such as aberrant
cytokine production, integrins, growth factors and cytokine
receptors activate the IKK complex for the phosphorylation
and ubiquitylation of IκBs, resulting in subsequent proteasomal
degradation of p105 to p50. This is the canonical NF-κB pathway
(Fig. 1A), which leads to the
nuclear entry of NF-κB1/RelA (p50/p65) (10,11).
IKKα also forms IKKα/IKKα homodimers to stimulate an
alternative NF-κB pathway by transformation of NF-κB2/p100/RELB
into p52/RELB heterodimers, which translocate into the nucleus and
exert transcriptional control (12)
(Fig. 1B). Furthermore, IKKα
may directly translocate to the nucleus and bind to histone 3 (H3)
(13) (Fig. 1C). Ultimately, these processes
contribute to gene expression and regulation of proliferation,
apoptosis, migration, tumorigenesis, inflammation, angiogenesis and
innate immunity (Fig. 1).
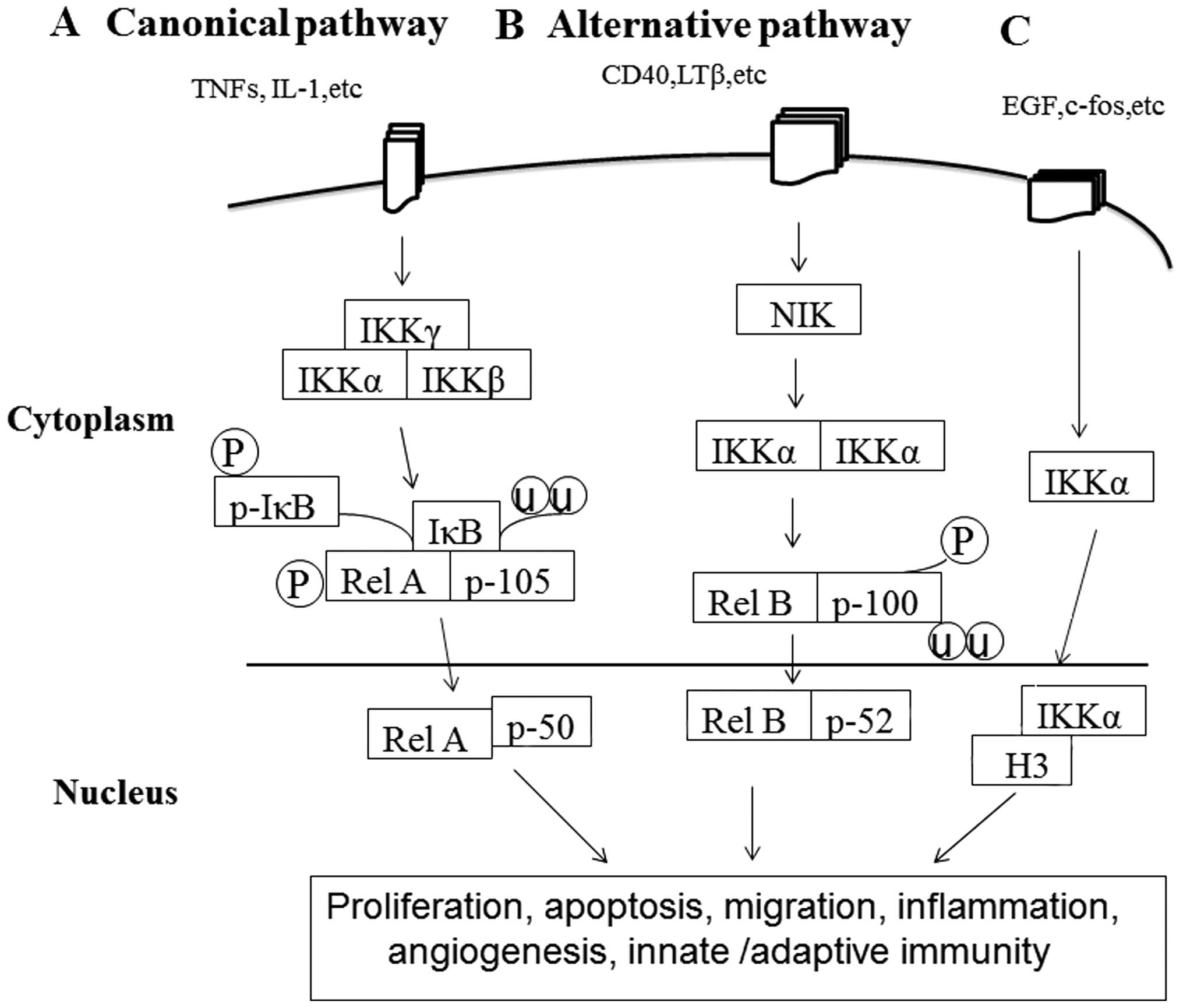 | Figure 1A proposed schematic diagram for
activation of pathways involved in the function of IKKα in cancer
pathogenesis. (A) Agents activating the canonical NF-κB pathway
include aberrant cytokine production, integrins, such as tumor
necrosis factor (TNF)-α, IL-1 and corresponding cytokine receptors.
Stimuli-induced activation of the multiprotein IKK complex results
in the phosphorylation of NH2-terminal serine residues in IκBs and
rapid separation from p105/RELA through a ubiquitin-26 S proteasome
pathway. Subsequently, proteasome degradation of p105 to p50 and
phosphorylation of RelA (p65) is stimulated, leading to nuclear
translocation of the NF-κB1/RelA (p50/p65) complex, which activates
gene expression regulating proliferation, apoptosis, migration,
inflammation, angiogenesis and innate immunity. (B) The alternative
pathway is triggered by other aberrant TNF cytokines and
lymphotoxin β (LTβ). Stimuli-induced activation of kinase NIK
phosphorylates and activates IKKα/IKKα homodimers, leading to
degradation and phosphorylation of NF-κB2/p100 precursor protein to
p52 and nuclear translocation of the p52/RelB complex. The
alternative pathway is the major provider of adaptive immunity in
mammals. (C) IKKα may also translocate to the nucleus and modulate
global levels of histone H3 phosphorylation on Ser10 in response to
mitogenic EGF stimuli. |
3. Role of IKKα in normal epithelial
and skin squamous cell carcinoma (SCC)
IKKα functions as a major regulator in
keratinocyte proliferation and differentiation
Calcium (Ca2+) is essential for the
induction and maintenance of the terminal differentiation status in
the epidermis (14,15). However, primary cultured
IKKα−/− keratinocytes fail to undergo terminal
differentiation, which is also not induced by Ca2+.
Conversely, reintroduction of wild-type (WT) or kinase-inactivated
IKKα induces terminal differentiation of keratinocytes,
while this does not occur when IKKβ is reintroduced, even
though it exhibits a similar structure and function with
IKKα. Furthermore, compared to IKKβ in the cytoplasm
exclusively, IKKα exists both in the cytoplasm and nucleus
(4,16). IKKα shows nuclear
localization in the normal epidermis of humans and mice (3,17). In
the nucleus, IKKα does not exert kinase activity and has a
special role different from that of IKKβ. Moreover, mutant
IKKα is not only incapable of moving to the nucleus, but it
also fails to induce terminal differentiation of
IKKα−/− keratinocytes (17). These findings indicate that nuclear
IKKα regulates keratinocyte proliferation and
Ca2+-dependent differentiation. However, how
Ca2+ is involved in the process of IKKα-induced
keratinocyte terminal differentiation remains to be determined.
Function of IKKα as a tumor suppressor in
skin SCC
Studies have demonstrated that IKKα mutations
are present in human SCC and that a marked reduction in IKKα
expression occurs in poorly differentiated human and mouse
cutaneous SCCs (18). In studies of
chemical carcinogen-induced (19)
or UVB-induced (20–22) skin carcinogenesis, the findings have
shown that lack of IKKα expression promotes the development
of skin papillomas and carcinomas in IKKα+/−
mice. In chemical carcinogen-induced skin carcinogenesis, reduced
levels of IKKα promote the oncogenic H-Ras pathway and
enhance the mitogenic activity, extracellular signal-regulated
kinase (ERK) activity and overexpression of growth factors
(19); and in addition, influence
UVB-related p53 mutations, upregulate the expression of monocyte
chemoattractant protein-1 (MCP-1/CCL2), TNF-α, IL-1 and elevate
macrophage migration, which are crucial for accelerating UVB skin
carcinogenesis (21,23). In contrast, increased IKKα
expression antagonizes mitogenesis and angiogenesis, thus
antagonizing the development of malignant carcinomas and metastases
(Fig. 2A).
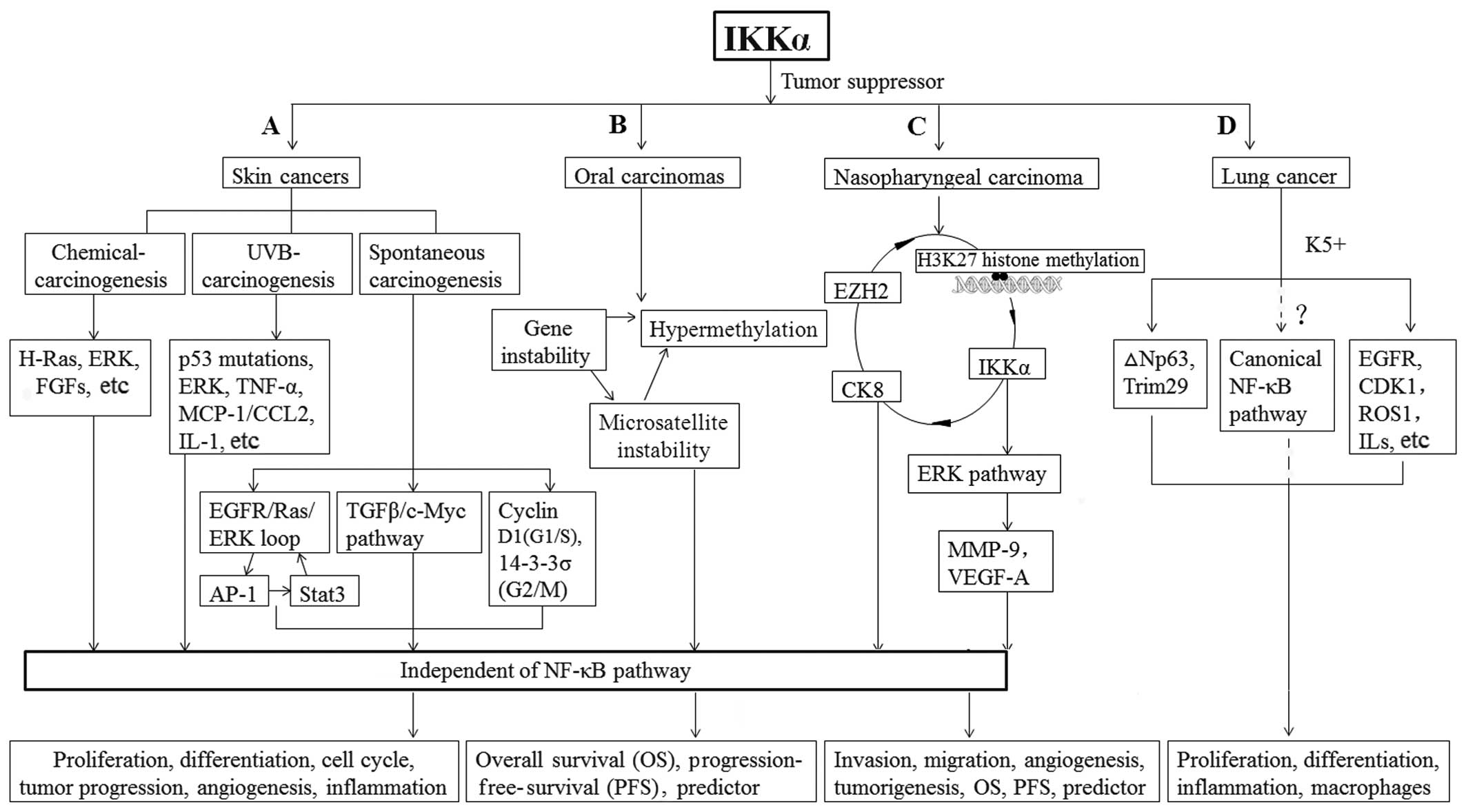 | Figure 2Mechanism of IKKα as a tumor
suppressor in epithelium-derived carcinoma development from tumor
initiation to progression. (A) In chemical carcinogen-induced skin
cancer, reintroduction of IKKα or kinase-inactive IKKα represses
DMBA-induced H-Ras mutations or TPA-induced excessive ERK activity
or excessive expression of EGF and FGFs. Reduction in IKKα
expression promotes expansion of cells carrying UVB-induced-p53
mutations and induces the recruitment of macrophages and
inflammation by upregulating expression of cytokines and
MCP-1/CCL2. In spontaneous carcinogenesis, IKKα suppresses the
transcription of EGFR ligands and their ligand activators,
resulting in inhibition of EGFR and ERK activities. IKKα
cross-talks with EGFR, AP-1 and Stat3 pathways in a loop. Moreover,
IKKα inhibits the TGF-β/Smad2/3/c-Myc pathway and contributes to
the regulation of cyclin D1 in the G1/S and 14-3-3σ in the G2/M
phase to maintain a normal balance in the cell cycle. (B) In oral
carcinoma, the genetic instability of IKKα and hypermethylation of
its CpG islands causes some promoter hypermethylation and
epigenetic inactivation of gene transcription. (C) Reduced EZH2
inhibits H3K27 histone methylation on the IKKα promoter and
relieves IKKα transcriptional repression. Overexpression of IKKα
reduces CK13 and increase CK8 expression to induce epithelial
differentiation. Through another manner, IKKα suppresses NPC
through the ERK pathway by regulating MMP-9. IKKα is independent of
the NF-κB pathway in the above process. (D) In spontaneous lung
SCCs, kinase-dead IKKα knock-in suppresses the expression of p63,
Trim29, K5 and EGFR/ERK activity and CDK1, ROS1, IL-1 levels to
keep balance between cell proliferation and differentiation and
prevents an excessive inflammatory microenvironment. However, the
increased canonical NF-κB pathway contributes to IKKα-related lung
SCC development and this requires further elucidation. |
Overall, IKKα plays an important role in skin
cancer, and the maintenance of an adequate level of IKKα is
essential for protecting the skin from various harmful stimulations
and carcinogen attacks. It is necessary to investigate whether
certain mutations in IKKα convert its function from a tumor
suppressor to an oncogene.
Mechanism of IKKα in IKKα-mediated skin
tumorigenesis
The EGFR/ERK/EGF/HB-EGF/ADAM signaling
pathway
Inactivation of epidermal growth factor receptor
(EGFR) or reintroduction of IKKα prevents
IKKα-induced epidermal hyperplasia and skin cancer
development, indicating a cross-talk between IKKα and EGFR
(3,19). IKKα deletion causes elevation
in EGFR and ERK activity, EGF and HB-EGF levels and the downstream
expression of ADAM sheddases (such as, ADAM9, 10, 12, 17 and 19),
which could cleave EGF and HB-EGF precursors to generate their
active soluble forms (24). These
molecules are activated in the EGFR autocrine loop. The EGFR/Ras
pathway is important for keratinocyte differentiation, regulation
of proliferation and skin tumor development. This autocrine loop in
IKKα-lacking cells could ultimately result in excessive
proliferation and dedifferentiation of the epidermis. Conversely,
reintroduction of IKKα or inactivation of EGFR and/or EGFR
inhibitors, blocks IKKα loss-induced epidermal
hyperproliferation and skin tumors in mice. Furthermore,
IKKα represses the EGFR/Ras/ERK loop via the suppression of
the transcription of genes that encode EGFR ligands (3,25).
In addition, studies have shown that reintroduced
IKKα or inactivated EGFR also represses AP-1 and Stat3,
whose excessive activity accelerates skin tumor development
(26–28). However, the cross-talk between
IKKα and the EGFR, AP-1, and Stat3 pathways in a loop
remains to be investigated (25).
Collectively, IKKα contributes to an EGFR-mediated loop and
represses cell proliferation, resulting in induction of terminal
differentiation in skin keratinocytes (Fig. 2A).
The TGF-β/Smad2/3/c-Myc pathway
The transforming growth factor β (TGF-β)/Smad2/3
pathway is also important for skin homeostasis and skin tumor
development (Fig. 2A). Upon
treatment with TGF-β, IKKα forms a complex with Smad2 or
Smad3 on the promoters of Mad genes (16,29).
Studies have suggested that loss of IKKα downregulates Mad1,
Mad2, Mad3, Mad4 and Ovol1 expression and increases c-Myc activity,
which is associated with cell differentiation in keratinocytes.
Excessive c-Myc activity inhibits the prevention of cell cycle exit
and cell apoptosis, thereby destroying regular cell
differentiation. Moreover, a reduced level of Max/Mad dimers
induces the competition with c-Myc, which can form dimers with Max
as well, thus abnormally enhancing c-Myc/Max dimer-induced cell
proliferation (30,31).
Although TGF-β acts as a tumor suppressor at the
early stage of tumor development and affects cell proliferation,
differentiation and inflammation (32), the levels of TGF-β are high in SCC.
Accumulating evidence has revealed that TGF-β plays a bidirectional
role in cancer progression (33,34).
Studies suggest that TGF-β works as a tumor promoter by modifying
tumor promoter-induced cell proliferation and inflammation,
stimulating angiogenesis and promoting epithelial-mesenchymal
transition (EMT) (35). In skin
cancer, TGF-β fails to exert its tumor suppressor activity in
IKKα-null or IKKα-mutated tumor cells, owing to the
fact that IKKα is a major target of mutagenesis in
tumorigenesis. Thus, IKKα is crucial for
TGF-β-induced tumor suppressor activity during skin
tumorigenesis.
The role of IKKα in the cell cycle
IKKα and cyclin D1
Several studies suggest that IKKα regulates
the turnover and subcellular distribution of cyclin D1 by inducing
its phosphorylation of cyclin D1 (36) (Fig.
2A). Cyclin D1 is an important regulator of cell-cycle
initiation and G1/S transition. Cyclin D1 binds to Cdk4 and Cdk6 to
form a pRB kinase, which phosphorylates and deactivates the
tumor-suppressor protein pRB. Upon phosphorylation, pRB loses its
repressive activity for the E2F transcription factor, which
activates the transcription of several genes required for
cell-cycle initiation and G1/S transition. Furthermore,
overexpression of cyclin D1 plays a critical role in tumor
development through stimulating various growth factors, such as
anchorage-independent growth and vascular endothelial growth factor
(VEGF) (37).
IKKα is critical for the phosphorylation of
cyclin D1 at the T286 residue, which is indispensable for the
transport of the protein out of the nucleus during the S phase of
the cell cycle and for its degradation (38). IKKα−/−cells have
cyclin D1 in the nucleus and therefore, abnormally activate CDK4/6
kinase activity and pRb phosphorylation, leading to the
deregulation of the G1/S checkpoint. This process may be enhanced
by the presence of cyclin D1 mutations (39); at the same the T286 residue remains
in the nucleus and promote tumorigenesis.
IKKα and 14-3-3σ
14-3-3σ, as a G2/M cell cycle checkpoint, regulates
the cell cycle and allows cells to repair genetic errors, thus
preventing mutagenesis and genomic instability (40). It is specifically and highly
expressed in keratinocytes and other epithelial cells. A large
number of studies have shown that 14-3-3σ is silenced in
many human epithelial cancer cells. Studies show that the level of
14-3-3σ is low in IKKα−/− cells but is restored
after the reintroduction of IKKα (Fig. 2A). Furthermore, the protein is a
downstream target of IKKα in the pathway of
IKKα-mediated cell cycle regulation and functions in
response to DNA damage (41,42).
Moreover, 14-3-3σ is also a target of IKKα
for maintaining genomic stability. 14-3-3σ contains many CpG
islands. Its hypermethylation and loss of final σ expression
are the most consistent molecular alterations in malignancies.
Studies have confirmed that 14-3-3σ CpG islands are
hypermethylated in IKKα−/− keratinocytes, which
is associated with trimethyl-H3-K9, a protein essential to DNA
methylation. An entire kinase domain (300 aa) deletion of
IKKα does not permit IKKα to bind to the N-terminal
tail of H3, which fails to prevent trimethylation of H3-K9 at the
14-3-3σ and fails to induce the expression of 14-3-3σ.
Conversely, binding of IKKα to H3 probably prevents H3-K9
trimethylation, thereby shielding the 14-3-3σ locus from
hypermethylation and maintaining genomic stability. Thus,
IKKα plays an important role in epigenetic regulatory
mechanisms of 14-3-3σ for cancer prevention (41). However, further studies may be
necessary to unveil whether IKKα utilizes this mechanism to
regulate a series of genes, providing the foundation for exploiting
its hypermethylation as a marker for the early detection of
malignancies.
Therefore, IKKα-mediated cell cycle
regulation mechanism via cyclinD1 or 14-3-3σ is important for
prevention of skin tumors.
4. The role of IKKα in lung SCC
tumorigenesis
Studies have revealed a decrease in IKKα RNA
expression level in human lung cancer cell lines (4). Recent findings have demonstrated the
pivotal role of IKKα in the development of spontaneous lung
SCC. Studies have established a kinase-dead IKKα knock-in
(IKKαK44A/K44A, IKKαKA/KA)
mouse model, in which the lysine (K) at amino acid 44, the
ATP-binding site, was replaced with alanine (A), to develop
spontaneous lung SCCs to determine the role of IKKα in
normal bronchial epithelium and its related carcinomas. The mice
did not display any obvious abnormalities at the time of birth,
indicating that IKKα kinase inactivation does not affect
mouse embryonic development. However, spontaneous lung tumors,
whose weight was timely increased, appeared in
IKKαKA/KA mice from 4 to 10 months of age and the
animals began to die after 6–10 months. In that case, IKKα
levels in the lungs of IKKαKA/KA newborns were
low and markedly decreased at 4 months of age. Therefore, reduction
in IKKα levels contributed to the development of spontaneous
lung tumors. Furthermore, atypical squamous hyperplasia development
in the fore-stomach, esophagus and skin was also investigated in
the IKKα-deficient mice. However, reintroduction of the
IKKα transgene prevented the development of the tumors
(43). These results demonstrated
that high levels of IKKα prevent lung tumor development.
Some studies suggest that lung SCCs are derived from
keratin 5-positive (K5+) basal cells of the
pseudostratified bronchial epithelium. K5+ epithelial
cells lacking IKKα may be targets for SCC development
(4,44). IKKαKA/KA mice
expressed high levels of K5, as well as transcription factor p63,
tripartite motif-containing 29 (Trim29) proteins, and Ki67, which
was similar to the expression profile in human lung SCCs (43).
p63, a member of the tumor suppressor
p53 family, is required for the formation of the epidermis,
other stratified epithelia and epithelial appendages (45). The N-terminal-truncated form of p63
(∆Np63) is predominately expressed in the epidermis and is
overexpressed in various epithelial cancers, where it exerts
oncogenic activities (46). In
addition, Trim29 overexpression has been studied in human
lung, bladder, colon, ovarian, endometrial and gastric cancers. In
these cell types, Trim29 promotes cell proliferation and inhibits
p53 activity (47,48). These findings highlighted that
increased epithelial cell-specific p63 and Trim29 may
contribute to lung SCC development. Compared to WT mice, ∆Np63 and
Trim29 protein levels were increased in the lungs of
IKKαKA/KA mice and further increased in the lung
affected by SCCs. Chromatin immunoprecipitation (ChIP) assay was
used to explain how IKKα regulates the expression of both
Trim29 and ∆Np63. The results showed that nuclear IKKα bound
to the promoter regions of Trim29 and p63, which was
associated with high levels of H3 lysine 27 trimethylation
(H3K27me3), a negative transcription modifier and low levels of H3
lysine 4 trimethylation (H3K4me3), a positive transcription
modifier, thus leading to the control of their expression. This
process occurred both in human and mouse lung epithelial cells
(43,49,50).
In consequence, nuclear IKKα regulates the expression of
both Trim29 and ∆Np63 at the transcription level by modifying the
chromatin structure of Trim29 and p63 in an
epigenetic manner to prevent tumor development (Fig. 2D).
In addition, the levels of insulin growth factor 1
(IGF-1), cyclin-dependent kinase 1 (CDK1) and the activities of
EGFR, ERK and p38 were elevated in the lungs of
IKKαKA/KA mice and were considerably increased in
SCC compared to WT lungs, which is similarly to human. Excessive
inflammatory cells and the microenvironment, cytokines and
chemokines were also present in the lungs of
IKKαKA/KA mice. Increased expression levels of
tumor necrosis factor (TNF)-α, interleukin (IL)-1b, IL-6, chemokine
(C-C motif) ligand 2 (CCL2), chemokine (C-X-C motif) ligand 5,
CCL11 and CCL8 were observed in the lungs of
IKKαKA/KA mice as well. Excessive amounts of
macrophages also caused increased inflammation, epithelial cell
proliferation, DNA damage and activity of many pathways that may
contribute to lung carcinogenesis in IKKαKA/KA
mice (43,51) (Fig.
2D).
Unlike the malfunction of the nuclear factor κB
(NF-κB) pathway in skin cancer (52), increased activity of the canonical
NF-κB pathway was found to contribute to lung SCC development in
IKKαKA/KA mice. Whether the NF-κB pathway can be
used as a therapeutic target to prevent and treat lung SCCs remains
to be elucidated (Fig. 2D).
5. The novel role of IKKα in head and
neck cancer
IKKα and oral carcinomas
Since IKKα is expressed in normal oral
keratinocytes but its expression is frequently decreased in
carcinoma cells, further studies have been conducted to investigate
the role of IKKα in oral carcinoma tissues and analyze its
prognostic significance in comparison with clinicopathological
parameters and patient survival. Results indicated that IKKα
levels decreased in 44.9% of carcinoma patients and the expression
of the protein was closely associated with progression-free
survival (PFS), independent of other risk factors. Therefore,
IKKα was considered to be a significant independent
predictor of mortality due to carcinomas (53).
In vitro, IKKα is located in the nucleus and
is upregulated upon differentiation in oral carcinomas.
Furthermore, IKKα shows genetic instability in its locus and
hypermethylation of its CpG islands (53). Studies have also shown that the
genetic instability is frequently associated with promoter
hypermethylation, resulting in epigenetic inactivation of gene
transcription (54). Otherwise,
there is a close correlation between the promoter hypermethylation
and microsatellite instability (MSI). MSI, also referred to as
simple sequence repeats (SSRs), is originally associated with DNA
mismatch repair (MMR), which plays a prominent role in the
correction of errors made during DNA replication, genetic
recombination and in the repair of small deletions and loops in
DNA. Moreover, it closely associates with gene silencing through
promoter hypermethylation. Thus, MSI may be important in
IKKα-related oral tumorigenesis (55,56).
In fact, most of the MSI carcinomas were methylated in a specific
site critical for IKKα expression, which is important for
the activity of IKKα as a tumor-suppressor gene in oral
carcinoma regulation (Fig. 2B).
However, since in some carcinomas, the methylation status did not
correlate with immunoreactivity, other mechanisms regulating the
gene expression remain to be investigated (53,57).
Similar to skin cancer, in oral carcinoma, IKKα in the
nucleus suppresses malignancy by acting on cell differentiation
independent of canonical NF-κB activation in oral carcinoma.
IKKα and nasopharyngeal carcinoma
Nasopharyngeal carcinoma (NPC) is one of the common
epithelium-derived carcinomas in the head and neck. The effect of
IKKα as the tumor suppressor in NPC cells or the survival of
NPC patients has recently been investigated.
IKKα is upregulated upon differentiation in
NPC cell lines in vitro. Differential expression of
IKKα exists in NPC cells, which is negatively correlated
with invasiveness, migration and angiogenesis, but not with
proliferation. Lack of IKKα could lead to increased cancer
invasion, migration and angiogenesis; on the contrary,
reintroduction of IKKα abrogated these biological behaviors
(58). Moreover, IKKα
inhibited tumorigenesis in mice inoculated with
IKKα-transfected NPC cells in vivo. These processes
were found to be independent of IKKα kinase activity and the
conventional effect of IKKα on NF-κB pathways (58,65).
Furthermore, this study (58) showed that 26.8% of a total of 157
NPC patients exhibited low expression or deletion of IKKα.
Although the IKKα expression levels had no correlation with
tumor stage, recurrence, distant metastasis, age and gender, the
survival (OS or DFS) of NPC patients was significantly associated
with IKKα expression. Similar to T stage, lymph node
metastasis, locoregional recurrence and distant metastasis,
IKKα expression significantly influenced clinical prognosis.
Its high expression could serve as an independent favorable
predictor for NPC patients.
Similar to oral carcinoma cell lines, the CpG
islands of the IKKα gene were heavily methylated in NPC cell
lines and clinical specimens. Studies have shown that increased
IKKα expression could induce differentiation and prevent NPC
development by reducing enhancer of zeste homologue 2
(EZH2)-related H3K27 histone methylation of the IKKα
promoter (59). Importantly,
overexpression of IKKα results in fusiform morphological
change, reduction in CK13 and increase in involucrin and CK8
without activating the NF-κB pathway, leading to differentiation of
NPC cells (59–61). These findings suggest an important
role of IKKα for NPC differentiation in an epigenetic
mechanism.
Furthermore, what is the downstream signaling
pathway or mechanism that regulates this newly recognized
suppressive effect of IKKα on NPC? Findings have shown that
IKKα inhibits NPC development through inactivation of ERK1/2
and its phosphorylation (pERK1/2) in the ERK pathway, but it is
independent of EGFR, although it usually acts as the upstream
protein in the ERK pathway, thus resulting in aberrant activation
of the ERK signaling pathway (65).
This finding is consistent with the malfunction of IKKα in
the proliferation of NPC cell lines (62). Furthermore, matrix
metalloproteinase-9 (MMP-9), but not MMP-2, is an essential
downstream molecule in the IKKα-related ERK pathway for repressing
NPC progression (Fig. 2C).
EBV is ubiquitously associated with the
carcinogenesis of NPC (63). In
addition, research has shown that Epstein-Barr nuclear antigen 1
(EBNA1) inhibits the canonical NF-κB pathway and contributes to the
pathogenesis of NPC, by inhibiting IKKα/IKKβ phosphorylation
(64). Thus, the correlation
between IKKα, EBNA1 and NF-κB in NPC was investigated,
although the expression of EBNA1 was high and undifferentiated
expressed in all EBV-positive NPC cell lines. Studies strongly
suggest that IKKα plays a crucial role as a tumor suppressor
in NPC and is not related to tumor-associated protein EBNA1
(65).
6. A different role of IKKα in
hormone-related breast and prostate cancer
As stated above, IKKα plays a pivotal role
as a tumor suppressor in epithelial-derived tumors, particularly in
SCCs; however, it has been determined that IKKα also
functions as a tumor promoter in hormone-related breast (66) and prostate cancers (67). One study showed that it provides an
important linking effect between RANK signaling and cyclin D1
expression in the development of the mammary gland (68). It was also demonstrated that the
C57BL/6 female mouse expresses a mutant IKKαAA
knock-in allele in which alanine replaces serine to inactivate
IKKα kinase activity, impairing the proliferation of mammary
epithelial cells and thus leading to a severe lactation defect.
However, this defect is completely reversed by the reintroduction
of a mammary specific cyclin D1 in the
IKKαAA/AA mammary epithelium. Furthermore, it has
been found that IKKα kinase activity is involved in cyclin
D1 expression and this process is activated by RANK ligand (RANKL),
an efficient NF-κB activator (68).
In breast cancer, IKKα phosphorylates ERα
and the nuclear hormone receptor co-activator AIB1/SRC-3 (amplified
in breast 1/steroid receptor coactivator-3) and thus activates the
transcription of estrogen-responsive genes, including cyclin
D1 and c-myc, to enhance proliferation of ER(+) breast
cancer cells (66). In addition,
another finding showed that the epidermal growth factor receptor
(Her-2) activated IKKα to induce tumor invasion through the
NF-κB canonical pathway in HER2+/ER− breast
cancer cells (69).
Except for breast cancer, a study demonstrated that
nuclear IKKα activation by the RANK ligand inhibits maspin
expression and leads to metastatic progression of mouse and human
prostate cancers (67). Therefore,
IKKα functions as an oncogenic factor, in conjunction with
the hormone-related genes, to contribute to the development of
breast and prostate cancers through NF-κB-dependent or -independent
pathways.
7. Conclusions
As a novel and potential suppressor, IKKα
activates or inactivates comprehensive pathways that prevent the
development of a variety of epithelium-derived carcinomas from
tumor initiation to progression (Fig.
2). Except for the cytoplasmic effect of IKKα on the
NF-κB pathway, the role of nuclear IKKα is much more
essential to epithelium-derived carcinomas. Although an increasing
amount of research has explored the function and mechanism of
IKKα in such carcinomas, the cross-talk among them has not
yet been established. It is still unclear whether IKKα
deletion targets stage-specific squamous epithelial cells to
initiate tumors or it turns on oncogenes or represses tumor
suppressors and whether IKKα plays a role in stem cell
maintenance, renewal or division. On the other hand, IKKα is
also suggested as a tumor promoter. Therefore, whether IKKα
functions differently in different types of tumors warrants further
investigation. Collectively, IKKα is a major regulator to
participate in the downstream gene cascade, leading to repression
of cell hyperproliferation, angiogenesis, epithelial-mesenchymal
transition and malignant progression. Furthermore, IKKα may
be a potential therapeutic strategy for preventing carcinoma.
Acknowledgments
The present study was supported by a grant from the
National Natural Science Foundation of China (no. 81072217) (to
C.N.).
Abbreviations:
|
IKKα
|
IκB kinase α
|
|
NF-κB
|
nuclear factor-κB
|
|
TSGs
|
tumor-suppressor genes
|
|
HLH
|
helix-loop-helix
|
|
SCC
|
squamous cell carcinoma
|
|
ERK
|
extracellular signal-regulated
kinase
|
|
MCP-1
|
monocyte chemoattractant
protein-1
|
|
EGFR
|
epidermal growth factor receptor
|
|
TGF-β
|
transforming growth factor β
|
|
EMT
|
epithelial-mesenchymal transition
|
|
VEGF
|
vascular endothelial growth
factor
|
|
CDK1
|
cyclin-dependent kinase 1
|
|
PFS
|
progression-free-survival
|
|
MSI
|
microsatellite instability
|
|
SSRs
|
simple sequence repeats
|
|
MMR
|
mismatch repair
|
|
NPC
|
nasopharyngeal carcinoma
|
|
MMP-9
|
matrix metalloproteinase-9
|
|
EBNA1
|
Epstein-Barr nuclear antigen 1
|
|
AIB1/SRC-3
|
amplified in breast 1/steroid
receptor coactivator-3
|
|
RANKL
|
RANK ligand
|
References
|
1
|
Berger AH, Knudson AG and Pandolfi PP: A
continuum model for tumour suppression. Nature. 476:163–169. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Stovall DB, Cao P and Sui G: SOX7: From a
developmental regulator to an emerging tumor suppressor. Histol
Histopathol. 29:439–445. 2014.
|
|
3
|
Liu B, Xia X, Zhu F, Park E, Carbajal S,
Kiguchi K, DiGiovanni J, Fischer SM and Hu Y: IKKalpha is required
to maintain skin homeostasis and prevent skin cancer. Cancer Cell.
14:212–225. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kwak YT, Radaideh SM, Ding L, Li R,
Frenkel E, Story MD, Girard L, Minna J and Verma UN: Cells lacking
IKKalpha show nuclear cyclin D1 overexpression and a neoplastic
phenotype: Role of IKKalpha as a tumor suppressor. Mol Cancer Res.
9:341–349. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Marinari B, Ballaro C, Koster MI,
Giustizieri ML, Moretti F, Crosti F, Papoutsaki M, Karin M, Alema
S, Chimenti S, et al: IKKalpha is a p63 transcriptional target
involved in the pathogenesis of ectodermal dysplasias. J Invest
Dermatol. 129:60–69. 2009. View Article : Google Scholar
|
|
6
|
Marinari B, Moretti F, Botti E,
Giustizieri ML, Descargues P, Giunta A, Stolfi C, Ballaro C,
Papoutsaki M, Alemà S, et al: The tumor suppressor activity of
IKKalpha in stratified epithelia is exerted in part via the
TGF-beta antiproliferative pathway. Proc Natl Acad Sci USA.
105:17091–17096. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
McKenzie FR, Connelly MA, Balzarano D,
Muller JR, Geleziunas R and Marcu KB: Functional isoforms of
IkappaB kinase alpha (IKKalpha) lacking leucine zipper and
helix-loop-helix domains reveal that IKKalpha and IKKbeta have
different activation requirements. Mol Cell Biol. 20:2635–2649.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Connelly MA and Marcu KB: CHUK, a new
member of the helix-loop-helix and leucine zipper families of
interacting proteins, contains a serine-threonine kinase catalytic
domain. Cell Mol Biol Res. 41:537–549. 1995.PubMed/NCBI
|
|
9
|
Ghosh S and Karin M: Missing pieces in the
NF-kappaB puzzle. Cell. 109(Suppl): S81–S96. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sakurai H, Suzuki S, Kawasaki N, Nakano H,
Okazaki T, Chino A, Doi T and Saiki I: Tumor necrosis
factor-alpha-induced IKK phosphorylation of NF-kappaB p65 on serine
536 is mediated through the TRAF2, TRAF5, and TAK1 signaling
pathway. J Biol Chem. 278:36916–36923. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Van Waes C, Yu M, Nottingham L and Karin
M: Inhibitor-kappaB kinase in tumor promotion and suppression
during progression of squamous cell carcinoma. Clin Cancer Res.
13:4956–4959. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Van Waes C: Nuclear factor-kappaB in
development, prevention, and therapy of cancer. Clin Cancer Res.
13:1076–1082. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Anest V, Cogswell PC and Baldwin AS Jr:
IkappaB kinase alpha and p65/RelA contribute to optimal epidermal
growth factor-induced c-fos gene expression independent of
IkappaBalpha degradation. J Biol Chem. 279:31183–31189. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Elias PM, Ahn SK, Denda M, Brown BE,
Crumrine D, Kimutai LK, Kömüves L, Lee SH and Feingold KR:
Modulations in epidermal calcium regulate the expression of
differentiation-specific markers. J Invest Dermatol. 119:1128–1136.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu B, Zhu F, Xia X, Park E and Hu Y: A
tale of terminal differentiation: IKKalpha, the master keratinocyte
regulator. Cell Cycle. 8:527–531. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu B, Park E, Zhu F, Bustos T, Liu J,
Shen J, Fischer SM and Hu Y: A critical role for I kappaB kinase
alpha in the development of human and mouse squamous cell
carcinomas. Proc Natl Acad Sci USA. 103:17202–17207. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sil AK, Maeda S, Sano Y, Roop DR and Karin
M: IkappaB kinase-alpha acts in the epidermis to control skeletal
and craniofacial morphogenesis. Nature. 428:660–664. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Park E, Liu B, Xia X, Zhu F, Jami WB and
Hu Y: Role of IKKalpha in skin squamous cell carcinomas. Future
Oncol. 7:123–134. 2011. View Article : Google Scholar
|
|
19
|
Park E, Zhu F, Liu B, Xia X, Shen J,
Bustos T, Fischer SM and Hu Y: Reduction in IkappaB kinase alpha
expression promotes the development of skin papillomas and
carcinomas. Cancer Res. 67:9158–9168. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhu F, Park E, Liu B, Xia X, Fischer SM
and Hu Y: Critical role of IkappaB kinase alpha in embryonic skin
development and skin carcinogenesis. Histol Histopathol.
24:265–271. 2009.
|
|
21
|
Xia X, Park E, Liu B, Willette-Brown J,
Gong W, Wang J, Mitchell D, Fischer SM and Hu Y: Reduction of
IKKalpha expression promotes chronic ultraviolet B exposure-induced
skin inflammation and carcinogenesis. Am J Pathol. 176:2500–2508.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Peinado C, Kang X, Hardamon C, Arora S,
Mah S, Zhang H, Ngolab J and Bui JD: The nuclear factor-kappaB
pathway down-regulates expression of the NKG2D ligand H60a in
vitro: Implications for use of nuclear factor-kappaB inhibitors in
cancer therapy. Immunology. 139:265–274. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fujimoto H, Sangai T, Ishii G, Ikehara A,
Nagashima T, Miyazaki M and Ochiai A: Stromal MCP-1 in mammary
tumors induces tumor-associated macrophage infiltration and
contributes to tumor progression. Int J Cancer. 125:1276–1284.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Huovila AP, Turner AJ, Pelto-Huikko M,
Karkkainen I and Ortiz RM: Shedding light on ADAM
metalloproteinases. Trends Biochemical Sci. 30:413–422. 2005.
View Article : Google Scholar
|
|
25
|
Liu S, Chen Z, Zhu F and Hu Y: IkappaB
kinase alpha and cancer. J Interferon Cytokine Res. 32:152–158.
2012. View Article : Google Scholar :
|
|
26
|
Liu B, Willette-Brown J, Liu S, Chen X,
Fischer SM and Hu Y: IKKalpha represses a network of inflammation
and proliferation pathways and elevates c-Myc antagonists and
differentiation in a dose-dependent manner in the skin. Cell Death
Differ. 18:1854–1864. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zenz R, Eferl R, Scheinecker C, Redlich K,
Smolen J, Schonthaler HB, Kenner L, Tschachler E and Wagner EF:
Activator protein 1 (Fos/Jun) functions in inflammatory bone and
skin disease. Arthritis Res Ther. 10:2012008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sano S, Chan KS and DiGiovanni J: Impact
of Stat3 activation upon skin biology: A dichotomy of its role
between homeostasis and diseases. J Dermatol Sci. 50:1–14. 2008.
View Article : Google Scholar
|
|
29
|
Descargues P, Sil AK, Sano Y, Korchynskyi
O, Han G, Owens P, Wang XJ and Karin M: IKKalpha is a critical
coregulator of a Smad4-independent TGFbeta-Smad2/3 signaling
pathway that controls keratinocyte differentiation. Proc Natl Acad
Sci USA. 105:2487–2492. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gandarillas A: The mysterious human
epidermal cell cycle, or an oncogene-induced differentiation
checkpoint. Cell Cycle. 11:4507–4516. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pulverer B, Sommer A, McArthur GA,
Eisenman RN and Luscher B: Analysis of Myc/Max/Mad network members
in adipogenesis: Inhibition of the proliferative burst and
differentiation by ectopically expressed Mad1. J Cell Physiol.
183:399–410. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Drabsch Y and ten Dijke P: TGF-β
signalling and its role in cancer progression and metastasis.
Cancer Metastasis Rev. 31:553–568. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mordasky Markell L, Perez-Lorenzo R,
Masiuk KE, Kennett MJ and Glick AB: Use of a TGFbeta type I
receptor inhibitor in mouse skin carcinogenesis reveals a dual role
for TGFbeta signaling in tumor promotion and progression.
Carcinogenesis. 31:2127–2135. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ikushima H and Miyazono K: TGFbeta
signalling: A complex web in cancer progression. Nat Rev Cancer.
10:415–424. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ravindran A, Mohammed J, Gunderson AJ, Cui
X and Glick AB: Tumor-promoting role of TGFbeta1 signaling in
ultraviolet B-induced skin carcinogenesis is associated with
cutaneous inflammation and lymph node migration of dermal dendritic
cells. Carcinogenesis. 35:959–966. 2014. View Article : Google Scholar :
|
|
36
|
Kwak YT, Li R, Becerra CR, Tripathy D,
Frenkel EP and Verma UN: IkappaB kinase alpha regulates subcellular
distribution and turnover of cyclin D1 by phosphorylation. J Biol
Chem. 280:33945–33952. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tashiro E, Tsuchiya A and Imoto M:
Functions of cyclin D1 as an oncogene and regulation of cyclin D1
expression. Cancer Sci. 98:629–635. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Diehl JA, Cheng M, Roussel MF and Sherr
CJ: Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis
and subcellular localization. Genes Dev. 12:3499–3511. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Alt JR, Cleveland JL, Hannink M and Diehl
JA: Phosphorylation-dependent regulation of cyclin D1 nuclear
export and cyclin D1-dependent cellular transformation. Genes Dev.
14:3102–3114. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chan TA, Hwang PM, Hermeking H, Kinzler KW
and Vogelstein B: Cooperative effects of genes controlling the
G(2)/M checkpoint. Genes Dev. 14:1584–1588. 2000.PubMed/NCBI
|
|
41
|
Zhu F, Xia X, Liu B, Shen J and Hu Y,
Person M and Hu Y: IKKalpha shields 14-3-3sigma, a G(2)/M cell
cycle checkpoint gene, from hypermethylation, preventing its
silencing. Mol Cell. 27:214–227. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Dellambra E, Golisano O, Bondanza S,
Siviero E, Lacal P, Molinari M, D'Atri S and De Luca M:
Downregulation of 14-3-3sigma prevents clonal evolution and leads
to immortalization of primary human keratinocytes. J Cell Biol.
149:1117–1130. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Xiao Z, Jiang Q, Willette-Brown J, Xi S,
Zhu F, Burkett S, Back T, Song NY, Datla M and Sun Z: The pivotal
role of IKKalpha in the development of spontaneous lung squamous
cell carcinomas. Cancer Cell. 23:527–540. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hackett NR, Shaykhiev R, Walters MS, Wang
R, Zwick RK, Ferris B, Witover B, Salit J and Crystal RG: The human
airway epithelial basal cell transcriptome. PloS One. 6:e183782011.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ye S, Lee KB, Park MH, Lee JS and Kim SM:
p63 regulates growth of esophageal squamous carcinoma cells via the
Akt signaling pathway. Int J Oncol. 44:2153–2159. 2014.PubMed/NCBI
|
|
46
|
Koster MI, Dai D, Marinari B, Sano Y,
Costanzo A, Karin M and Roop DR: p63 induces key target genes
required for epidermal morphogenesis. Proc Natl Acad Sci USA.
104:3255–3260. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Cambiaghi V, Giuliani V, Lombardi S,
Marinelli C, Toffalorio F and Pelicci PG: TRIM proteins in cancer.
Adv Exp Med Biol. 770:77–91. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Sho T, Tsukiyama T, Sato T, Kondo T, Cheng
J, Saku T, Asaka M and Hatakeyama S: TRIM29 negatively regulates
p53 via inhibition of Tip60. Biochim Biophys Acta. 1813:1245–1253.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Hayashi A, Yamauchi N, Shibahara J, Kimura
H, Morikawa T, Ishikawa S, Nagae G, Nishi A, Sakamoto Y and Kokudo
N: Concurrent activation of acetylation and tri-methylation of
H3K27 in a subset of hepatocellular carcinoma with aggressive
behavior. PloS One. 9:e913302014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Tie F, Banerjee R, Saiakhova AR, Howard B,
Monteith KE, Scacheri PC, Cosgrove MS and Harte PJ: Trithorax
monomethylates histone H3K4 and interacts directly with CBP to
promote H3K27 acetylation and antagonize Polycomb silencing.
Development. 141:1129–1139. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ring BZ, Seitz RS, Beck RA, Shasteen WJ,
Soltermann A, Arbogast S, Robert F, Schreeder MT and Ross DT: A
novel five-antibody immunohistochemical test for subclassification
of lung carcinoma. Mod Pathol. 22:1032–1043. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Hu Y, Baud V, Oga T, Kim KI, Yoshida K and
Karin M: IKKalpha controls formation of the epidermis independently
of NF-kappaB. Nature. 410:710–714. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Maeda G, Chiba T, Kawashiri S, Satoh T and
Imai K: Epigenetic inactivation of IkappaB Kinase-alpha in oral
carcinomas and tumor progression. Clin Cancer Res. 13:5041–5047.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Choi JD and Lee JS: Interplay between
epigenetics and genetics in cancer. Genomics Inform. 11:164–173.
2013. View Article : Google Scholar
|
|
55
|
Ahuja N, Mohan AL, Li Q, Stolker JM,
Herman JG, Hamilton SR, Baylin SB and Issa JP: Association between
CpG island methylation and microsatellite instability in colorectal
cancer. Cancer Res. 57:3370–3374. 1997.PubMed/NCBI
|
|
56
|
Bairwa NK, Saha A, Gochhait S, Pal R,
Gupta V and Bamezai RN: Microsatellite instability: an indirect
assay to detect defects in the cellular mismatch repair machinery.
Methods Mol Biol. 1105:497–509. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Gu L, Zhu N, Findley HW, Woods WG and Zhou
M: Identification and characterization of the IKKalpha promoter:
Positive and negative regulation by ETS-1 and p53, respectively. J
Biol Chem. 279:52141–52149. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Deng L, Li Y, Ai P, Xie Y, Zhu H and Chen
N: Increase in IkappaB kinase alpha expression suppresses the tumor
progression and improves the prognosis for nasopharyngeal
carcinoma. Mol Carcinog. 54:156–165. 2015. View Article : Google Scholar
|
|
59
|
Yan M, Zhang Y, He B, Xiang J, Wang ZF,
Zheng FM, Xu J, Chen MY, Zhu YL, Wen HJ, et al: IKKalpha
restoration via EZH2 suppression induces nasopharyngeal carcinoma
differentiation. Nat Commun. 5:36612014. View Article : Google Scholar
|
|
60
|
van Dorst EB, van Muijen GN, Litvinov SV
and Fleuren GJ: The limited difference between keratin patterns of
squamous cell carcinomas and adenocarcinomas is explicable by both
cell lineage and state of differentiation of tumour cells. J Clin
Pathol. 51:679–684. 1998. View Article : Google Scholar
|
|
61
|
Huang WG, Cheng AL, Chen ZC, Peng F, Zhang
PF, Li MY, Li F, Li JL, Li C, Yi H, et al: Targeted proteomic
analysis of 14-3-3sigma in nasopharyngeal carcinoma. Int J Biochem
Cell Biol. 42:137–147. 2010. View Article : Google Scholar
|
|
62
|
Sullu Y, Demirag GG, Yildirim A, Karagoz F
and Kandemir B: Matrix metalloproteinase-2 (MMP-2) and MMP-9
expression in invasive ductal carcinoma of the breast. Pathol Res
Pract. 207:747–753. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Busson P, Ooka T and Corbex M:
Nasopharyngeal carcinomas and Epstein-Barr virus: From epidemiology
and detection to therapy. Med Sci (Paris). 20:453–457. 2004.In
French. View Article : Google Scholar
|
|
64
|
Valentine R, Dawson CW, Hu C, Shah KM,
Owen TJ, Date KL, Maia SP, Shao J, Arrand JR and Young LS:
Epstein-Barr virus-encoded EBNA1 inhibits the canonical NF-kappaB
pathway in carcinoma cells by inhibiting IKK phosphorylation. Mol
Cancer. 9:12010. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Xie Y, Li Y, Peng X, Henderson F Jr, Deng
L and Chen N: Ikappa B kinase alpha involvement in the development
of nasopharyngeal carcinoma through a NF-kappaB-independent and
ERK-dependent pathway. Oral Oncol. 49:1113–1120. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Park KJ, Krishnan V, O'Malley BW, Yamamoto
Y and Gaynor RB: Formation of an IKKalpha-dependent transcription
complex is required for estrogen receptor-mediated gene activation.
Mol Cell. 18:71–82. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Luo JL, Tan W, Ricono JM, Korchynskyi O,
Zhang M, Gonias SL, Cheresh DA and Karin M: Nuclear
cytokine-activated IKKalpha controls prostate cancer metastasis by
repressing Maspin. Nature. 446:690–694. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Cao Y, Bonizzi G, Seagroves TN, Greten FR,
Johnson R, Schmidt EV and Karin M: IKKalpha provides an essential
link between RANK signaling and cyclin D1 expression during mammary
gland development. Cell. 107:763–775. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Merkhofer EC, Cogswell P and Baldwin AS:
Her2 activates NF-kappaB and induces invasion through the canonical
pathway involving IKKalpha. Oncogene. 29:1238–1248. 2010.
View Article : Google Scholar :
|














