Introduction
Breast cancer is a common cancer in women worldwide.
Many physiological conditions, including hormone secretion and
metabolic homeostasis, influence breast cancer progression
(1). Breast cancer patients with
metabolic dysregulation are associated with poor response to
current chemotherapy (2). The
growth of tumor cells is coupled by metabolic reprogramming
(3,4). The metabolic shift is observed during
carcinogenesis and has been considered to be a reliable marker for
tumors. The intermediary metabolism can also fuel cell growth. For
example, cancer cells are addicted to glutamine due to its usage as
a supplement. The glucose metabolites, serine and glycine, mediate
one-carbon metabolism which is important in tumorigenesis (5,6). The
semi-essential amino acid arginine plays an important role in
nitric oxide production and the urea cycle, and is a precursor for
glutamate, proline, polyamones and agmatine (7). The concentration of plasma arginine is
lower in breast, colon and pancreatic cancer patients (8–10). A
dietary supplement with arginine increases colonic carcinogenesis
(11). Compared to breast cancer
patients fed a standard diet, patients with dietary L-arginine
supplementation have higher tumor protein synthesis. Furthermore,
the protein synthesis rate was found to be highly correlated with
Ki67 expression (12). In contrast,
deprivation of dietary arginine inhibits cancer metastasis
(13,14). Arginine depletion by ADI is used as
an approach in cancer therapy (15,16).
Arginine is synthesized from citrulline by
argininosuccinate synthase (ASS) and argininosuccinate lyase (ASL).
ASS catalyzes the conversion of citrulline and aspartate into
argininosuccinate. Argininosuccinate is then converted into
arginine and fumarate by ASL. A high level of ASS is observed in
malignant lung, ovarian, gastric and colonic epithelium compared to
corresponding normal epithelium (17–19).
In contrast, tumors usually express reduced ASS including breast
cancer, hepatocellular carcinoma, melanoma, renal cell carcinoma
and pancreatic cancer, and the ASS level is inversely correlated
with survival (20–23). Tumors with loss of ASS are dependent
on extracellular arginine for growth, characteristic of arginine
auxotrophy. Breast cancer and melanoma with deficiency of ASS are
sensitive to arginine deprivation via arginine deiminase (4,24). The
complex of ASS and ASL with NOS contributes to the efficient
channeling for NO production (25,26).
ASL-deficient mice and argininosuccinic aciduria patients have a
deficiency in the production of NO (25). ASL is highly expressed in
hepatocellular carcinoma and downregulation of ASL by shRNA
attenuates tumor growth in vivo (27).
Endoplasmic reticulum (ER), which plays a major role
in membrane and secretory protein synthesis, has been associated
with metabolic disease (28–30).
Endoplasmic reticulum stress, which emanates from the accumulation
of unfolded protein, has a profound impact on the pathogenesis of
many diseases, including liver disease, diabetes and cancer
(31–34). In response to ER stress, the
unfolded protein response (UPR) initiates signaling cascades and
restores the protein-folding homeostasis. The amino acid metabolism
genes are also activated under ER stress condition (35,36).
Given the relationship between ER stress, metabolism and cancer
progression, it is predictable that ER stress may affect metabolic
enzymes in cancer cells. ASL upregulation by an ER stress inducer
was observed in liver cancer cells (27). Arginine metabolic enzyme is usually
expressed in liver cells. It is uncertain whether the arginine
metabolic enzyme, ASL, exerts functions in other types of tissues.
The present study aimed to determine the relationship between ER
stress and ASL in breast cancer cells and to ascertain whether the
arginine-NO complex mediates breast cancer growth.
Our results revealed that ASL is elevated by ER
stress and is highly expressed in breast tumor tissues.
Downregulation of ASL by ASL shRNA decreased tumor growth in
vivo and in vitro. ASL knockdown induced cyclin A2
degradation and the cell growth was rescued by exogenous cyclin A2.
Furthermore, ASL downregulation inhibited NO expression and induced
autophagy.
Materials and methods
Reagents, chemicals and antibodies
The anti-ASL antibody was purchased from Abnova
(Taipei, Taiwan). The anti-GRP78 antibody was purchased from BD
(Erembodegem, Belgium). The anti-β-actin antibody was purchased
from Chemicon (Pittsburgh, PA, USA). The anti-GAPDH antibody was
purchased from GeneTex (Irvine, CA, USA). The anti-cyclin A2,
anti-CDK4 and anti-CDK2 antibodies were purchased from Santa Cruz
Biotechnology (Santa Cruz, CA, USA). The anti-cyclin B1 and
anti-cyclin E1 antibodies were purchased from Epitomics
(Burlingame, CA, USA). The anti-cyclin D1 antibody was purchased
from Cell Signaling (Beverly, MA, USA). The anti-LC3B antibody was
purchased from Sigma (St. Louis, MO, USA). The anti-HA antibody was
purchased from Roche Applied Science (Mannheim, Germany). The
powder of G418, thiazolyl blue tetrazolium bromide (MTT),
3-methyladenine (3-MA), bafilomycin A1, sodium nitrite, 2-amino
purine and arginine were purchased from Sigma. The plasmid
containing ASL was purchased from OriGene Technologies, Inc.
(Rockville, MD, USA). The plasmid containing cyclin A2 was provided
by Dr Ih-Jen Su (Division of Clinical Research, National Health
Research Institute, Taiwan).
Cell culture
MCF-7 and MDA MB-231 cells and the stable
transfectants were cultured in Dulbecco's modified Eagle's medium
(DMEM) (HyClone, Logan, UT, USA) containing 10% fetal bovine serum
(FBS) (Biological Industries, Kibbutz Beit-Haemek, Israel), 100
U/ml penicillin and 100 µg/ml streptomycin (Invitrogen
Corporation, Carlsbad, CA, USA) at 37°C in 5% CO2.
Reverse transcription-polymerase chain
reaction (RT-PCR) analysis
Total RNA was extracted with TRIzol (MDBio, Taiwan).
cDNA was synthesized using M-MLV transcriptase (Promega, Madison,
MI, USA). PCR was performed using Pro Taq polymerase
(PROtech Technology Enterprise Co., Ltd., Taipei, Taiwan) on a
thermocycler (ABI, Foster City, CA, USA). The 5′ and 3′ human ASL
gene-specific primers were: 5′-TGA TGC CCC AGA AGA AAA AC-3′
(sense) and 5′-TTT GCG GAC CAG GTA ATA GG-3′ (antisense); the 5′
and 3′ human GRP78 gene-specific primers were: 5′-CGC CTC ATC GGA
CGC ACT TG-3′ (sense) and 5′-AGG TTC CAC CGC CCA GGT CA-3′
(antisense); the 5′ and 3′ human CCND1 gene-specific primers were:
5′-AAC TAC CTG GAC CGC TTC CT-3′ (sense) and 5′-TGA GGC GGT AGT AGG
ACA GG-3′ (antisense); the 5′ and 3′ human CCNE1 gene-specific
primers were: 5′-ATC CCC ACA CCT GAC AAA GA-3′ (sense) and 5′-AGG
GGA CTT AAA CGC CAC TT-3′ (antisense); the 5′ and 3′ human CCNA2
gene-specific primers were: 5′-GCA CCC CTT AAG GAT CTT CC-3′
(sense) and 5′-CCT CTC AGC ACT GAC ATG GA-3′ (antisense); the 5′
and 3′ human CCNB1 gene-specific primers were: 5′-GGC CAA AAT GCC
TAT GAA GA-3′ (sense) and 5′-AA CAT GGC AGT GAC ACC AA-3′
(antisense).
Western blotting
The cell lysates were prepared in RIPA lysis buffer.
The protein concentration was determined with the Micro BCA™
protein assay kit (Millipore, Billerica, MA, USA). Cell lysates
were loaded onto acrylamide gels and were then transferred onto
polyvinylidene fluoride membranes (Amersham Biosciences,
Piscataway, NJ, USA) after electrophoresis. The membranes were
incubated with the indicated antibody for the specific protein,
probed with ECL Western Blotting Detection system (Millipore) and
visualized using the BioSpectrum AC imaging system.
Tissue samples
The specimens of breast cancer and corresponding
normal liver were obtained from the Human Biobank within the
Research Center of Clinical Medicine of the National Cheng Kung
University Hospital (Tainan, Taiwan) following the approval of the
Institutional Review Board.
Oncomine database analysis
The expression of ASL in clinical specimens of
cancer vs. normal patients was analyzed using Oncomine database
(https://www.oncomine.org/resource/login.html). We
analyzed the results of fold-change, cancer subtypes and p-values
with a threshold of p<0.05.
Kaplan-Meier plotter analysis
The overall survival of the patients with high and
low ASL expression was analyzed using Kaplan-Meier plotter
(http://www.kmplot.com/). We analyzed the
relapse-free survival and ASL expression with probe 204608 in upper
tertile patients. The number-at-risk, the hazard ratio and the
log-rank p were indicated.
RNA interference and lentiviral
production
The shRNA targeting ASL was obtained from the
National RNAi Core Facility (Academia Sinica, Taipei, Taiwan). The
target sequence of shRNA was 5′-AGGAGGCTGCTGTGTGTTT-3′ (shASL1669).
The lentiviral production was managed according to the protocol
provided by the National RNAi Core Facility.
Colony formation assay
Colony formation was performed by seeding cells into
6-well plates. The colonies were stained with 2% methylene blue and
counted after incubation for 10 days in 5% CO2 and
37°C.
Anchorage-independent growth ability
Agar (0.6%) in DMEM was prepared as an under layer
in a plastic Petri dish. Five thousand cells were suspended in 0.3%
agar in DMEM containing 10% FBS and added over the upper layer. The
plates were placed in a 5% CO2 atmosphere humidified
incubator at 37°C for 14 days and the colonies were quantified.
Tumorigenicity in NOD/SCID mice
NOD/SCID mice were obtained from the Animal Center
of the National Cheng Kung University. All study protocols were
approved by the Animal Welfare Committee of the National Cheng Kung
University. MDA MB-231 cells (5×105) were subcutaneously
implanted into the NOD/SCID mice. For our model of inhibiting tumor
growth by lentiviral ASL shRNA, the NOD/SCID mice implanted with
MDA MB-231 cells for 10 days were intratumorally injected with
lentiviral particles.
Monodansylcadaverine (MDC) staining of
autophagy
The monodansylcadaverine (MDC) staining was analyzed
by Cayman autophagy/cytotoxicity dual staining kit (Cayman Chemical
Company, Ann Arbor, MI, USA) and detected by fluorescence
microscopy.
Measurement of intracellular arginine
content
The intracellular arginine concentration was
analyzed by HPLC analysis using Agilent ZORBAX Eclipse AAA column
(Agilent PN 993400-902) (Agilent Technologies, Inc., Santa Clara,
CA, USA).
Statistical analysis
All statistical analyses were performed using the
Student's t-test. The error bars in the graphs represent the
SEM.
Results
ASL expression is induced by ER stress
and is highly expressed in breast cancer
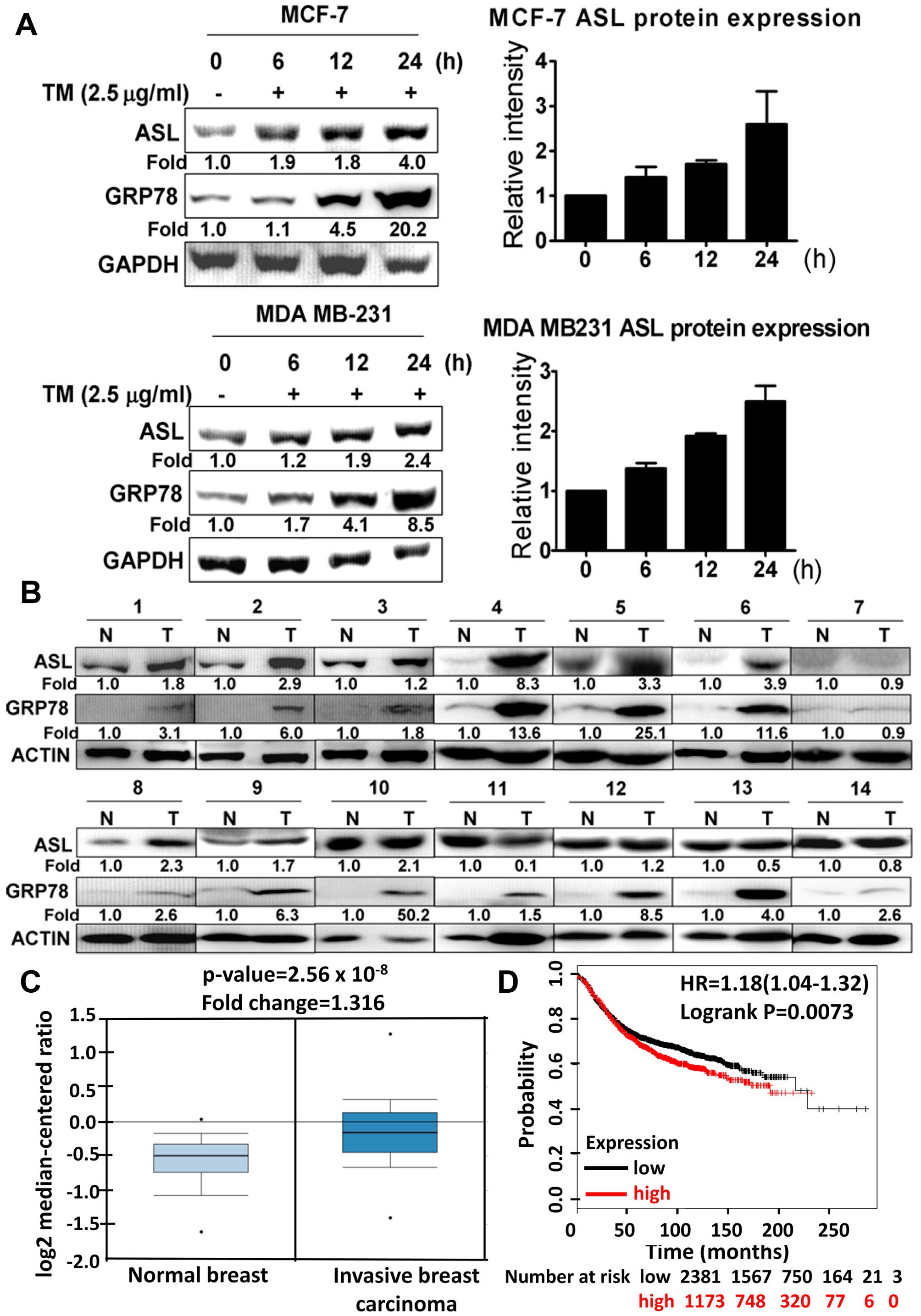
To analyze whether ASL is induced by ER stress, the
breast cancer cell lines, MCF-7 and MDA MB-231, were incubated with
tunicamycin. ASL expression was increased after tunicamycin
treatment as demonstrated by western blotting (Fig. 1A). We further examined whether ASL
expression was increased in human breast cancer. Western blot
analysis was used to detect ASL and GRP78 expression in 14 paired
breast cancer and adjacent normal breast tissues. The results
indicated that ASL was overexpressed in the breast cancer tissues
(Fig. 1B). From the Oncomine
database, ASL expression was upregulated in different subtypes of
breast cancer (Fig. 1C, Tables I and II). Kaplan-Meier plotter analysis in
breast cancer showed a correlation between overexpression of ASL
and lower overall survival rates (Fig.
1D).
 | Table IASL expression of the normal and
breast cancer tissues from the Oncomine database. |
Table I
ASL expression of the normal and
breast cancer tissues from the Oncomine database.
| Tissue (no.) | P-value | Fold-change | Ref. |
|---|
Normal breast
(61)
Mucinous breast carcinoma (4) | 0.004 | 1.601 | TCGA |
Normal breast
(61)
Invasive breast carcinoma (76) | 2.56E-08 | 1.316 | |
Normal breast
(61)
Invasive ductal and lobular carcinoma (3) | 2.20E-02 | 1.298 | |
Normal breast
(61)
Invasive lobular breast carcinoma (36) | 3.62E-05 | 1.359 | |
Normal breast
(61)
Invasive ductal breast carcinoma (389) | 9.21E-10 | 1.287 | |
| Table IIASL expression of the normal and
breast cancer tissues from the Oncomine database. |
Table II
ASL expression of the normal and
breast cancer tissues from the Oncomine database.
| Tissue (no.) | P-value | Fold-change | Ref. |
|---|
Normal breast
(144)
Invasive lobular breast carcinoma (148) | 2.36E-12 | 1.108 | Curtis et
al(41) |
Normal breast
(144)
Invasive ductal breast carcinoma (1,556) | 1.85E-28 | 1.111 | |
Normal breast
(144)
Breast carcinoma (14) | 8.00E-03 | 1.056 | |
Normal breast
(144)
Medullary breast carcinoma (32) | 9.13E-04 | 1.137 | |
Normal breast
(144)
Invasive ductal and invasive lobular breast carcinoma (90) | 3.45E-06 | 1.07 | |
Normal breast
(144)
Mucinous breast carcinoma (46) | 7.00E-03 | 1.049 | |
Normal breast
(144)
Ductal breast carcinoma in situ (10) | 7.70E-02 | 1.067 | |
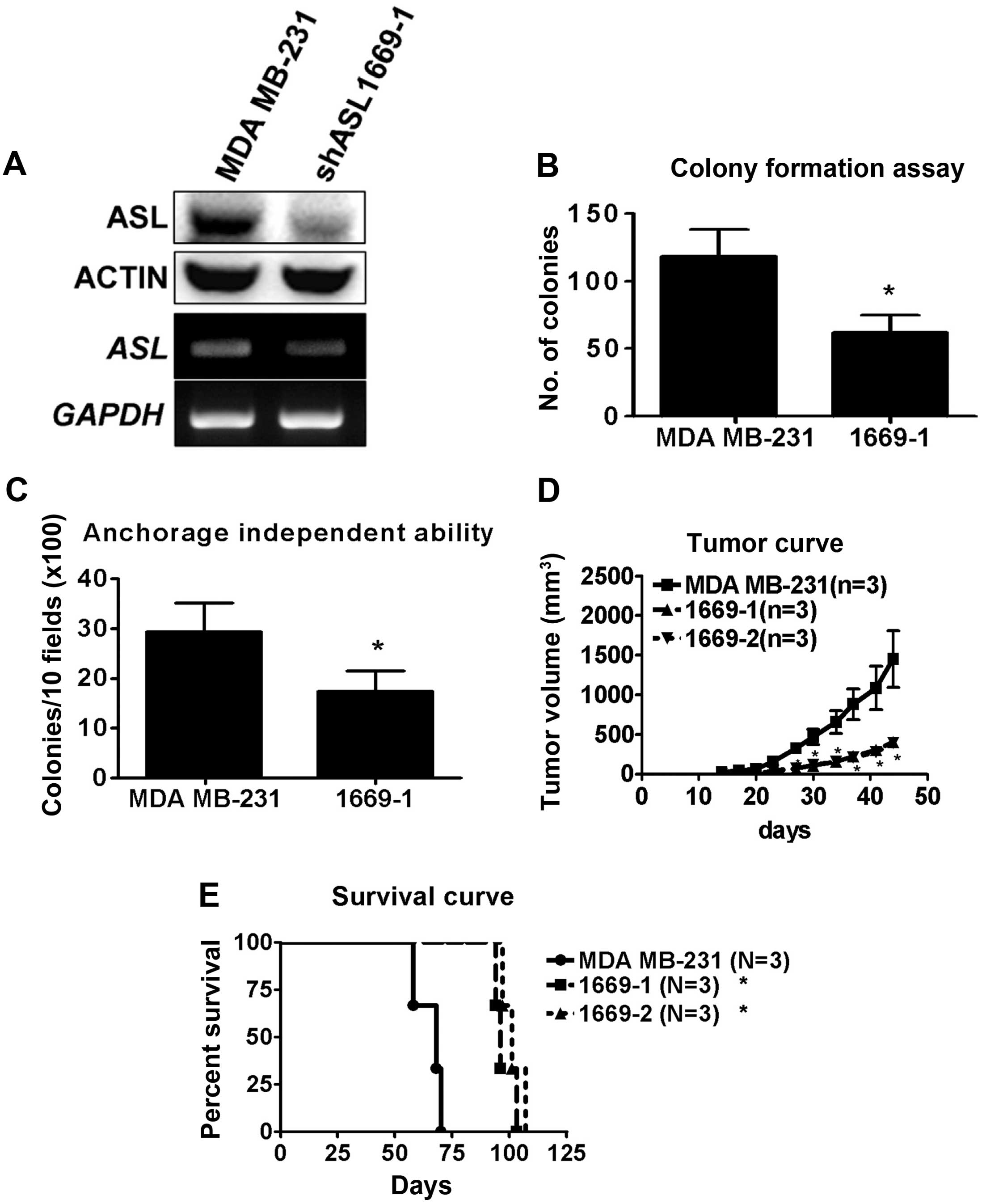
ASL shRNA inhibits breast cancer cell
growth
To study the role of ASL in breast cancer, MDA
MB-231 cells were transfected with ASL shRNA and stable
transfectants were selected by puromycin. The mRNA and protein
levels of the ASL knockdown stable transfectants were determined by
RT-PCR and western blotting (Fig.
2A). The proliferation of the ASL knockdown stable
transfectants was determined by colony formation assay. ASL
knockdown stable transfectants exhibited a significantly reduced
proliferation in vitro (Fig.
2B). To investigate the effect of ASL on tumorigenic ability,
the anchorage-independent growth ability of the ASL knockdown
stable transfectants was analyzed. Decreased anchorage-independent
growth ability was observed in the ASL knockdown stable
transfectants of the MDA MB-231 cells (Fig. 2C). We further investigated the
tumorigenicity of ASL knockdown stable transfectants in vivo
and found that tumor growth was decreased by ASL shRNA (Fig. 2D). Moreover, there was a significant
increase in the survival rate of the ASL knockdown stable
transfectants when compare to that of the parental MDA MB-231 cells
(Fig. 2E). The results indicated
that ASL shRNA decreased cell growth in vivo and in
vitro.
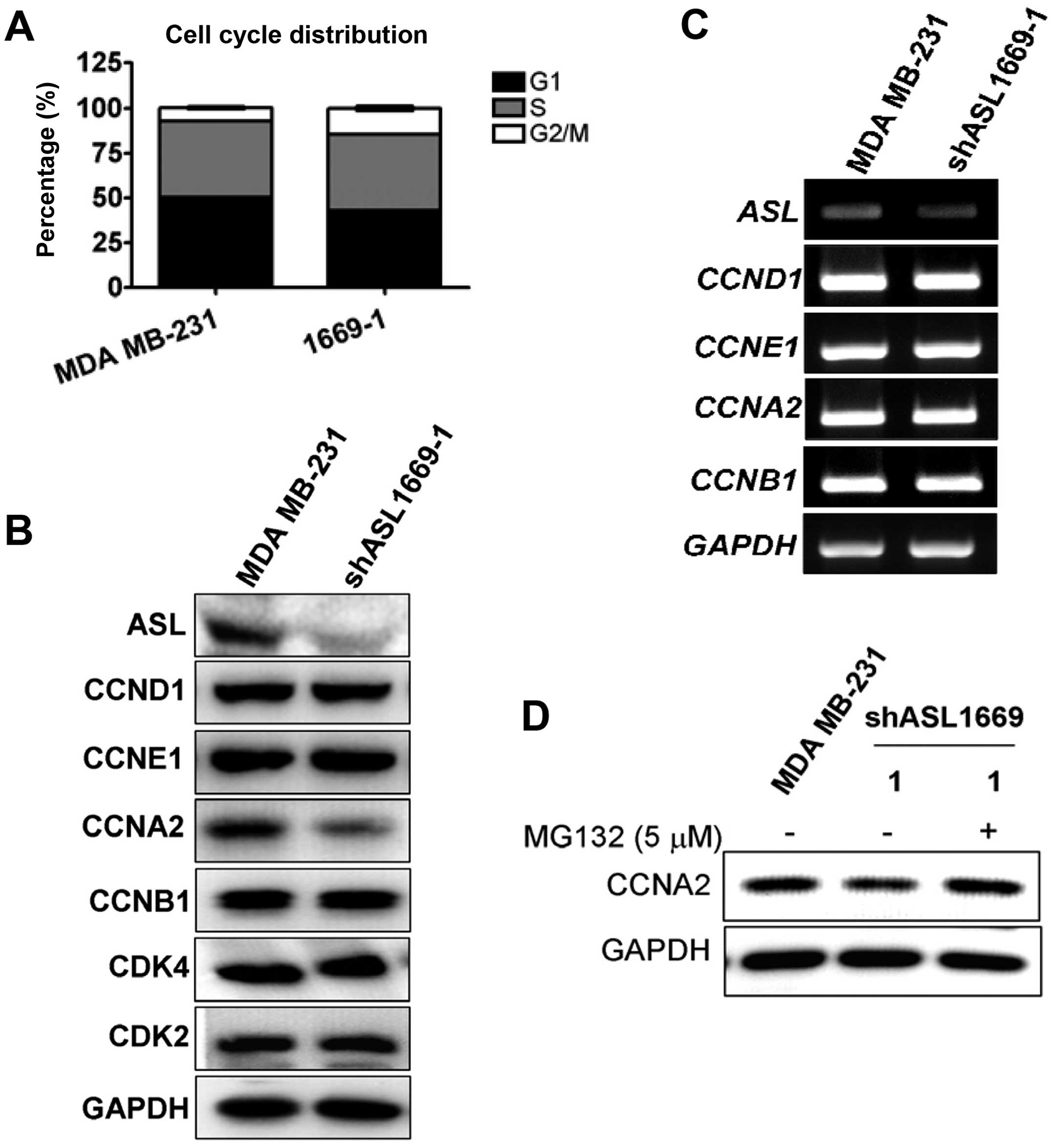
ASL shRNA inhibits cyclin A2 expression
and causes G2/M cell cycle delay
To further study ASL shRNA-induced growth
inhibition, we examined the changes in cell cycle progression using
flow cytometric methods. A delay in G2/M phase was observed in the
ASL knockdown stable transfectants (Fig. 3A). We then determined whether ASL
shRNA affects the changes in cell cycle-associated molecules.
Cyclin A2 was significantly reduced in the ASL knockdown
transfectants, while cyclin D1 E1 and B1, CDK2 and CDK4 were not
decreased (Fig. 3B). The mRNA
expression of cyclins was not altered (Fig. 3C). Since the cyclins are frequently
regulated by protein degradation, we ascertained whether the
downregulation of cyclins by ASL shRNA could be restored by a
proteasome inhibitor. Addition of MG132, a proteasome inhibitor,
restored cyclin A2 protein expression (Fig. 3D).
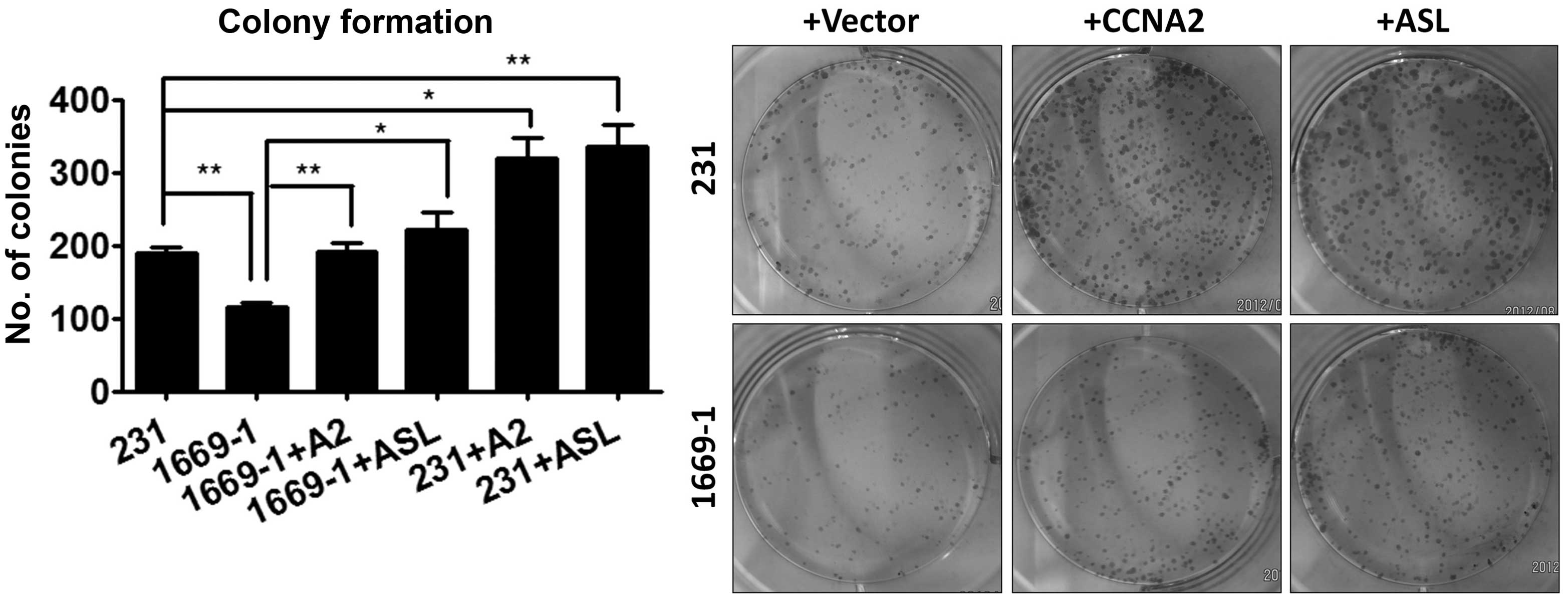
Ectopic expression of cyclin A2 restores
ASL shRNA-induced growth inhibition
Since ASL shRNA reduced cyclin A2 expression and
cell growth, we next examined the role of cyclin A2 in the
proliferation by ectopically expressing cyclin A2 in the ASL
knockdown transfectants. Ectopic cyclin A2 reversed the cell growth
inhibition by ASL shRNA, indicating that cyclin A2 plays an
important role in the inhibition of cell growth by ASL shRNA
(Fig. 4).
ASL shRNA induces autophagy in breast
cancer cells
Autophagy is required for amino acid maintenance and
responds to nitrogen deprivation in breast cancer (37). We next examined whether ASL shRNA
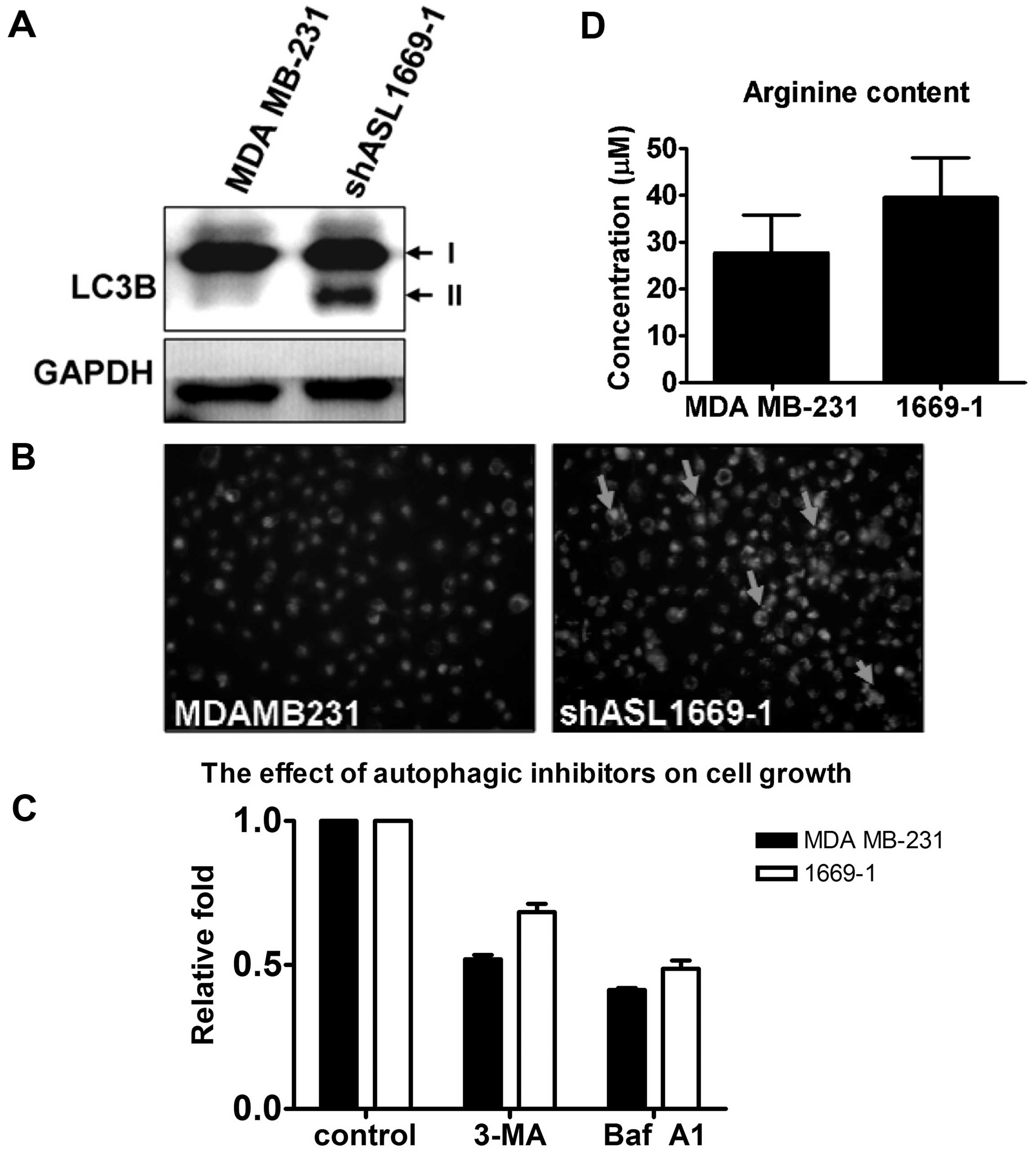
induces autophagy. LC3B expression and autophagic vacuoles stained
by MDC were increased in the ASL knockdown transfectants (Fig. 5A and B). To study the correlation
between autophagy and cell growth, the autophagic inhibitors, 3-MA
and bafilomycin A1, were incubated with the parental and
ASL-knockdown MDA MB-231 cells.
The cell growth of the ASL knockdown transfectants
was higher than that of the parental MDA MB-231 cells after
treatment with the autophagic inhibitors, indicating the
autophagy-induced pro-survival role by ASL shRNA (Fig. 5C). The cellular arginine level was
analyzed by HPLC analysis. There was no significant difference in
the arginine level between parental cells and the ASL knockdown
transfectants (Fig. 5D). These data
indicate that autophagy is induced independent of total cellular
arginine content.
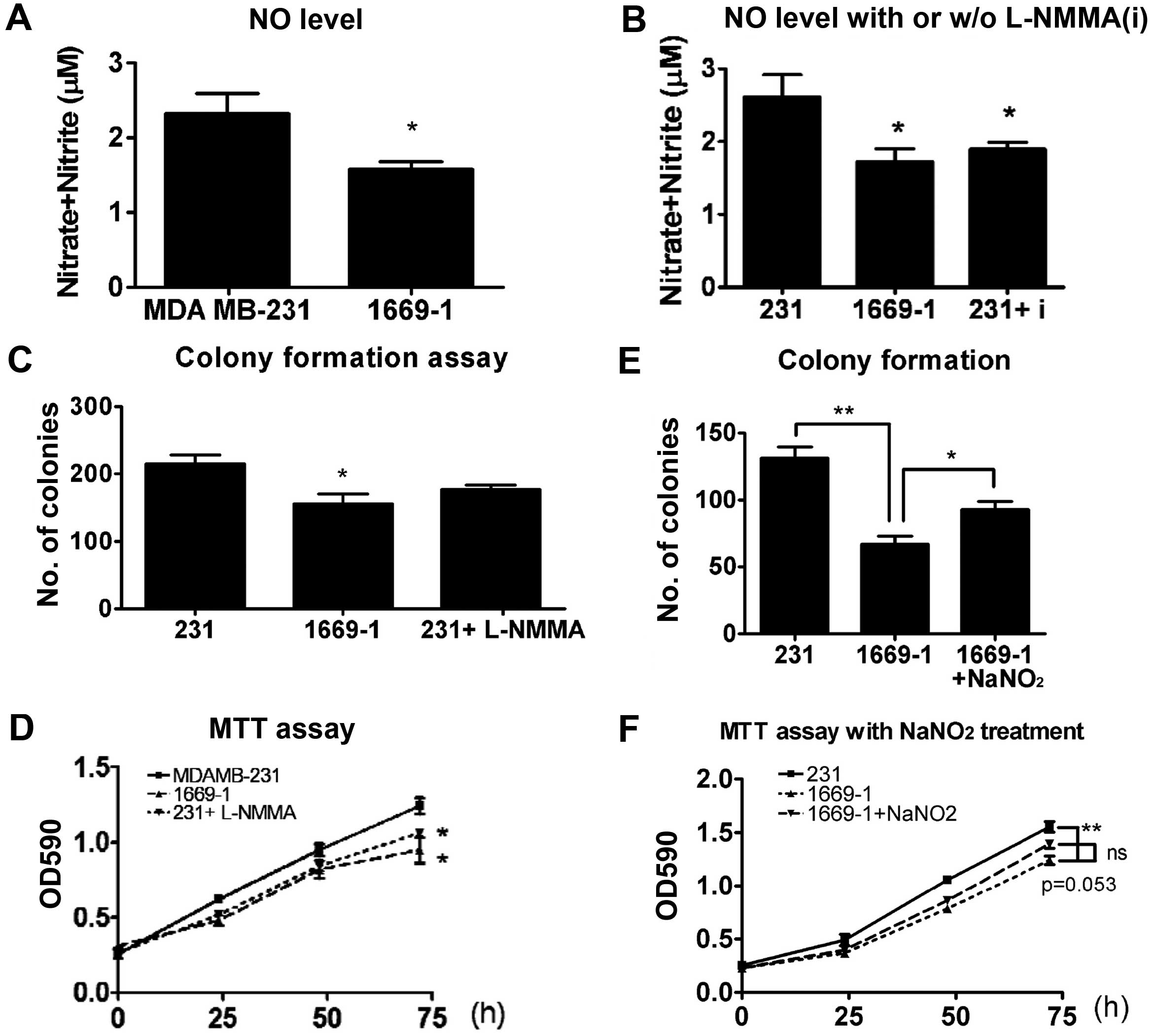
ASL shRNA attenuates NO content in breast
cancer cells
Nitric oxide synthase (NOS) and ASL constitute the
citrulline-argininosuccinate-arginine cycle and permit nitric oxide
production. Therefore, we examined the NO content in ASL knockdown
transfectants. The NO content was significantly decreased by ASL
shRNA (Fig. 6A). The NO inhibitor,
L-NMMA, also attenuated the NO level in the MDA MB-231 cells
(Fig. 6B). Since NO production is
implicated in cancer progression, we analyzed whether an NOS
inhibitor attenuates breast cancer growth. The proliferation was
inhibited by L-NMMA in MDA-MB-231 cells as demonstrated by colony
formation (Fig. 6C) and MTT assays
(Fig. 6D). The NO donor, sodium
nitrite (NaNO2), partially restored the ASL
shRNA-induced growth inhibition as demonstrated by colony formation
(Fig. 6E) and MTT assays (Fig. 6F), indicating that reduction in the
NO level may in part be responsible for the growth inhibition by
ASL shRNA in breast cancer.
Discussion
In the present study, we demonstrated that ASL
expression was induced by ER stress and was overexpressed in breast
cancer. ASL shRNA attenuated cell proliferation and
anchorage-independent growth. The breast cancer cells with low ASL
expression had lower ability to form tumors in NOD/SCID mice.
Furthermore, ASL downregulation induced autophagy. The cyclin A2
and NO levels in the ASL knockdown transfectants were decreased and
ectopic cyclin A2 and NO donor restored the inhibition of cell
growth by ASL shRNA. A similar effect of NO on cell proliferation
was further observed using an NOS inhibitor, L-NMMA, suggesting
that NO played an important role in the ASL knockdown
transfectants.
ASL plays an important role in liver cancer
progression (27). This finding
demonstrating the effect of ASL on cancer cell growth in
vivo and in vitro is consistent with the present study.
Kaplan-Meier plotter database indicated that the breast cancer
patients with high ASL expression were associated with a poor
clinical outcome. ASL expression was also overexpressed in the
breast cancer tissues in the Cancer Genome Anatomy Project (CGAP)
database. Downregulation of ASL was found to contribute to tumor
regression in both liver and breast cancer. These data indicate
that ASL may play a tumorigenic role in human cancer, suggesting
that ASL may serve as a therapeutic target. The role of ASL in
other types of cancers warrants further study.
The mouse model with ASL deficiency has an effect on
NO synthesis (25). The complex of
ASL, ASS and NOS is responsible for NO recycling (38). Our data support the notion that the
metabolic enzyme, ASL, contributes to NO production. Excessive NO
production has been implicated in cancer progression (39). The NO donor restored the cell
inhibition by ASL shRNA and the NOS inhibitor attenuated cell
growth, indicating the oncogenic role of NO in cancer development.
However, a previous study found that macrophage and natural killer
cell-derived NO exerts an antitumor effect (40). The mechanism by which NO mediates
cancer growth warrants further investigation.
In conclusion, ASL is overexpressed in breast cancer
and ASL downregulation decreases tumor growth by inhibiting cyclin
A2 and NO. Administration of ASL shRNA may be a novel treatment to
prevent cancer cell proliferation and induce cancer cell death.
Acknowledgments
The present study was supported by a grant (to M.D.
Lai) NSC-100-2325-B-006-008 from the National Science Council,
Taiwan, and NHRI-EX100-9927B1 from the National Health Research
Institute, Taiwan; to Establish Centers of Excellence for Cancer
Research in Taiwan, DOH101-TD-C-111-003 Department of Health,
Executive Yuan, Taiwan.
References
|
1
|
McDonnell DP, Park S, Goulet MT, Jasper J,
Wardell SE, Chang CY, Norris JD, Guyton JR and Nelson ER: Obesity,
cholesterol metabolism, and breast cancer pathogenesis. Cancer Res.
74:4976–4982. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Stebbing J, Sharma A, North B, Athersuch
TJ, Zebrowski A, Pchejetski D, Coombes RC, Nicholson JK and Keun
HC: A metabolic phenotyping approach to understanding relationships
between metabolic syndrome and breast tumour responses to
chemotherapy. Ann Oncol. 23:860–866. 2012. View Article : Google Scholar
|
|
3
|
Hirayama A, Kami K, Sugimoto M, Sugawara
M, Toki N, Onozuka H, Kinoshita T, Saito N, Ochiai A, Tomita M, et
al: Quantitative metabolome profiling of colon and stomach cancer
microenvironment by capillary electrophoresis time-of-flight mass
spectrometry. Cancer Res. 69:4918–4925. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Qiu F, Chen YR, Liu X, Chu CY, Shen LJ, Xu
J, Gaur S, Forman HJ, Zhang H, Zheng S, et al: Arginine starvation
impairs mitochondrial respiratory function in ASS1-deficient breast
cancer cells. Sci Signal. 7:ra312014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Possemato R, Marks KM, Shaul YD, Pacold
ME, Kim D, Birsoy K, Sethumadhavan S, Woo HK, Jang HG, Jha AK, et
al: Functional genomics reveal that the serine synthesis pathway is
essential in breast cancer. Nature. 476:346–350. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Amelio I, Cutruzzolá F, Antonov A,
Agostini M and Melino G: Serine and glycine metabolism in cancer.
Trends Biochem Sci. 39:191–198. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wu G and Morris SM Jr: Arginine
metabolism: Nitric oxide and beyond. Biochem J. 336:1–17. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Vissers YL, Dejong CH, Luiking YC, Fearon
KC, von Meyenfeldt MF and Deutz NE: Plasma arginine concentrations
are reduced in cancer patients: Evidence for arginine deficiency?
Am J Clin Nutr. 81:1142–1146. 2005.PubMed/NCBI
|
|
9
|
Daly JM, Reynolds J, Thom A, Kinsley L,
Dietrick-Gallagher M, Shou J and Ruggieri B: Immune and metabolic
effects of arginine in the surgical patient. Ann Surg. 208:512–523.
1988. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Talamonti MS, Kim SP, Yao KA, Wayne JD,
Feinglass J, Bennett CL and Rao S: Surgical outcomes of patients
with gastric carcinoma: The importance of primary tumor location
and microvessel invasion. Surgery. 134:720–729. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yerushalmi HF, Besselsen DG, Ignatenko NA,
Blohm-Mangone KA, Padilla-Torres JL, Stringer DE, Guillen JM,
Holubec H, Payne CM and Gerner EW: Role of polyamines in
arginine-dependent colon carcinogenesis in
ApcMin/+ mice. Mol Carcinog. 45:764–773.
2006. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Park KG, Heys SD, Blessing K, Kelly P,
McNurlan MA, Eremin O and Garlick PJ: Stimulation of human breast
cancers by dietary L-arginine. Clin Sci. 82:413–417. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yeatman TJ, Risley GL and Brunson ME:
Depletion of dietary arginine inhibits growth of metastatic tumor.
Arch Surg. 126:1376–1382. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ma Q, Wang Y, Gao X, Ma Z and Song Z:
L-arginine reduces cell proliferation and ornithine decarboxylase
activity in patients with colorectal adenoma and adenocarcinoma.
Clin Cancer Res. 13:7407–7412. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Feun L, You M, Wu CJ, Kuo MT, Wangpaichitr
M, Spector S and Savaraj N: Arginine deprivation as a targeted
therapy for cancer. Curr Pharm Des. 14:1049–1057. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Phillips MM, Sheaff MT and Szlosarek PW:
Targeting arginine-dependent cancers with arginine-degrading
enzymes: Opportunities and challenges. Cancer Res Treat.
45:251–262. 2013. View Article : Google Scholar
|
|
17
|
Delage B, Fennell DA, Nicholson L, McNeish
I, Lemoine NR, Crook T and Szlosarek PW: Arginine deprivation and
arginino-succinate synthetase expression in the treatment of
cancer. Int J Cancer. 126:2762–2772. 2010.PubMed/NCBI
|
|
18
|
Shen LJ and Shen WC: Drug evaluation:
ADI-PEG-20 - a PEGylated arginine deiminase for
arginine-auxotrophic cancers. Curr Opin Mol Ther. 8:240–248.
2006.PubMed/NCBI
|
|
19
|
Shen LJ, Beloussow K and Shen WC:
Modulation of arginine metabolic pathways as the potential
anti-tumor mechanism of recombinant arginine deiminase. Cancer
Lett. 231:30–35. 2006. View Article : Google Scholar
|
|
20
|
Dillon BJ, Prieto VG, Curley SA, Ensor CM,
Holtsberg FW, Bomalaski JS and Clark MA: Incidence and distribution
of argininosuccinate synthetase deficiency in human cancers: A
method for identifying cancers sensitive to arginine deprivation.
Cancer. 100:826–833. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ensor CM, Holtsberg FW, Bomalaski JS and
Clark MA: Pegylated arginine deiminase (ADI-SS PEG20,000
mw) inhibits human melanomas and hepatocellular carcinomas in
vitro and in vivo. Cancer Res. 62:5443–5450. 2002.PubMed/NCBI
|
|
22
|
Bowles TL, Kim R, Galante J, Parsons CM,
Virudachalam S, Kung HJ and Bold RJ: Pancreatic cancer cell lines
deficient in argininosuccinate synthetase are sensitive to arginine
deprivation by arginine deiminase. Int J Cancer. 123:1950–1955.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yoon CY, Shim YJ, Kim EH, Lee JH, Won NH,
Kim JH, Park IS, Yoon DK and Min BH: Renal cell carcinoma does not
express argininosuccinate synthetase and is highly sensitive to
arginine deprivation via arginine deiminase. Int J Cancer.
120:897–905. 2007. View Article : Google Scholar
|
|
24
|
Feun LG, Marini A, Walker G, Elgart G,
Moffat F, Rodgers SE, Wu CJ, You M, Wangpaichitr M, Kuo MT, et al:
Negative argininosuccinate synthetase expression in melanoma
tumours may predict clinical benefit from arginine-depleting
therapy with pegylated arginine deiminase. Br J Cancer.
106:1481–1485. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Erez A, Nagamani SC, Shchelochkov OA,
Premkumar MH, Campeau PM, Chen Y, Garg HK, Li L, Mian A, Bertin TK,
et al: Requirement of argininosuccinate lyase for systemic nitric
oxide production. Nat Med. 17:1619–1626. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Turner MA, Simpson A, McInnes RR and
Howell PL: Human argininosuccinate lyase: A structural basis for
intragenic complementation. Proc Natl Acad Sci USA. 94:9063–9068.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Huang HL, Hsu HP, Shieh SC, Chang YS, Chen
WC, Cho CY, Teng CF, Su IJ, Hung WC and Lai MD: Attenuation of
argininosuccinate lyase inhibits cancer growth via cyclin A2 and
nitric oxide. Mol Cancer Ther. 12:2505–2516. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cunard R and Sharma K: The endoplasmic
reticulum stress response and diabetic kidney disease. Am J Physiol
Renal Physiol. 300:F1054–F1061. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lee AH, Scapa EF, Cohen DE and Glimcher
LH: Regulation of hepatic lipogenesis by the transcription factor
XBP1. Science. 320:1492–1496. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang X, Eno CO, Altman BJ, Zhu Y, Zhao G,
Olberding KE, Rathmell JC and Li C: ER stress modulates cellular
metabolism. Biochem J. 435:285–296. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Clarke HJ, Chambers JE, Liniker E and
Marciniak SJ: Endoplasmic reticulum stress in malignancy. Cancer
Cell. 25:563–573. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang G, Yang ZQ and Zhang K: Endoplasmic
reticulum stress response in cancer: Molecular mechanism and
therapeutic potential. Am J Transl Res. 2:65–74. 2010.PubMed/NCBI
|
|
33
|
Malhi H and Kaufman RJ: Endoplasmic
reticulum stress in liver disease. J Hepatol. 54:795–809. 2011.
View Article : Google Scholar
|
|
34
|
Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi
NN, Ozdelen E, Tuncman G, Görgün C, Glimcher LH and Hotamisligil
GS: Endoplasmic reticulum stress links obesity, insulin action, and
type 2 diabetes. Science. 306:457–461. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Barbosa-Tessmann IP, Chen C, Zhong C,
Schuster SM, Nick HS and Kilberg MS: Activation of the unfolded
protein response pathway induces human asparagine synthetase gene
expression. J Biol Chem. 274:31139–31144. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Okada T, Yoshida H, Akazawa R, Negishi M
and Mori K: Distinct roles of activating transcription factor 6
(ATF6) and double-stranded RNA-activated protein kinase-like
endoplasmic reticulum kinase (PERK) in transcription during the
mammalian unfolded protein response. Biochem J. 366:585–594. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Liang XH, Jackson S, Seaman M, Brown K,
Kempkes B, Hibshoosh H and Levine B: Induction of autophagy and
inhibition of tumorigenesis by beclin 1. Nature. 402:672–676. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Oyadomari S, Gotoh T, Aoyagi K, Araki E,
Shichiri M and Mori M: Coinduction of endothelial nitric oxide
synthase and arginine recycling enzymes in aorta of diabetic rats.
Nitric Oxide. 5:252–260. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Korde Choudhari S, Sridharan G, Gadbail A
and Poornima V: Nitric oxide and oral cancer: A review. Oral Oncol.
48:475–483. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lechner M, Lirk P and Rieder J: Inducible
nitric oxide synthase (iNOS) in tumor biology: The two sides of the
same coin. Semin Cancer Biol. 15:277–289. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Curtis C, Shah SP, Chin SF, Turashvili G,
Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, et
al: The genomic and transcriptomic architecture of 2,000 breast
tumours reveals novel subgroups. Nature. 486:346–352.
2012.PubMed/NCBI
|