Introduction
Acute myeloid leukemia (AML) is a disorder of
hematopoietic stem cells, accompanied by obstruction in
hematopoietic cell differentiation and increased clonal neoplastic
proliferation (1). Acute
promyelocytic leukemia (APL) is a subtype of AML, characterized by
a block of granulocytic differentiation and accumulation of
promyelocytes in the bone marrow and blood (2). APL is believed to be one of the most
fatal forms of AML with poor patient outcomes (2). Despite extensive investigations, the
pathogenesis of AML is still far from being completely understood
(3).
PBK/TOPK is a protein kinase derived from
PDZ-binding kinase (PBK)/T-lymphokine-activated killer (T-LAK)
cell-originated protein kinase (TOPK) (4,5).
Although PBK/TOPK was originally cloned differentially by Gaudet
et al (4) and Abe et
al (5), the sequences of both
genes were later found to be the same. PBK/TOPK is a 322 amino acid
serine-threonine kinase that is reported to be expressed in
proliferative cells and tissues and to play an important role in
spermatogenesis (6). It has been
found that PBK/TOPK is expressed in hematologic tumors such as
leukemia, lymphoma and myeloma, and its expression is correlated
with the malignant potential of these tumors (7–9). In
interphase cells, TOPK is expressesed in the cytosol and nucleus
without any significant association with microtubule networks
(10). During mitosis, expression
of PBK/TOPK was found to be upregulated and phosphory-lated
(4,10). In addition, PBK/TOPK acts as a
substrate of cdc2/cyclin B and it possesses a possible
phosphorylation site by cdc2, S/T-PX-K/R, at N-terminus (4,10).
Once phosphorylated at Thr-9, PBK/TOPK functions to play an
important role in the formation of spindle midzone and in
cytokinesis. Compared with non-transformed cells, PBK is expressed
at high levels in tumor cell lines (11). PBK/TOPK was shown to promote tumor
cell proliferation through activation of p38 MAPK activity
(5,11) and regulation of the DNA damage
response (11). Moreover, PBK/TOPK
expression was found to be decreased during tetradecanoyl phorbol
acetate-induced HL-60 leukemic cell differentiation.
In the present study, we aimed to investigate the
role of PBK/TOPK in regulating the proliferation of promyelocytes
and the possible mechanism. The results showed that knockdown of
PBK/TOPK inhibited the proliferation of promyelocytes, induced G2/M
cell cycle arrest and apoptosis. Downregulation of nuclear factor
(erythroid-derived 2)-like 2 (Nrf2) was identified as being
responsible for PBK/TOPK KD-induced G2/M cell cycle arrest,
apoptosis and inhibition of cell proliferation.
Materials and methods
Chemicals and materials
α-tubulin and Nrf2 antibodies were purchased from
Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). Cleaved
caspase-3 and -9, cdc2 and cyclin B antibodies were purchased from
Cell Signaling Technology (Danvers, MA, USA). DCFH-DA, propodium
iodide (PI) and trypan blue were procured from Sigma. All of the
other chemicals used were of the highest grade available
commercially.
Cell culture
The human APL NB4 and HL-60 cell lines were
maintained in RPMI-1640 medium supplemented with 10% fetal bovine
serum (FBS), 100 U/ml penicillin and 100 mg/ml streptomycin (Gibco,
Grand Island, NY, USA) in a humidified incubator at 37°C with 5%
CO2.
Transfection of lentivirus and
plasmids
The PBK/TOPK-RNAi-lentivirus was constructed by
Shanghai GeneChem Co., Ltd. (Shanghai, China). NB4 and HL-60 cells
were transduced with the PBK/TOPK-RNAi-lentivirus and purified by
puromycin treatment. Transduction efficiency of the cells was
examined for expression of PBK/TOPK by real-time PCR. In some
experiments, cells were transfected with the plasmid vector or
plasmid expressing Nrf2 (Shanghai GeneChem) using Lipofectamine
2000 reagent (Invitrogen) according to the manufacturer's
instructions.
Analysis of cell cycle distribution
For cell cycle analysis, cells were centrifuged for
5 min at 1,000 rpm. The pellet was resus-pended in cold
phosphate-buffered saline (PBS) containing 200 µg/ml RNAse
A, and kept on ice. Five minutes before the analysis, NP-40 and PI
(Sigma) were added at a final concentration of 0.1% and 50
µg/ml, respectively. DNA content was measured in the FL-2
channel using flow cytometry monitored by CellQuest software (BD
Biosciences).
Cell proliferation and viability
Cell proliferation and viability were determined by
trypan blue exclusion and
3-(4,5-dimethyl-thiazoyl-2-yl)-2,5-diphenyltetrazolium bromide
(MTT) assays, respectively. For trypan blue determination, the
cells were cultured in a 24-well plate. To monitor cell growth at
intervals, attached cells were removed from quadruplicate wells
using trypsin-ethylenediaminetetraacetic acid, and the viable cells
were counted in a hemocytometer by trypan blue exclusion. For MTT
evaluation, the cells were seeded into 96-well plates. After the
treatment, the cell viability was determined by the MTT assay. In
brief, the supernatant was discarded, and the cells were rinsed
with PBS. After that, the cells were treated with 0.5 mg/ml MTT
(dissolved in water and filtered through a 0.2-mm membrane) at
37°C. Four hours later, the formazan crystals were dissolved in
DMSO, and the absorption values were determined using a Bio-Rad
microplate reader.
Measurement of apoptosis
Cell apoptosis was determined by terminal
deoxynucleotidyl transferase-mediated dUTP nick end labeling
(TUNEL) assay according to the manufacturer's instructions (Roche).
Positive apoptotic cells were counted and the results are shown as
a fold of the control.
Oxygen consumption rate
After the treatment, mitochondrial function was
assessed by determination of the oxygen consumption rate. In brief,
after the treatment, mitochondria were isolated from cells using a
commercial kit (Thermo Fisher Scientific). Oxygen consumption rate
was measured with a Clark oxygen electrode and is expressed as a
percentage of the oxygen consumption.
ROS determination
After the experiment, cells were harvested and
resuspended in serum-free medium. DCFH-DA was added to a final
concentration of 10 µM and cells were cultured at 37°C for
30 min in the dark. Subsequently, the cells were washed three times
and analyzed using flow cytometry in the FL-1 channel. The ROS
level is expressed as a percentage of the control.
Reporter gene assay
Cells were transfected with pGL6-ARE-luciferase
(Beyotime Institute of Biotechnology, Haimen, China) and
Renilla TK (Promega) plasmids using a transfection reagent
(TurboFect; Thermo Fisher Scientific). After the experiments, the
cells were harvested in passive lysis buffer (Promega), and the
reporter assay was performed using the Dual-luciferase reporter
assay system (Promega). Firefly luciferase activity was normalized
to Renilla luciferase and is shown as a ratio of relative
light units.
RNA isolation and real-time polymerase
chain reaction
In some experiments, total RNA was isolated using a
commercial RNA isolation kit (Tiangen Biotech Co., Ltd., Beijing,
China) according to the manufacturer's protocols. Subsequently, the
concentration of total RNA was determined and then RNA was
reverse-transcribed to cDNA using a cDNA synthesis kit (Takara).
The samples were analyzed by real-time polymerase chain reaction
(PCR) and 1 µl of cDNA was amplified with SYBR Premix Ex Taq
(Takara). The results were analyzed using the Bio-Rad RT-PCR System
for quantitative evaluation.
Western blot analysis
Briefly, after the treatment, the cells were lysed
with cell lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1%
Triton X-100, 1 mM EDTA, 10 mM NaF, 1 mM
Na3VO4, and protease inhibitor cocktail) on
ice for 30 min. After centrifugation at 20,000 × g for 20 min at
4°C, the protein contents were determined by the BCA assay kit
(Pierce Biotechnology, Inc., Rockford, IL, USA). After boiling for
5 min in a 2X SDS loading buffer, 20 µg of total proteins
were subjected to SDS-PAGE, and transferred onto a PVDF membrane.
Then, the membrane was blocked and probed with the indicated
primary antibodies overnight at 4°C. After washing for 4 times, the
membrane was incubated in the appropriate horseradish
peroxidase-conjugated secondary antibody at 37°C for 30 min. The
protein bands were visualized using chemiluminescent reagents
according to the manufacturer's instructions and quantified using
an image analyzer Quantity One System (Bio-Rad Laboratories Inc.,
Richmond, CA, USA).
Statistical analysis
Results are expressed as the means ± SD. The
statistical analysis was performed by GraphPad Prism software.
Statistical analysis was carried out by one-way ANOVA followed by
Newman-Keuls multiple-comparison post hoc test. A P-value <0.05
was considered to indicate a statistically significant result.
Results
PBK/TOPK knockdown decreases the cell
growth and viability of the promyelocytes
In the present study, NB4 and HL-60 cells were used
as in vitro models of APL, to investigate the role of
PBK/TOPK in the proliferation of promyelocytes. NB4 and HL-60 cells
were transduced with the PBK/TOPK-RNAi-lentivirus. Through
purification by puromycin treatment, we established cell lines with
stable low expression of PBK/TOPK using the NB4 and HL-60 cell
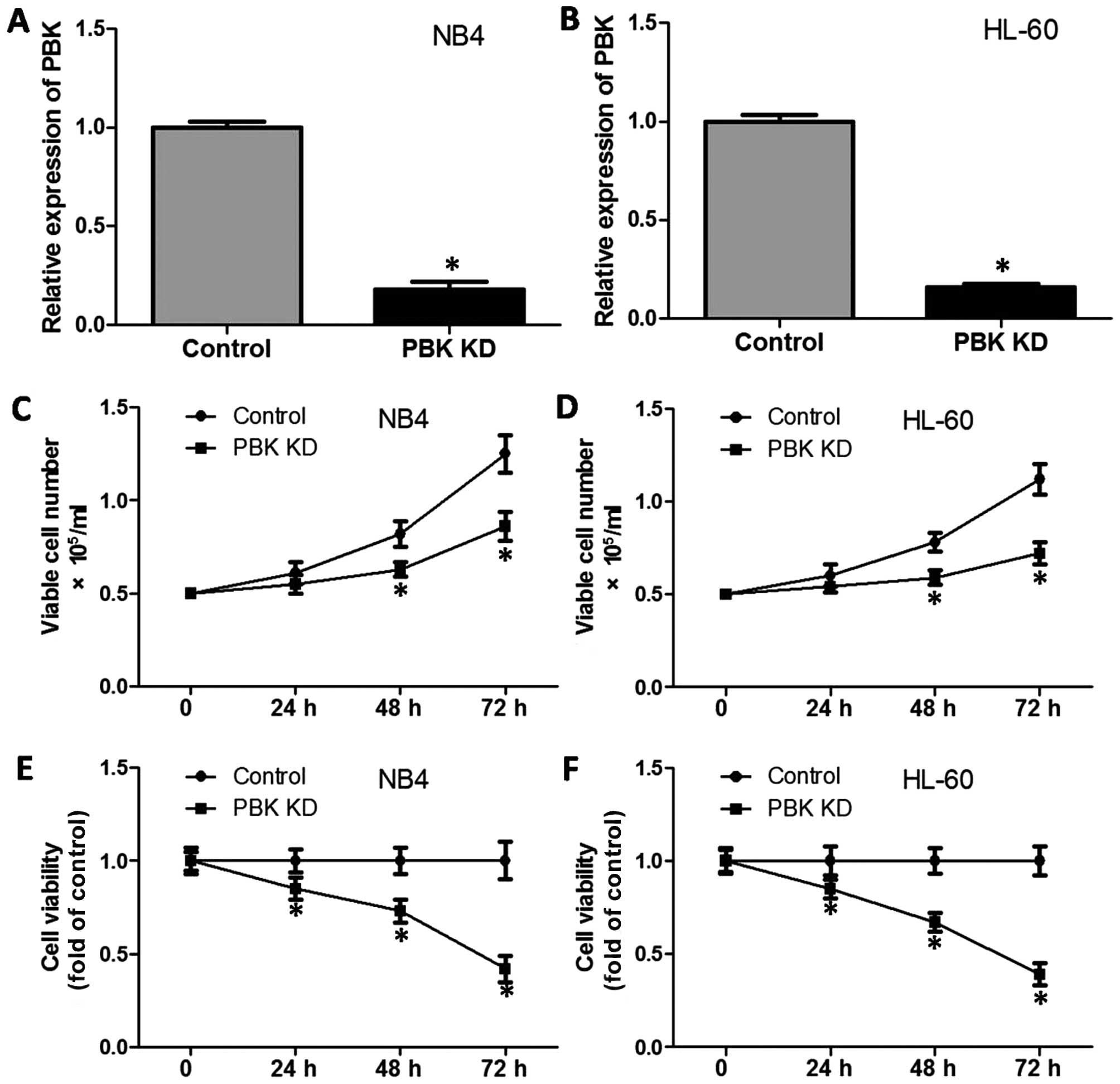
lines. Fig. 1A and B shows that the
interference effectively decreased PBK/TOPK expression. Then, we
evaluated the effect of the knockdown (KD) of PBK/TOPK on cell
growth and viability. As shown in Fig.
1C and D, after 48–72 h of incubation, PBK/TOPK KD
significantly decreased the proportion of viable cells.
Consistently, during a 72-h incubation, PBK/TOPK KD significantly
decreased the cell viability in the NB4 and HL-60 cells. After
culture for 72 h, PBK/TOPK KD significantly decreased the cell
viability to <50% of the respective control (Fig. 1E and F).
PBK/TOPK knockdown induces G2/M cell
cycle arrest in the promyelocytes
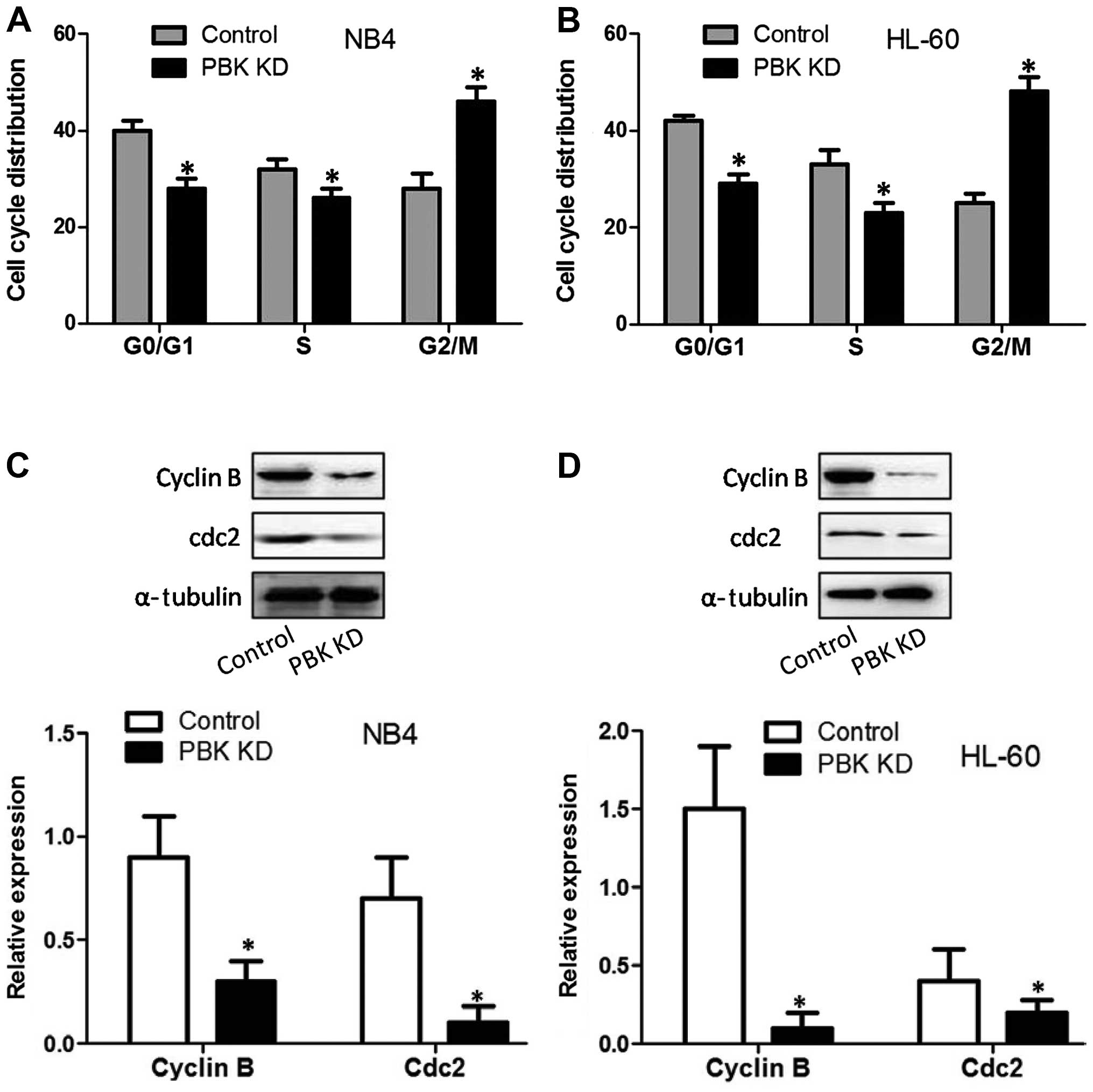
To test the effect of PBK/TOPK KD on cell cycle
progression, cell cycle distribution was analyzed. As reflected in
Fig. 2A and B, in both NB4 and
HL-60 cells, PBK/TOPK KD decreased the percentage of the cell
population in the G0/G1 phase and S phase, and significantly
increased the percentage of cell population in G2/M. The results
indicated that PBK/TOPK KD resulted in G2/M cell cycle arrest of
the NB4 and HL-60 cells. We next assessed the effect of PBK/TOPK KD
on cdc2 and cyclin B expression which are checkpoints of G2/M cell
cycle progression. The results showed that in the NB4 and HL-60
cells, PBK/TOPK KD significantly decreased cdc2 and cyclin B
expression (Fig. 2C and D).
PBK/TOPK knockdown induces the apoptosis
of the promyelocytes
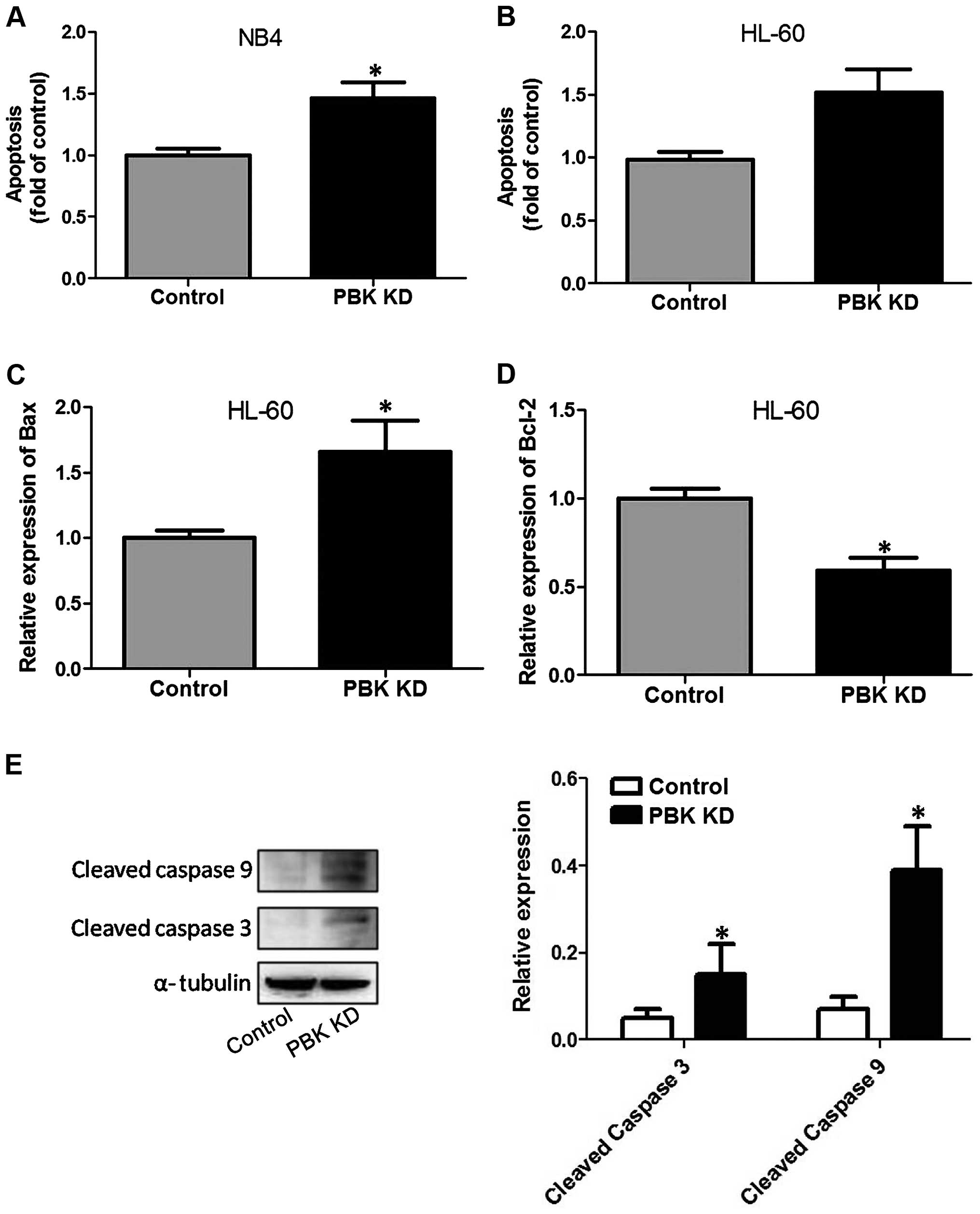
To test the effect of PBK/TOPK KD on apoptosis,
TUNEL assay was conducted. As shown in Fig. 3A and B, in both the NB4 and HL-60
cells, PBK/TOPK KD notably increased the proportion of
TUNEL-positive cells, indicating the occurrence of apoptosis.
Moreover, we assessed the effect of PBK/TOPK KD on molecular
cascades in the HL-60 cells. As illustrated in Fig. 3C, Bax mRNA expression was markedly
enhanced by PBK/TOPK KD. In addition, PBK/TOPK KD markedly reduced
Bcl-2 mRNA expression in the HL-60 cells (Fig. 3D). Furthermore, KD of PBK/TOPK in
the HL-60 cells resulted in a significant increase in the cleavage
of caspase-3 and -9 (Fig. 3E).
PBK/TOPK knockdown induces mitochondrial
dysfunction and ROS generation in promyelocytes
Research has shown that apoptosis is closely
associated with mitochondrial dysfunction and reactive oxygen
species (ROS) generation. To evaluate the possible role of the
mitochondrial pathway in PBK/TOPK KD-enhanced apoptosis,
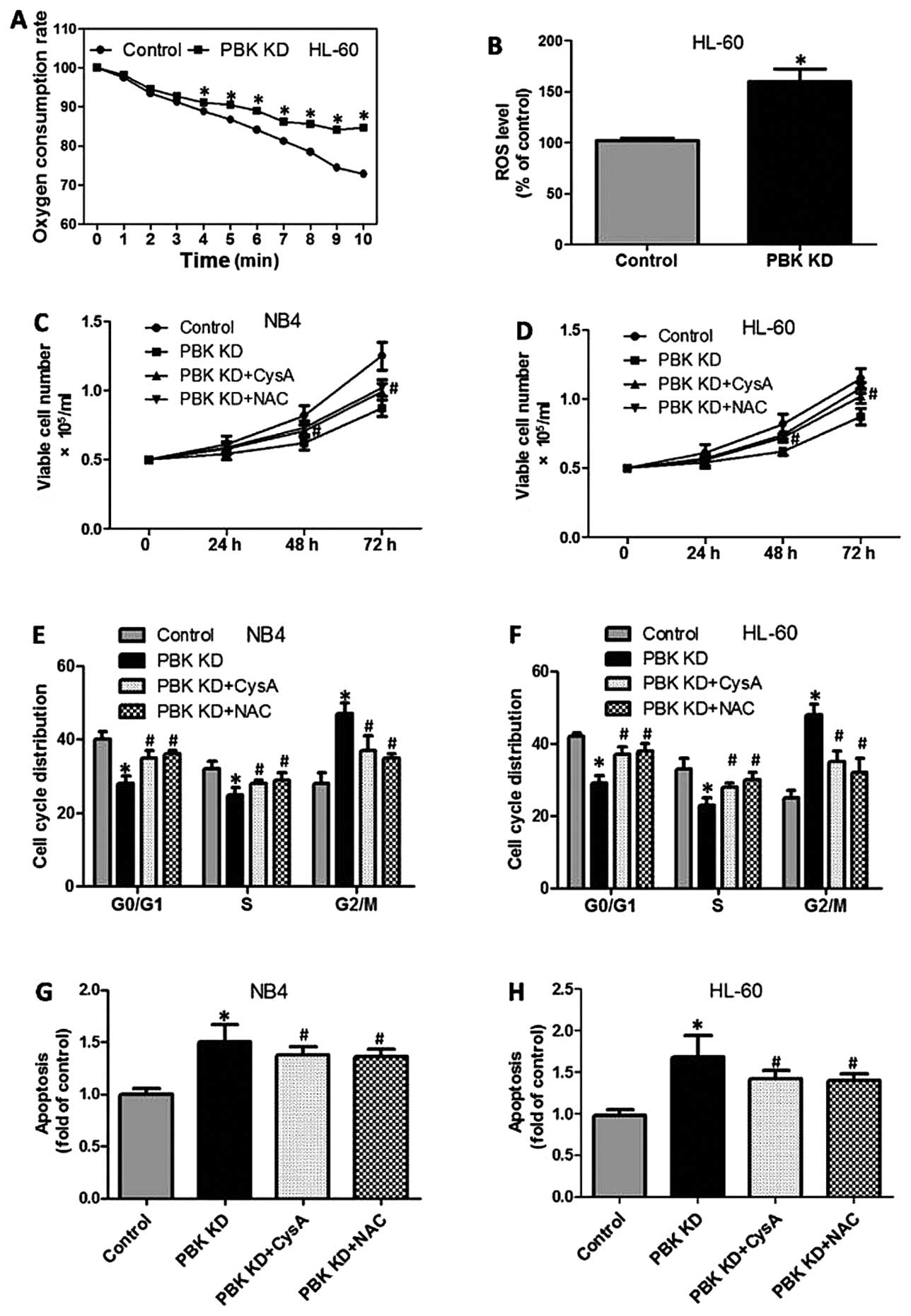
mitochondrial function and the ROS level were determined. As
illustrated in Fig. 4A, PBK/TOPK KD
in the HL-60 cells resulted in a significant decrease in oxygen
consumption ability, as evidenced by decreased oxygen consumption.
Moreover, PBK/TOPK KD markedly increased DCFH-DA fluorescence in
the HL-60 cells, indicating elevation in the ROS level (Fig. 4B). To test the role of mitochondrial
dysfunction and ROS generation in PBK/TOPK KD-induced inhibition of
proliferation of the promyelocytes, NB4 and HL-60 cells with
PBK/TOPK KD were incubated with cyclosporine (CysA, a mitochondrial
protective agent) or N-acetylcysteine (NAC, a potent antioxidant).
The results showed that in the presence of CysA and NAC, the
inhibitory effect of PBK/TOPK KD on cell growth in the NB4 and
HL-60 cells was significantly suppressed (Fig. 4C and D). PBK/TOPK KD-induced G2/M
cell cycle arrest was significantly blocked following treatment of
CysA and NAC, as evidenced by a decreased proportion of G2/M phase
cells and increased proportion of G0/G1 and S phase cells (Fig. 4E and F). In addition, the increase
in TUNEL-positive cell numbers induced by PBK/TOPK KD was notably
prohibited by CysA and NAC incubation (Fig. 4G and H).
Role of the reduction in Nrf2 expression
and activity in PBK/TOPK KD-induced inhibition of proliferation of
the promyelocytes
We next evaluated the effect of PBK/TOPK KD on the
expression and activity of Nrf2, an important upstream redox
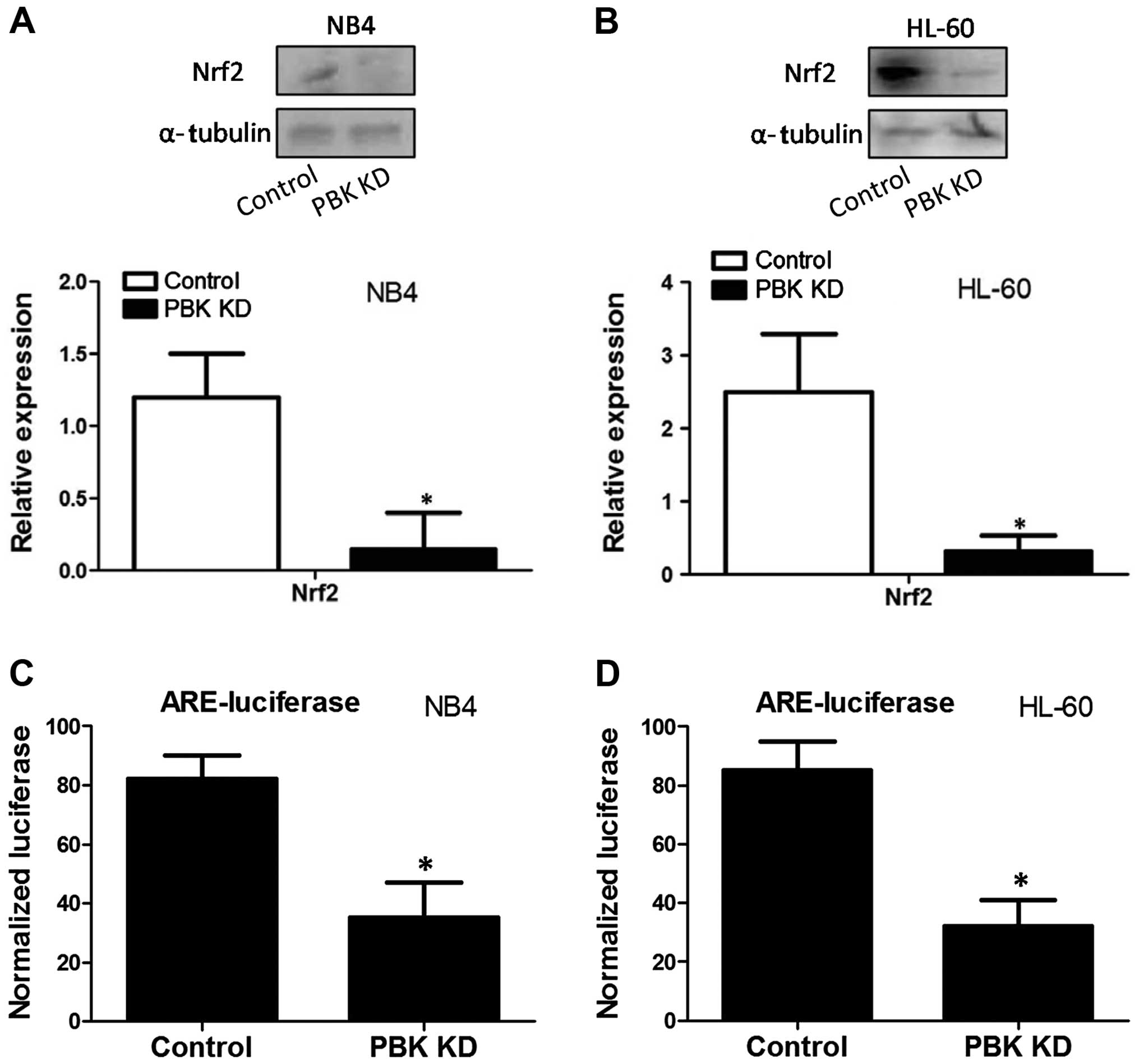
director. The results showed that in both NB4 and HL-60 cells,
PBK/TOPK KD notably decreased the protein expression of Nrf2
(Fig. 5A and B). Reporter gene
assay was conducted to evaluate the effect of PBK/TOPK KD on Nrf2
transcription activity. As shown in Fig. 5C and D, PBK/TOPK KD significantly
decreased ARE-luciferase activity in the NB4 and HL-60 cells,
indicating the reduction of Nrf2 transcription activity.
To test the role of the reduction in Nrf2
transcription activity in PBK/TOPK KD-induced inhibition of
proliferation of promyelocytes, NB4 and HL-60 cells with PBK/TOPK
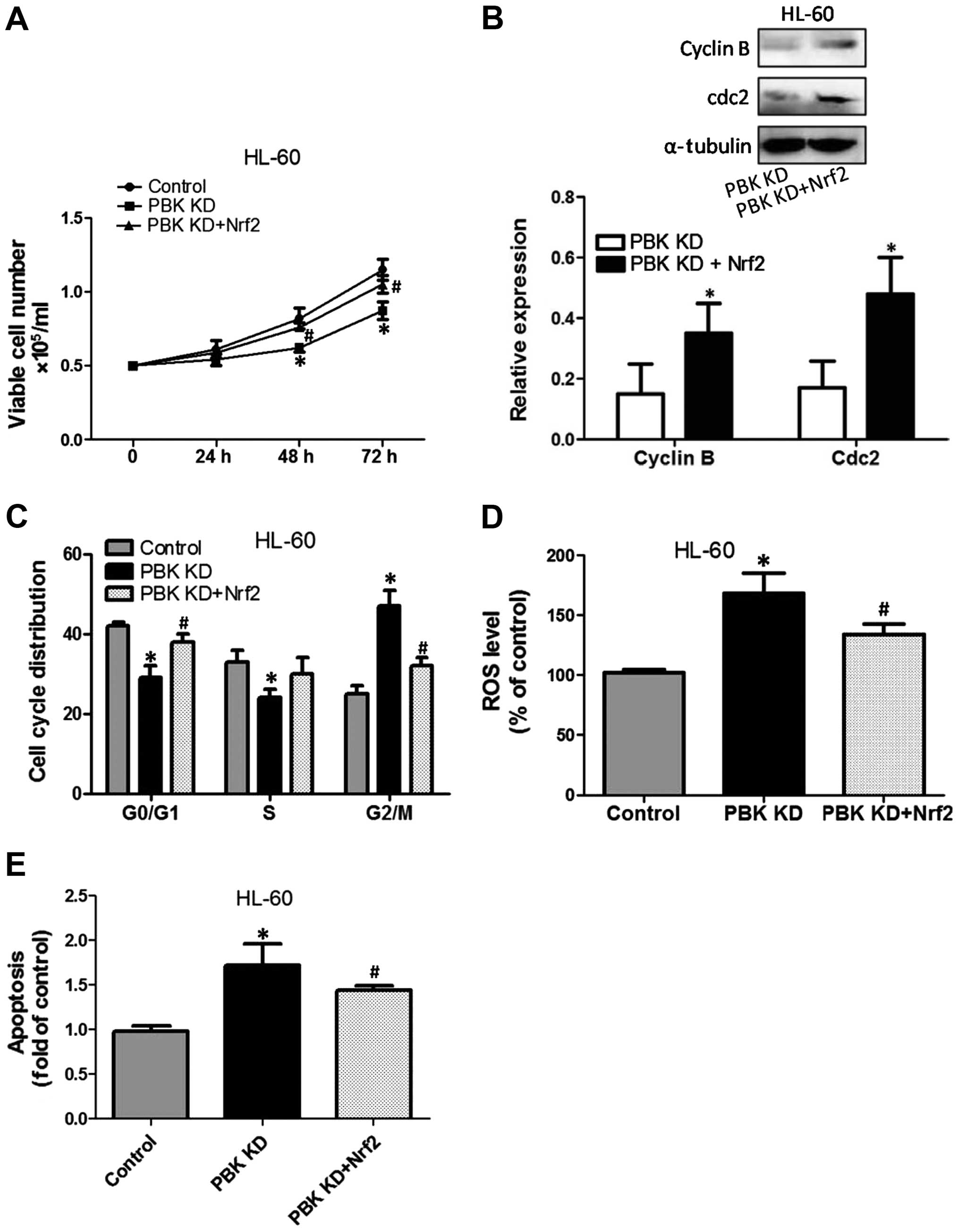
KD were transfected with a plasmid expressing Nrf2. The results
showed that overexpression of Nrf2 significantly suppressed
PBK/TOPK KD-induced decreased cell growth, as evidenced by an
increase in the viable cell numbers compared with that of the
PBK/TOPK KD cells (Fig. 6A). In
addition, the effect of the overexpression of Nrf2 on cell cycle
checkpoints and distribution was determined. As shown in Fig. 6B, compared with that of the PBK/TOPK
KD cells, overexpression of Nrf2 significantly increased cdc2 and
cyclin B protein expression. Overexpression of Nrf2 notably
decreased the proportion of G2/M phase cells and increased the
proportion of G0/G1 and S phase cells (Fig. 6C). Furthermore, the increase in ROS
level induced by PBK/TOPK KD was suppressed by overexpression of
Nrf2 (Fig. 6D). PBK/TOPK KD-induced
apoptosis was inhibited by enhancement of Nrf2 expression (Fig. 6E).
Discussion
Results from our and other laboratories have shown
that PBK/TOPK is pivotal for proliferation and malignant
transformation of hematologic tumors (7–9). In
the present study, we examined the molecular mechanism of
PBK/TOPK-mediated proliferation and viability of hematologic cells.
We showed that PBK/TOPK KD significantly inhibited cell
proliferation and viability in the promyelocytes.
Cell cycle progression is essential for cell
proliferation and growth and is controlled by a series of
cyclin-cdk complexes. cdc2/cyclin B is a complex controlling G2/M
cell cycle transition (12,13). Previous studies have shown that
PBK/TOPK is a substrate of cdc2/cyclin B and facilitates mitosis
(4,10). Our results showed that PBK/TOPK KD
significantly resulted in G2/M cell cycle arrest in promyelocytes,
as reflected by a notable increase in the G2/M phase cell
proportion. Moreover, the results showed that PBK/TOPK KD
contributed to the reduction in cdc2/cyclin B expression, which may
be responsible for G2/M cell cycle arrest. These results
demonstrated that PBK/TOPK and cdc2/cyclin B complex may form a
feedback circle in the regulation of cell cycle progression and
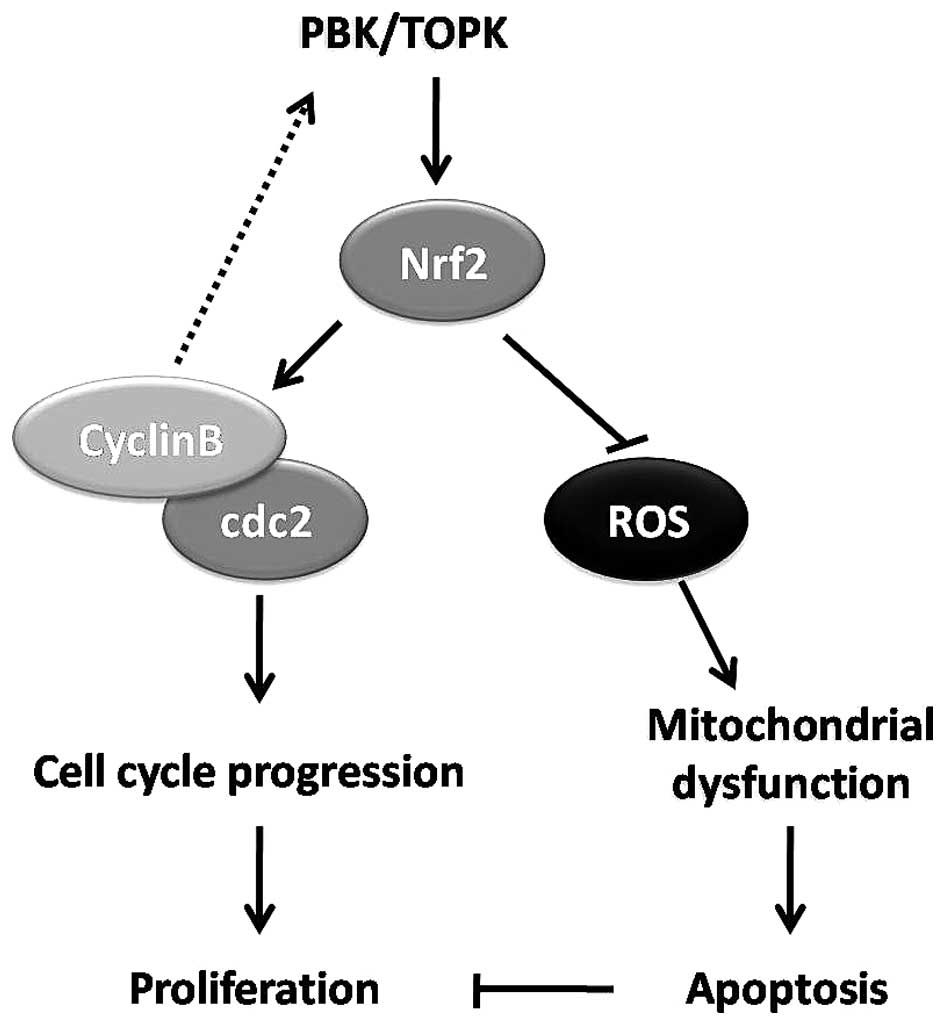
thus cell proliferation in promyelocytes (Fig. 7).
Apoptosis, also called programmed cell death, is a
common pathway for cell death which is inhibited in tumor
development (14,15). In the present study, we also tested
the effect of PBK/TOPK KD on apoptosis. The results showed that
PBK/TOPK KD significantly increased Bax expression, decreased Bcl-2
expression, promoted cleavage of caspase-3 and -9, and increased
apoptotic cell death in the NB4 and HL-60 cells. These results
indicated that potential blunting of the apoptotic pathway may be
involved in the PBK/TOPK-mediated cell proliferation in
promyelocytes.
ROS are an important stimuli of the activation of
the apoptotic pathway under both physiological and pathological
conditions (16). In cells, ROS are
mainly generated by the mitochondria (17,18)
and the generation of ROS contributes to mitochondrial damage,
release of pro-apoptotic molecules and the activation of caspase
cascades (19,20). Mitochondrial dysfunction has been
implicated in cellular senescence mainly by promoting oxidative
damage-induced cell cycle arrest (21). In addition, mitochondrial ROS
generation may influence cell cycle checkpoints resulting in cell
cycle arrest (22). In the present
study, we evaluated the effect of PBK/TOPK KD on mitochondrial
function and ROS generation. The results showed that PBK/TOPK KD
significantly increased ROS production and reduced mitochondrial
function. Moreover, we used the CysA, a mitochondrial protective
agent (23,24), and NAC, a potent antioxidant
(25), to test the role of ROS
production and mitochondrial dysfunction in PBK/TOPK KD-induced
inhibition of proliferation in promyelocytes. The results showed
that CysA and NAC significantly inhibited PBK/TOPK KD-induced G2/M
cell cycle arrest, apoptosis and blockage of cell proliferation in
promyelocytes. These results demonstrated that mitochondrial
dysfunction and ROS generation were involved in the PBK/TOPK
KD-induced G2/M cell cycle arrest, apoptosis and inhibition of
proliferation of promyelocytes (Fig.
7).
Antioxidant response element (ARE)-mediated
transcription of cytoprotective genes is essential for cellular
protection against excessive ROS generation and related disorders
(26,27). With ARE-binding activity, Nrf2
serves as a sensor and director of ROS insult and redox balance
(28). In addition, a large amount
of evidence supports that Nrf2 is a tumor promoter that promotes
oncogenesis under certain circumstances. For example,
overexpression of several oncogenes in mice led to increased Nrf2
transcription, increased basal expression of Nrf2, and decreased
ROS, leading to oncogenesis (29).
It has also been reported that Nrf2 upregulates transcription of
anti-apoptotic genes encoding Bcl-2 and Bcl-xL (30). Moreover, the cell cycle is
considered to be a redox cycle which is regulated by ROS and Nrf2
(31). It is noted that Nrf2
activity is upregulated in several types of leukemia where it
contributes to leukemogenesis (32). In AML, ROS usually mediate the
cytotoxic effect of therapeutic agents (33). Thus, high basal nuclear expression
of Nrf2 in AML reduces sensitivity to proteasome inhibitors
(33). In the present study, we
showed that PBK/TOPK KD caused a decrease in Nrf2 expression and
its binding activity with ARE. On the one hand, a decrease in Nrf2
and activity resulted in an increase in ROS generation, which
contributed to elevation of the ROS level and activation of the
mitochondrial apoptotic pathway. On the other hand, downregulation
of Nrf2 decreased cdc2 and cyclin B expression, leading to G2/M
cell cycle arrest. However, whether cdc2 and cyclin B were
regulated by Nrf2 itself or ROS indirectly and how did PBK/TOPK
regulate Nrf2 remain to be elucidated.
Based on previous literature and our results, it was
demonstrated that Nrf2 may be a crucial regulator that mediates
PBK/TOPK-exerted promotion of cell proliferation. PBK/TOPK
stabilizes Nrf2, strictly regulates the ROS level, promotes cell
cycle progression and inhibits apoptosis, contributing to
proliferation of promyelocytes. Our results provide new insights
into the molecular mechanism of PBK/TOPK-mediated promyelo-cyte
proliferation and the pathogenesis of AML.
Abbreviations:
|
AML
|
acute myeloid leukemia
|
|
APL
|
acute promyelocytic leukemia
|
|
ARE
|
antioxidant response element
|
|
CysA
|
cyclosporine
|
|
KD
|
knockdown
|
|
NAC
|
N-acetylcysteine
|
|
Nrf2
|
nuclear factor (erythroid-derived
2)-like 2
|
|
PBK
|
PDZ-binding kinase
|
|
ROS
|
reactive oxygen species
|
|
TOPK
|
T-lymphokine-activated killer (T-LAK)
cell-originated protein kinase
|
|
TUNEL
|
terminal deoxynucleotidyl
transferase-mediated dUTP nick end labeling
|
References
|
1
|
Rampal R and Mascarenhas J: Pathogenesis
and management of acute myeloid leukemia that has evolved from a
myeloproliferative neoplasm. Curr Opin Hematol. 21:65–71. 2014.
View Article : Google Scholar
|
|
2
|
Nasr R, Lallemand-Breitenbach V, Zhu J,
Guillemin MC and de Thé H: Therapy-induced PML/RARA proteolysis and
acute promyelocytic leukemia cure. Clin Cancer Res. 15:6321–6326.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Coombs CC, Tavakkoli M and Tallman MS:
Acute promyelocytic leukemia: Where did we start, where are we now,
and the future. Blood Cancer J. 5:e3042015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gaudet S, Branton D and Lue RA:
Characterization of PDZ-binding kinase, a mitotic kinase. Proc Natl
Acad Sci USA. 97:5167–5172. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Abe Y, Matsumoto S, Kito K and Ueda N:
Cloning and expression of a novel MAPKK-like protein kinase,
lymphokine-activated killer T-cell-originated protein kinase,
specifically expressed in the testis and activated lymphoid cells.
J Biol Chem. 275:21525–21531. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhao S, Dai J, Zhao W, Xia F, Zhou Z, Wang
W, Gu S, Ying K, Xie Y and Mao Y: PDZ-binding kinase participates
in spermatogenesis. Int J Biochem Cell Biol. 33:631–636. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nandi A, Tidwell M, Karp J and Rapoport
AP: Protein expression of PDZ-binding kinase is up-regulated in
hematologic malignancies and strongly down-regulated during
terminal differentiation of HL-60 leukemic cells. Blood Cells Mol
Dis. 32:240–245. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Côté S, Simard C and Lemieux R: Regulation
of growth-related genes by interleukin-6 in murine myeloma cells.
Cytokine. 20:113–120. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Simons-Evelyn M, Bailey-Dell K, Toretsky
JA, Ross DD, Fenton R, Kalvakolanu D and Rapoport AP: PBK/TOPK is a
novel mitotic kinase which is upregulated in Burkitt's lymphoma and
other highly proliferative malignant cells. Blood Cells Mol Dis.
27:825–829. 2001. View Article : Google Scholar
|
|
10
|
Matsumoto S, Abe Y, Fujibuchi T, Takeuchi
T, Kito K, Ueda N, Shigemoto K and Gyo K: Characterization of a
MAPKK-like protein kinase TOPK. Biochem Biophys Res Commun.
325:997–1004. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ayllón V and O'connor R: PBK/TOPK promotes
tumour cell proliferation through p38 MAPK activity and regulation
of the DNA damage response. Oncogene. 26:3451–3461. 2007.
View Article : Google Scholar
|
|
12
|
Kumagai A and Dunphy WG: Control of the
Cdc2/cyclin B complex in Xenopus egg extracts arrested at a G2/M
checkpoint with DNA synthesis inhibitors. Mol Biol Cell. 6:199–213.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Clarke PR, Klebe C, Wittinghofer A and
Karsenti E: Regulation of cdc2/cyclin b activation by ran, a
ras-related GTPase. J Cell Sci. 108:1217–1225. 1995.PubMed/NCBI
|
|
14
|
Mohammad RM, Muqbil I, Lowe L, Yedjou C,
Hsu HY, Lin LT, Siegelin MD, Fimognari C, Kumar NB, Dou QP, et al:
Broad targeting of resistance to apoptosis in cancer. Semin Cancer
Biol. Apr 28–2015.Epub ahead of print. View Article : Google Scholar
|
|
15
|
Flusberg DA and Sorger PK: Surviving
apoptosis: Life-death signaling in single cells. Trends Cell Biol.
Apr 25–2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sinha K, Das J, Pal PB and Sil PC:
Oxidative stress: The mitochondria-dependent and
mitochondria-independent pathways of apoptosis. Arch Toxicol.
87:1157–1180. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Balaban RS, Nemoto S and Finkel T:
Mitochondria, oxidants, and aging. Cell. 120:483–495. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Turrens JF: Mitochondrial formation of
reactive oxygen species. J Physiol. 552:335–344. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bonora M and Pinton P: The mitochondrial
permeability transition pore and cancer: Molecular mechanisms
involved in cell death. Front Oncol. 4:3022014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chistiakov DA, Sobenin IA, Revin VV,
Orekhov AN and Bobryshev YV: Mitochondrial aging and age-related
dysfunction of mitochondria. Biomed Res Int. 2014:2384632014.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Correia-Melo C and Passos JF:
Mitochondria: Are they causal players in cellular senescence?
Biochim Biophys Acta. May 28–2015.Epub ahead of print. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Singh KK: Mitochondria damage checkpoint,
aging, and cancer. Ann NY Acad Sci. 1067:182–190. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Penna C, Perrelli MG and Pagliaro P:
Mitochondrial pathways, permeability transition pore, and redox
signaling in cardioprotection: Therapeutic implications. Antioxid
Redox Signal. 18:556–599. 2013. View Article : Google Scholar
|
|
24
|
Osman MM, Lulic D, Glover L, Stahl CE, Lau
T, van Loveren H and Borlongan CV: Cyclosporine-A as a
neuroprotective agent against stroke: Its translation from
laboratory research to clinical application. Neuropeptides.
45:359–368. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rushworth GF and Megson IL: Existing and
potential therapeutic uses for N-acetylcysteine: The need for
conversion to intracellular glutathione for antioxidant benefits.
Pharmacol Ther. 141:150–159. 2014. View Article : Google Scholar
|
|
26
|
Kaspar JW, Niture SK and Jaiswal AK:
Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic Biol
Med. 47:1304–1309. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hayes JD and McMahon M: NRF2 and KEAP1
mutations: Permanent activation of an adaptive response in cancer.
Trends Biochem Sci. 34:176–188. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang DD: Mechanistic studies of the
Nrf2-Keap1 signaling pathway. Drug Metab Rev. 38:769–789. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
DeNicola GM, Karreth FA, Humpton TJ,
Gopinathan A, Wei C, Frese K, Mangal D, Yu KH, Yeo CJ, Calhoun ES,
et al: Oncogene-induced Nrf2 transcription promotes ROS
detoxification and tumorigenesis. Nature. 475:106–109. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Niture SK and Jaiswal AK: Nrf2-induced
antiapoptotic Bcl-xL protein enhances cell survival and drug
resistance. Free Radic Biol Med. 57:119–131. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Burhans WC and Heintz NH: The cell cycle
is a redox cycle: Linking phase-specific targets to cell fate. Free
Radic Biol Med. 47:1282–1293. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rushworth SA, MacEwan DJ and O'Connell MA:
Lipo-polysaccharide-induced expression of NAD(P)H:quinone
oxidoreductase 1 and heme oxygenase-1 protects against excessive
inflammatory responses in human monocytes. J Immunol.
181:6730–6737. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rushworth SA, Bowles KM and MacEwan DJ:
High basal nuclear levels of Nrf2 in acute myeloid leukemia reduces
sensitivity to proteasome inhibitors. Cancer Res. 71:1999–2009.
2011. View Article : Google Scholar : PubMed/NCBI
|