Introduction
Esophageal cancer (EC) is the eighth most common
cancer worldwide and ranks sixth among all cancers in mortality due
to its high fatality rate (1). EC
is mainly comprised of two histologic subtypes, esophageal squamous
cell carcinoma (ESCC) and esophageal adenocarcinoma, which shows
striking variation by both geography and etiologic factors
(2). In China, often referred to as
the 'esophageal cancer belt', 90% of all cases are ESCC (3). According to the epidemiologic
statistics (4), cigarette smoking
is one of the major risk factors for ESCC development. Abundant
evidence has shown that the exposure of cancer cells to cigarette
smoke extracts (CSE) could affect cell proliferation, invasion,
metastasis, cell death and immune response by modulating several
critical signaling pathways (5).
Cyclooxygenase-2 (COX-2), an immediate-early
response gene that is induced by a variety of stimuli such as
mitogens, cytokines and growth factors. Although evidence strongly
supports that smoking extracts upregulate COX-2 expression, thereby
facilitating cell malignant transformation and cancer development,
the detailed mechanisms of how CSE elevates COX-2 expression are
largely unknown. To date, researchers have found that CSE function
along with β-adrenoceptors and α7-nAChR to stimulate COX-2 and its
derived prostanoids by activation of COX-2 gene regulators NF-κB
and CREB through p38MAPK, ERK and cAMP-dependent pathways (6). Another study also found that
methylation of COX-2 promoter regulates COX-2 expression in ESCC in
response to the stimulation of CSE (7). Those mechanisms mainly relate to
transcriptional regulation of COX-2 by CSE, yet,
post-transcriptional mechanisms by which CSE upregulates COX-2
expression have not been explored.
MicroRNAs (miRNAs) are endogenous, small (~22
nucleotide long) non-coding RNAs, which serve as key regulators of
gene expression at the post-transcriptional level by binding to the
3′-untranslated region (UTR) of corresponding target mRNA (8). Growing evidence has showed that
exposure of cells to CSE causes extensive alterations in miRNA
expression (9). Yet, the detailed
miRNAs and correspondent targets contributing to
CSE-induced-cancinogensis are not fully defined. In the present
study, through microarray analysis of differential microRNA
expression in human esophageal epithelial cell line treated with or
without CSE, we found 47 downregulated and 13 upregulated miRNAs.
Furthermore, we revealed CSE-miR-101(−)-COX-2(+) axis, from which
low concentration of CSE facilitated cancer cell proliferation,
thereby providing insights into the mechanisms of miR-101-3p
contributing to CSE-associated ESCC development.
Materials and methods
Cell line and culture
The human immortalized non-tumorigenic esophageal
epithelial cell line (Het-1A) was purchased from the American Type
Culture Collection (ATCC; lot no. CRL-2692) and was cultured in
BEGM culture medium including BPE, hydrocortisone, human epidermal
growth factor, epinephrine, insulin, triiodothyronine, transferrin,
gentamicin/amphotericin-B and retinoic acid. HEK293T cell line was
preserved in our laboratory. The human ESCC cell line (Eca109) was
obtained from the Shanghai Institute of Biochemistry and Cell
Biology (Shanghai, China). Eca109 cells were grown in RPMI-1640
containing 10% fetal bovine serum (both from Gibco, New York, NY,
USA) 100 units of penicillin/ml and 100 mg of streptomycin/ml
(Invitrogen, Carlsbad, CA, USA), and all cells were incubated at
37° C in a humidified chamber supplemented with 5%
CO2.
Cigarette smoke exact preparation
Cigarettes were purchased from Hunan Zhongyan
Industrial Co., Ltd. [Hongmei Brand; tar content, 12 mg; and
nicotine content, 1.3 mg/cigarette; smoke gas (CO): 13 mg]. CSE was
prepared by a modification based on the method of Su et al
(10). In brief, three cigarettes
with filters were combusted with a modified syringe-driven
apparatus. The smoke from cigarettes was bubbled through 30 ml of
sterile phosphate-buffered solution (PBS) which was pre-warmed to
37° C by application of a vacuum to the vessel containing the PBS.
Each cigarette was smoked for 5 min, and three cigarettes were used
per 30 ml of PBS to generate a CSE-PBS solution. Control solutions
were prepared with the same protocols used to generate CSE, except
that the cigarettes were unlit. CSE stock was adjusted to pH 7.4,
then filtered through a 0.20-µm pore filter. The
concentration of nicotine in the CSE stock solution was measured by
high-performance liquid chromatography (HPLC) at the Department of
Pharmacy, The Second Xiangya Hospital of Central South University,
and the concentration of nicotine was ~10,000 ng/l in CSE stock
solution. The CSE solutions were diluted with RPMI-1640 medium and
used immediately as subsequently described. Final concentrations of
these solutions are expressed as percent values, which were
calculated with the following equation: (ml CSE solution ÷ total
ml) × 100. Total milliliters in this equation are the sum of
milliliters of CSE solution and milliliters of RPMI-1640 (10). Solutions with concentration ranging
from 0.01 to 20% were used in the present study.
Establishment of the stable miR-101-3p
overexpressing cell lines
Commercial pLVX-IRES-ZsGreen1 for miR-101-3p
overexpression was purchased from Clontech (Palo Alto, CA, USA).
Lentiviral vectors of pLVX-pre-miRNA-101-3p which expressed
miR-101-3p precursor were constructed by Yinrunbio (Changsha,
China). A pre-mixed Lentiviral Packaging System (Biosettia, San
Diego, CA, USA) used for viral packaging was transfected into
HEK293T cells using Lipofectamine 2000 according to the
manufacturer's instructions (Invitrogen). Lentivirus was added to
Het-1A or Eca109 cells at 50% confluency in 100 mm dishes along
with Polybrene at a final concentration of 8 µg/ml. After 72
h transduction, Het-1A or Eca109 cells with Lenti-miR-101-3p or
Lenti-NC (negative control) expression were sorted by
fluorescence-activated cell sorting (FACS) using green fluorescence
protein ZsGreen1 as selecting marker, and then used for subsequent
experiments including proliferation, quantitative real-time
polymerase chain reaction (qRT-PCR) and western blotting.
Quantitative real-time polymerase chain
reaction (qRT-PCR) and miRNA microarray analysis
Total RNA from the frozen tissue specimens and
cultured cells was extracted using the TRIzol kit (Invitrogen)
according to the manufacturer's instructions. cDNA synthesis and
qPCR was performed with Invitrogen NCode™ miRNA
SYBR®-Green qRT-PCR analysis (Invitrogen). The forward
primer for miR-101-3p was 5′-CCGGTACAGTACTGTGATAACTGAA-3′.
Fold-change (2−ΔΔCt) normalized to control U6 small
nuclear RNA (snRNA) levels was used to compare differential miRNA
expression. For miRNA microassay analysis, total RNA was isolated
with TRIzol reagent from Het-1A cells with or without CSE
treatments. Samples were labeled and hybridized with miRCURY LNA™
Array v. 16.0 (Exiqon, Denmark). GenePix 4000B scanner and GenePix
Pro 6.0 software (Axon Instruments, Union City, CA, USA) were used
to scan images for the analysis. Each chip was normalized to the U6
signal intensity. miRNAs with a significant value of 0.05 or lower
and a fold-change value of 2 or higher were considered to be
differentially expressed.
Luciferase reporter gene assays
The putative miR-101 binding sites at the 3′-UTRs of
COX-2 mRNAs (NM_000963.1 nt 3481-3954 with seed sequence at nt
3689-3696) was cloned downstream of luciferase gene into the
pGL3-control Dual-Luciferase miRNA Target Expression Vector
(Promega, Madison, WI, USA). Primer used for cloning were:
5′-GGACTAGTGCTATCTGTAACCAAGATGG-3′ (forward) and
5′-CCCAAGCTTCACATAGGCCTATCCTAAGG-3′ (reverse) (11). The pGL3-COX-2-3-UTR-MU plasmid,
which carried the mutated sequence in the complementary sites for
the seed region of miR-101-3p, was generated based on
pGL3-COX-2-3-UTR-WT plasmid by site-specific mutagenesis
(QuikChange™ II; Stratagene, La Jolla, CA, USA).
Eca109 cells with Lenti-miR-101-3p and Lenti-NC
expression were plated into 24-well plates (3×104
cells/well). After 24 h, the cells were co-transfected with the 50
ng control pRL-TK plasmid containing the Renilla luciferase
gene (Promega) and 300 ng pGL3-COX-2-3-UTR (WT/MUT) plasmid DNA or
300 ng pGL3 control-luciferase plasmid using Lipofectamine 2000. At
48 h post-transfection, luciferase activity was detected using the
Dual-Luciferase Reporter Assay system (Promega). All transfection
experiments were performed in triplicate and repeated at least
three times.
Cell viability
Cell viability was measured using
3-(4,5-dimeth-ylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay. After incubation with CSE at different concentrations for 0,
24, 48 and 72 h, Het-1A or Eca109 cells with Lenti-miR-101-3p or
Lenti-NC overexpression were washed with PBS three times to remove
CSE and then incubated with 2.5% MTT solution (5 mg/ml) for another
4 h at 37° C. Thereafter, DMSO was added to solubilize the crystals
for 20 min at room temperature. The optical density was determined
with a spectrophotometer (Thermo Scientific Varioskan Flash, USA)
at a wavelength of 490 nm. All experiments were performed three
times in triplicate.
siRNA interference
To suppress expression of COX-2, the following
previously described pairs of oligonucleotides were used: siMock,
5′-GUAAGACACGACUUAUCGCdTdT-3′ and 5′-GCGAUAAGUCGUGUCUUACdTdT-3′;
siCOX-2, 5′-UGA AAGGACUUAUGGGUAAdTdT-3′ and 5′-UUACCCAUAAG
UCCUUUCAdTdT-3′ (12). Each pair of
oligonucleotides (5 µM each) was dissolved in an annealing
buffer (5 mM Tris-HCl, pH 7.5, 1 mM EDTA), heated at 65° C for 5
min and then slowly cooled to room temperature and stored at −80°
C. Eca109 cells were transfected with 100 pmol of each duplex/well
of a 24-well using oligofectamine reagent (Invitrogen) according to
the manufacturer's recommendation.
Protein extraction and western
blotting
All cells were rinsed with PBS (pH 7.4) and were
lysed on ice for 30 min in RIPA lysis buffer (Beyotime, China)
supplemented with a protease inhibitor cocktail (Roche,
Switzerland). The tissue samples were frozen solid with liquid
nitrogen, ground into powder, and lysed on ice for 30 min in RIPA
lysis buffer containing the protease and phosphatase inhibitor.
When necessary, sonication was used to facilitate lysis. Cell
lysates or tissue homogenates were centrifuged for 30 min (14,100 ×
g, 4° C). The supernatant was collected, and the protein
concentration was calculated using the BCA protein assay kit
(Conway Century, China). The protein levels were analyzed via
western blotting using the COX-2 antibody (CST, Danvers, MA, USA).
The protein levels were normalized by probing the same blots with a
β-actin antibody (Sigma, St. Louis, MO, USA). Protein bands were
analyzed using the ImageJ (National Institutes of Health, Bethesda,
MD, USA).
Flow cytometric analysis
Approximately 1−2×106 single cells were
harvested and washed in cold PBS twice, then fixed in 70% ethanol
overnight. Cells were washed the next day in cold PBS once and then
incubated in propidium iodide (PI) buffer (PBS containing 40
µg/ml PI and 100 µg/ml RNAase) at 37° C for 30 min
prior to analysis by flow cytometry (BD FACSCanto II analyzer; BD
Biosciences). The percentage of sub-G1 population indicative of
cell death was analyzed with WinMDI 2.9. The mean value was
calculated from three independent experiments.
Patients and samples
From October 2011, 8 pairs of fresh human samples of
ESCC and corresponding adjacent non-tumor tissue (3 cm from the
cancer tissue) were obtained from patients at The Second Xiangya
Hospital of Central South University. All the 8 patients in the
study had a long history of smoking but almost no drinking. None of
them received any chemotherapy or radiation therapy before surgery.
All the patients were informed of the purpose and procedure of the
present study and agreed to donate excess tissue. The study was
approved by the Ethics Committee of the Second Xiangya Hospital
[file no. 184 (2010)] and written informed consent was obtained
from all surgical patients to use resected samples for
research.
Statistical analysis
All statistical analyses were carried out using SPSS
version 18.0 statistical software (Aspire Software International,
Leesburg, VA, USA). Student's t-test was used to determine
statistical significance. All data represent mean ± SD. All
statistical tests were two-sided and P-values <0.05 were
considered to indicate a statistically significant result.
Results
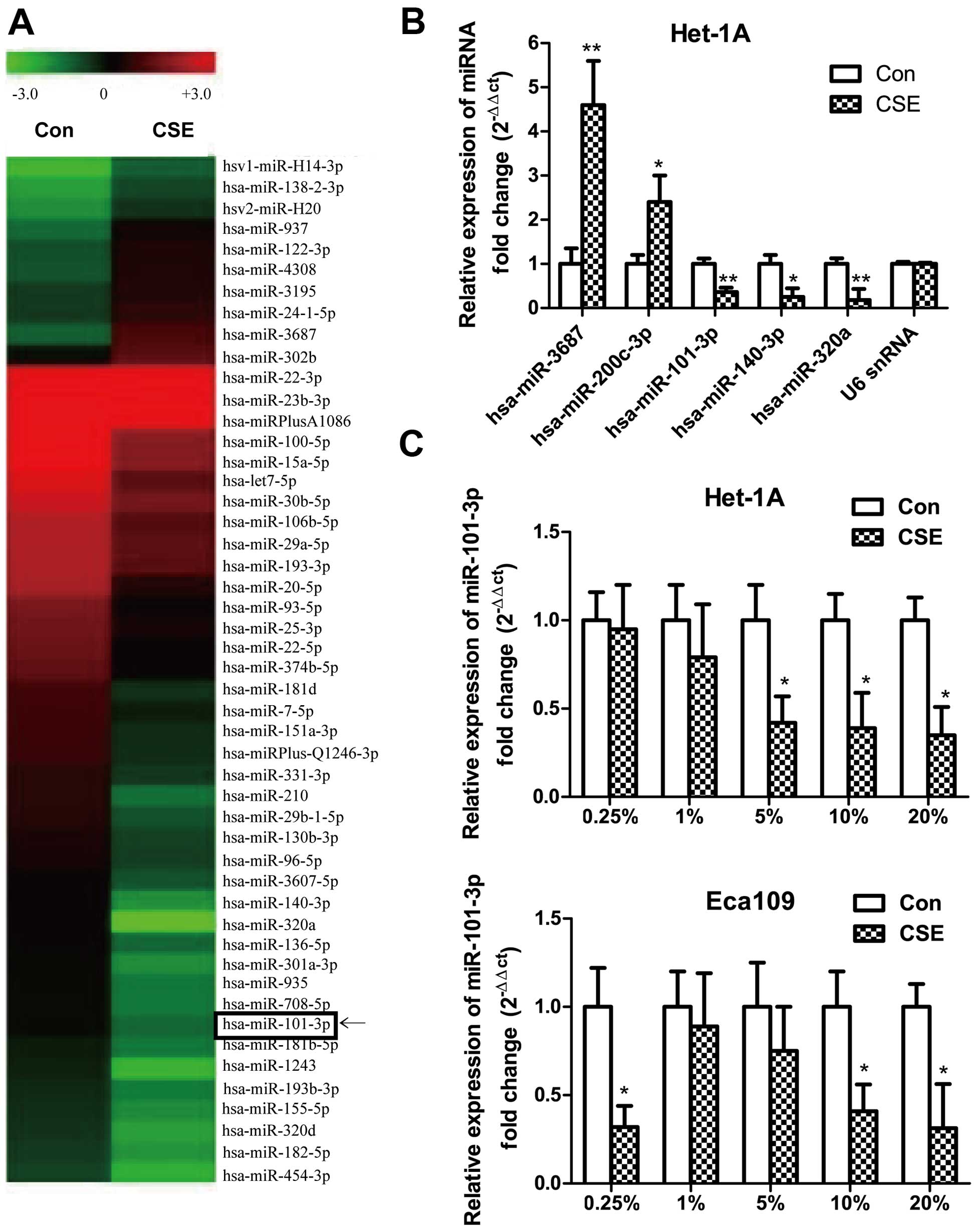
CSE induces a decrease in miR-101-3p
expression
Microarray analysis revealed 60 miRNAs that were
differentially regulated in 20% CSE treated immortalized
non-tumorigenic esophageal epithelial cell line Het-1A compared to
sham-exposed cells, among which 47 miRNAs are downregulated and 13
miRNAs are upregulated (Table I). A
representative heat map of differentially expressed miRNAs between
CSE-exposed and sham-exposed Het-1A cells is shown in Fig. 1A. Microarray results were validated
by qRT-PCR analysis of the following miRNAs: hsa-miR-3687,
hsa-miR-200c-3p (upregulated) and hsa-miR-101-3p, hsa-miR-140-3p
and hsa-miR-320a (downregulated) (Fig.
1B). Among those miRNAs, we focused on miR-101-3p, functions of
which have not been defined upon cells exposure to CSE, yet the
expression has been found downregulated in many types of cancers,
including ESCC (13). We then
studied the effects of different concentration of CSE on the
expression of miR-101-3p in Het-1A and cancer cell line Eca109, and
qRT-PCR results showed that higher concentrations of CSE could
induce a decrease in miR-101-3p expression in both cell lines
(Fig. 1C), notably, low
concentration of CSE (0.25%), not medium concentration of CSE, also
induced a significant decrease in miR-101-3p expression in Eca109,
but not Het1A cells (Fig. 1C). The
differences of miR-101-3p expression to CSE treatments facilitated
us to study phenotype changes of cells to different concentrations
of CSE solution.
 | Table IThe differential expression profiles
of miRNAs in CSE-induced Het-1A cells compared to sham-control
cells using miRNA array analysis. |
Table I
The differential expression profiles
of miRNAs in CSE-induced Het-1A cells compared to sham-control
cells using miRNA array analysis.
| ID | Name | Fold-changes | Normalized
|
|---|
| Co. | Exp. |
|---|
| Upregulated |
| 14808 | hsa-miR-3687 | 2.43 | 0.45 | 1.86 |
| 42551 | hsa-miR-122-3p | 2.26 | 0.58 | 1.28 |
| 145789 |
hsa-miR-550a-3-5p/hsa-miR-550a-5p | 2.68 | 0.14 | 0.31 |
| 147604 | hsa-miR-4285 | 2.64 | 0.16 | 0.43 |
| 147947 | hsa-miR-4308 | 2.16 | 0.49 | 1.30 |
| 42454 | hsa-miR-138-2-3p | 2.85 | 0.26 | 0.56 |
| 42514 | hsa-miR-937 | 3.0 | 0.41 | 1.18 |
| 17427 | hsa-miR-200c-3p | 2.15 | 0.12 | 0.35 |
| 148000 | hsa-miR-3195 | 2.31 | 0.63 | 1.35 |
| 146043 | hsa-miR-24-1-5p | 2.01 | 0.63 | 1.45 |
| 147739 | hsa-miR-3161 | 2.39 | 0.12 | 0.24 |
| 45764 | hsa-miR-302e | 2.77 | 0.89 | 2.12 |
| 145914 | hsa-miR-135b-5p | 2.43 | 0.09 | 0.24 |
| Downregulated |
| 145638 | hsa-miR-29a-5p | 0.48 | 4.62 | 2.23 |
| 31026 | hsa-miR-101-3p | 0.47 | 0.92 | 0.43 |
| 10943 | hsa-miR-136-5p | 0.45 | 0.96 | 0.43 |
| 17463 | hsa-miR-151a-3p | 0.44 | 1.62 | 0.71 |
| 10964 | hsa-miR-155-5p | 0.45 | 0.69 | 0.31 |
| 17810 | hsa-miR-29b-1-5p | 0.38 | 1.37 | 0.51 |
| 10975 | hsa-miR-182-5p | 0.45 | 0.63 | 0.29 |
| 29190 | hsa-miR-708-5p | 0.39 | 0.93 | 0.36 |
| 42663 | hsa-miR-20a-3p | 0.23 | 0.32 | 0.08 |
| 145852 | hsa-miR-210 | 0.28 | 1.40 | 0.40 |
| 145742 | hsa-miR-935 | 0.40 | 0.94 | 0.37 |
| 27536 | hsa-miR-190a | 0.47 | 0.44 | 0.21 |
| 10936 |
hsa-miR-130b-3p | 0.47 | 1.26 | 0.59 |
| 10986 |
hsa-miR-193a-3p | 0.47 | 4.59 | 2.14 |
| 42682 | hsa-miR-25-3p | 0.47 | 2.60 | 1.23 |
| 147735 | hsa-miR-4289 | 0.47 | 0.38 | 0.18 |
| 17885 |
hsa-miRPlus-A1086 | 0.49 | 27.97 | 13.21 |
| 28950 | hsa-miR-455-3p | 0.48 | 0.31 | 0.15 |
| 42630 | hsa-miR-140-3p | 0.28 | 1.05 | 0.30 |
| 27533 | hsa-miR-320a | 0.12 | 0.98 | 0.12 |
| 9938 | hsa-let-7i-5p | 0.32 | 6.90 | 2.20 |
| 11020 | hsa-miR-22-3p | 0.34 | 46.80 | 15.70 |
| 27720 | hsa-miR-15a-5p | 0.40 | 7.95 | 3.15 |
| 4610 | hsa-miR-126-3p | 0.10 | 0.32 | 0.03 |
| 17961 | hsa-miR-629-5p | 0.28 | 0.50 | 0.14 |
| 19582 |
hsa-miR-106b-5p | 0.44 | 4.63 | 2.04 |
| 13147 | hsa-miR-96-5p | 0.48 | 1.24 | 0.60 |
| 29490 | hsa-miR-7-5p | 0.48 | 1.69 | 0.81 |
| 42887 | hsa-miR-331-3p | 0.46 | 1.41 | 0.65 |
| 145670 | hsa-miR-18b-5p | 0.48 | 0.49 | 0.23 |
| 42532 | hsa-miR-22-5p | 0.41 | 2.45 | 1.01 |
| 148620 | hsa-miR-454-3p | 0.35 | 0.58 | 0.20 |
| 148418 |
hsa-miR-3607-5p | 0.48 | 1.07 | 0.51 |
| 10972 |
hsa-miR-181b-5p | 0.45 | 0.81 | 0.36 |
| 17918 | hsa-miR-222-5p | 0.24 | 0.31 | 0.07 |
| 30687 | hsa-miR-93-5p | 0.40 | 2.79 | 1.11 |
| 145943 | hsa-miR-100-5p | 0.40 | 7.97 | 3.16 |
| 145636 | hsa-miR-181d | 0.39 | 1.73 | 0.67 |
| 145845 | hsa-miR-20a-5p | 0.31 | 4.24 | 1.33 |
| 13143 |
hsa-miR-301a-3p | 0.32 | 0.94 | 0.30 |
| 17888 | hsa-let-7a-3p | 0.50 | 0.30 | 0.15 |
| 46870 | hsa-miR-320d | 0.38 | 0.67 | 0.26 |
| 146112 | hsa-miR-30b-5p | 0.47 | 5.84 | 2.74 |
| 148098 |
hsa-miR-374b-5p | 0.44 | 2.26 | 0.99 |
| 46439 | hsa-miR-1243 | 0.24 | 0.77 | 0.19 |
| 10987 |
hsa-miR-193b-3p | 0.49 | 0.73 | 0.35 |
| 145841 | hsa-miR-23b-3p | 0.43 | 27.31 | 11.86 |
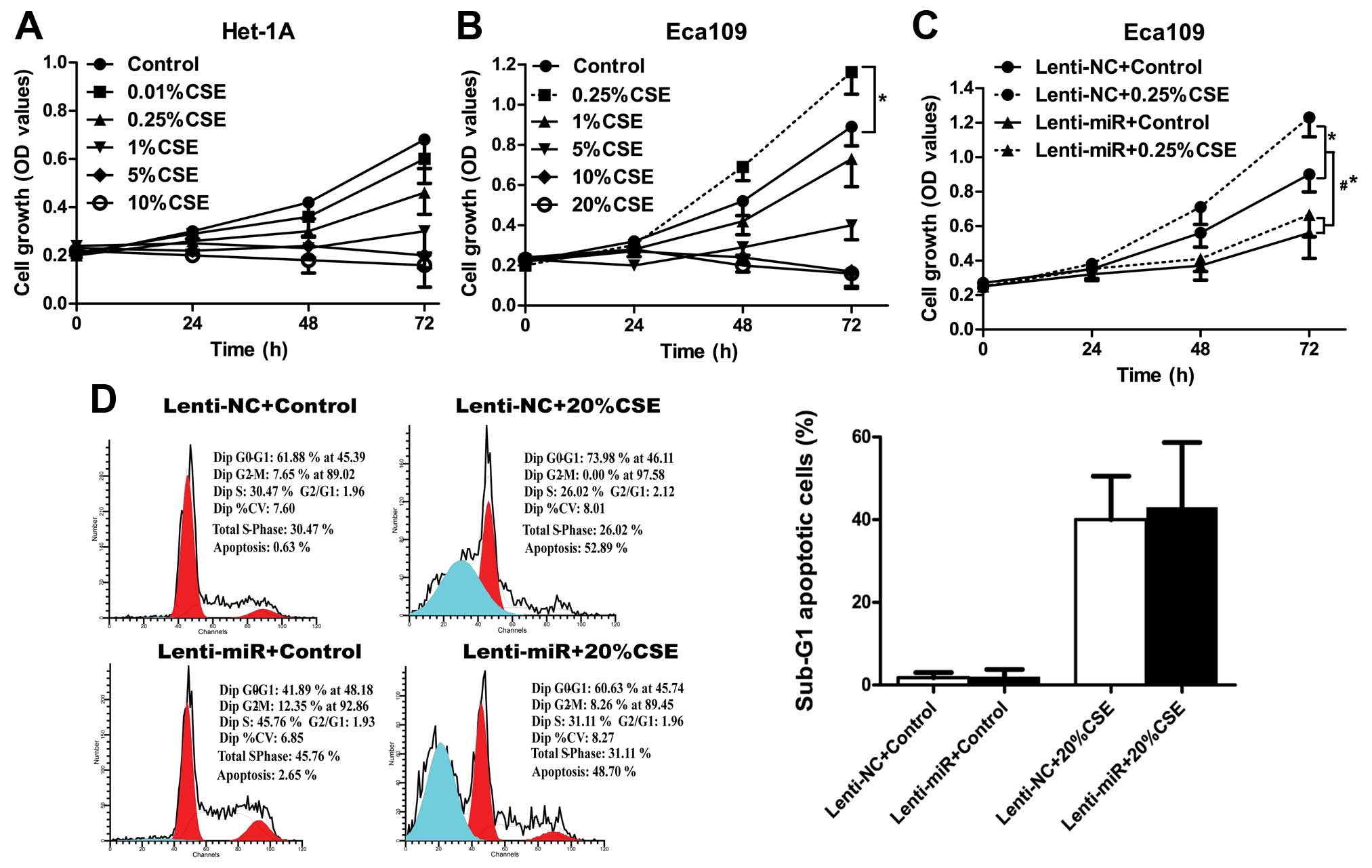
Cancer cell proliferation induced by
exposure to low concentration of CSE was mediated via suppression
of miR-101-3p
As contradictory results on cell growth and cell
death have been reported when cells were exposed to different
models and concentrations of CS-extract, we first applied MTT assay
to study cell proliferation effects in both Het-1A and Eca109 cells
under different concentrations of our CSE solution. Results showed
that CSE inhibited proliferation of the normal esophageal
epithelial Het-1A cells, and massive cell death was found at 5% and
the higher concentration of CSE treatment, which presented as a
plateau or decrease in cell proliferation curve (Fig. 2A). Eca109 cells are comparatively
resistant to CSE treatment with a plateau or decrease in cell
proliferation at 10% and higher concentrations. Notably, low
concentration of CSE (0.25%) treatment of Eca109 did not inhibit
cell growth but stimulated cell proliferation (Fig. 2B). Considering the miR-101-3p
regulation upon CSE exposure, which showed significant
downregulation at low and high concentration of CSE treatment in
Eca109 cells, we wondered whether promotion of Eca109 cell
proliferation under low concentration of CSE as well as induced
cell death under high concentration of CSE were depended on
miR-101-3p downregulation. First, we applied MTT assay to study
cell proliferation under low concentration of CSE exposure in
Eca109 cells, and results showed overexpression of miR-101-3p could
reverse the increased cell proliferation induced by low
concentration (0.25%) of CSE treatment (p<0.05) (Fig. 2C), indicating an important role of
miR-101-3p in promotion of cell proliferation under low
concentration of CSE treatment. Next, we studied cell apoptosis
under high concentration of CSE exposure by flow cytometric
analysis of sub-G1 population, however, overexpression of
miR-101-3p could not rescue cell death induced by high
concentration (20%) of CSE treatment (Fig. 2D). Collectively, our data suggested
that suppression of miR-101-3p plays a crucial role in
low-CSE-induced cell proliferation, yet the role of miR-101-3p in
response to high-CSE is not known.
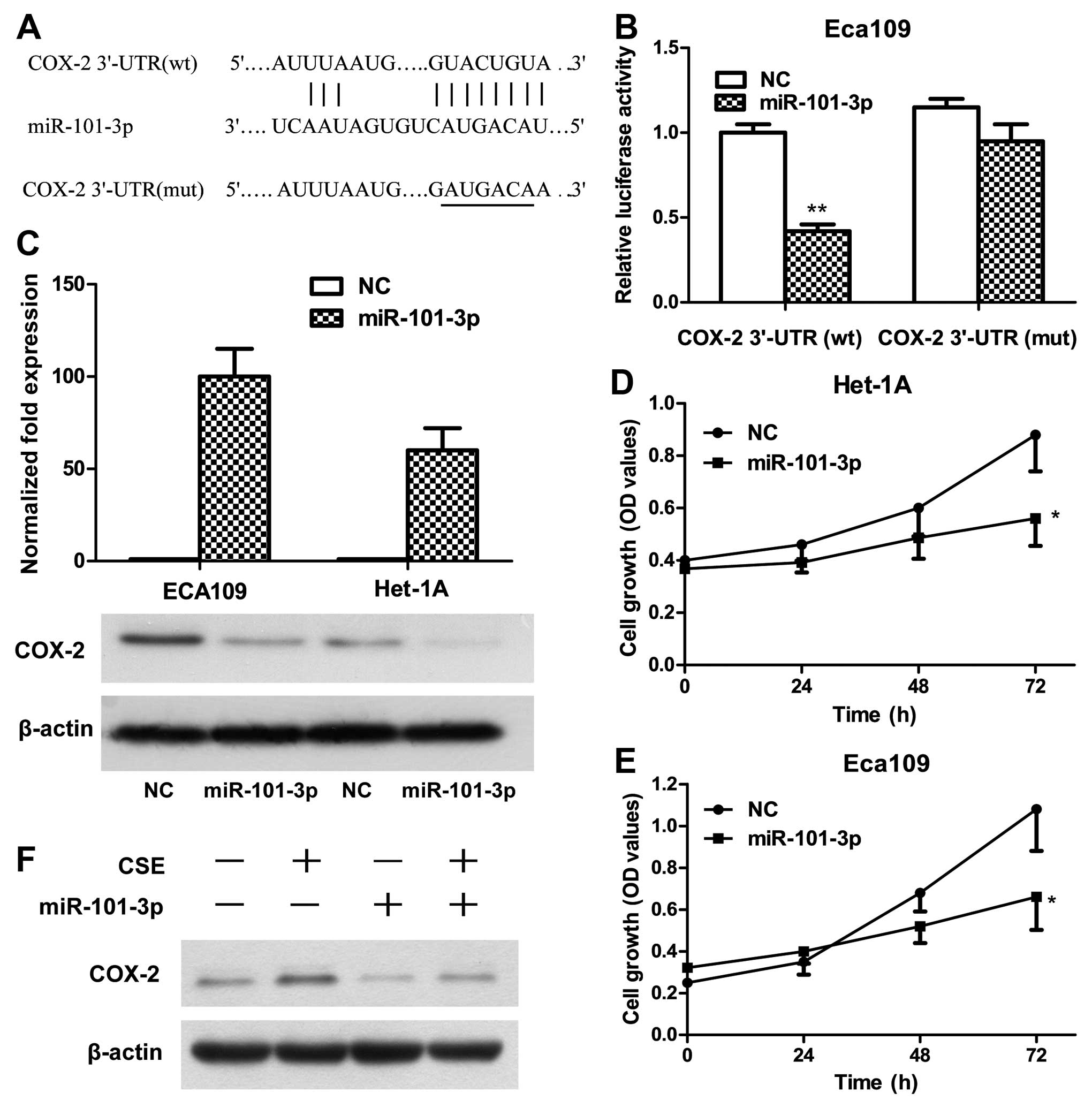
miR-101-3p inhibits COX-2 expression by
targeting 3′-UTR of COX-2 mRNA
As miR-101 was reported to inhibit COX-2
post-transcriptional expression by binding to the 3′-UTR of COX-2
mRNA in prostate and colon cancer cells (14,15),
to verify whether COX-2 is a direct target of miR-101-3p in
esophageal cancer cell lines, COX-2 3′-UTR containing the miR-101
binding sites (WT), and binding site mutant (Fig. 3A) were cloned downstream of the
luciferase open reading frame to obtain constructs for luciferase
activity assay. The assay results showed that increased expression
of miR-101-3p significantly downregulated luciferase activity in
Eca109 cells that co-transfected with COX-2 3′-UTR-WT construct,
while the luciferase activities of binding site mutants were
unaffected by lentiviral transduction of miR-101-3p (Fig. 3B). Without exogenous expression of
miR-101-3p, the luciferase activity in cells with luciferase
construct of COX-2 3′-UTR-WT was lower than that in cells with
construct of binding site mutant, suggesting a functional effect of
endogenous miR-101-3p (Fig. 3B).
The protein expression of COX-2 was also showed downregulated upon
lentiviral transduction of miR-101-3p in both esophageal epithelial
cell line Het-1A and esophageal squamous cancer cell line Eca109
(Fig. 3C), confirming miR-101-3p
negative regulation of COX-2 protein expression in esophageal
cells. MTT analysis showed proliferation of Het-1A and Eca109 cells
were significantly inhibited concomitant with miR-101-3p
overexpression (Fig. 3D and E).
Furthermore, the level of COX-2 protein in Eca109 cells
significantly increased under the stimulus of 0.25% CSE for 48 h,
and attenuated by miR-101-3p overexpression (Fig. 3F), indicating COX-2 is a direct
target of miR-101-3p under low concentration of CSE.
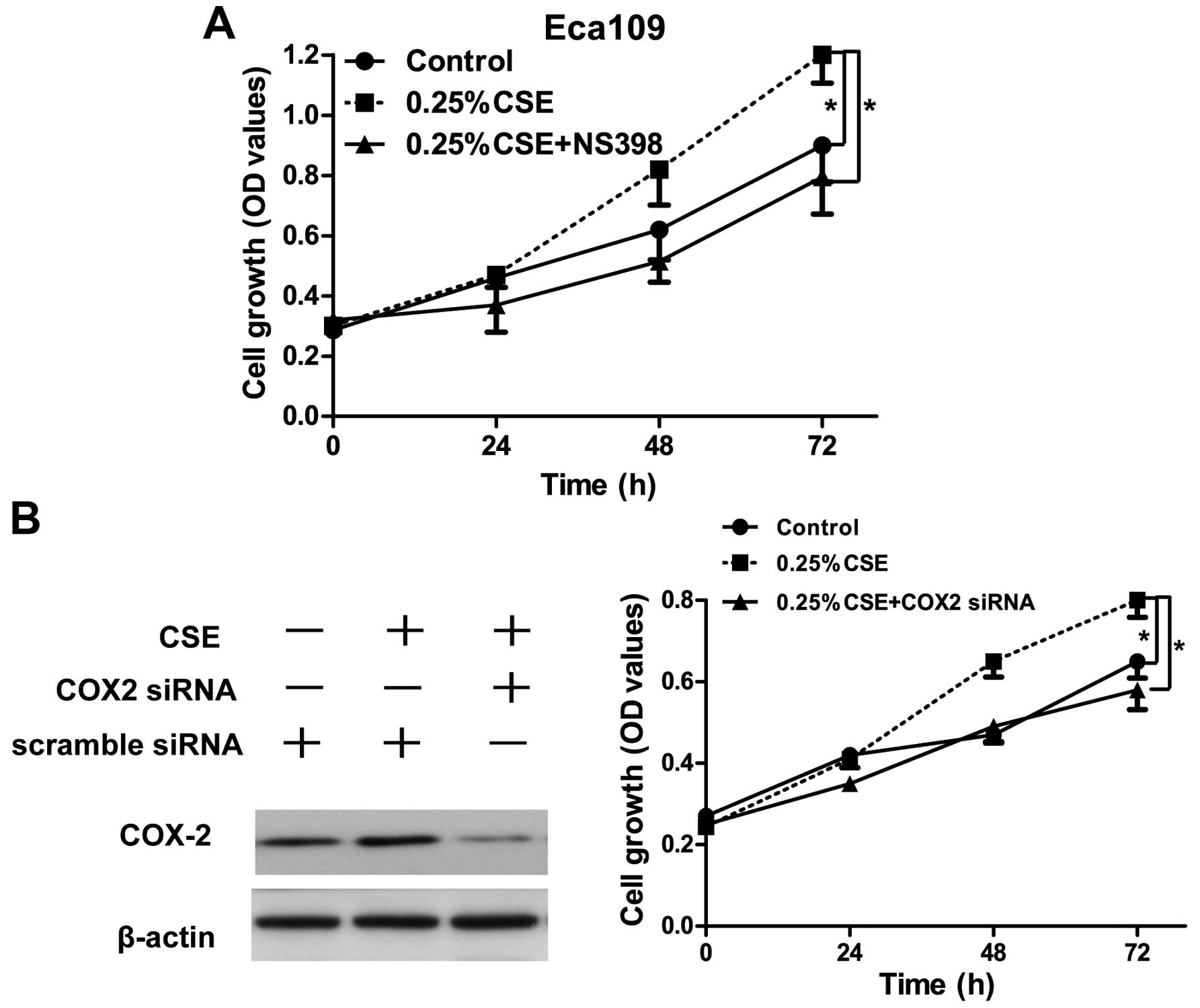
Promotion of cancer cell proliferation
induced by low concentration of CSE is dependent on COX-2
expression
As COX-2 is a direct target of miR-101-3p under low
concentration of CSE, we wondered whether promotion of Eca109 cell
proliferation under low concentration of CSE were depended on COX-2
activity. Indeed, COX-2 inhibitor (NS398) reversed 0.25%
CSE-induced cancer cell proliferation (p<0.05) (Fig. 4A). Moreover, knockdown of COX-2 also
inhibited 0.25% CSE-induced cancer cell proliferation (Fig. 4B). Collectively, promotion of cancer
cell proliferation induced by low concentration of CSE was
dependent on COX-2 upregulation.
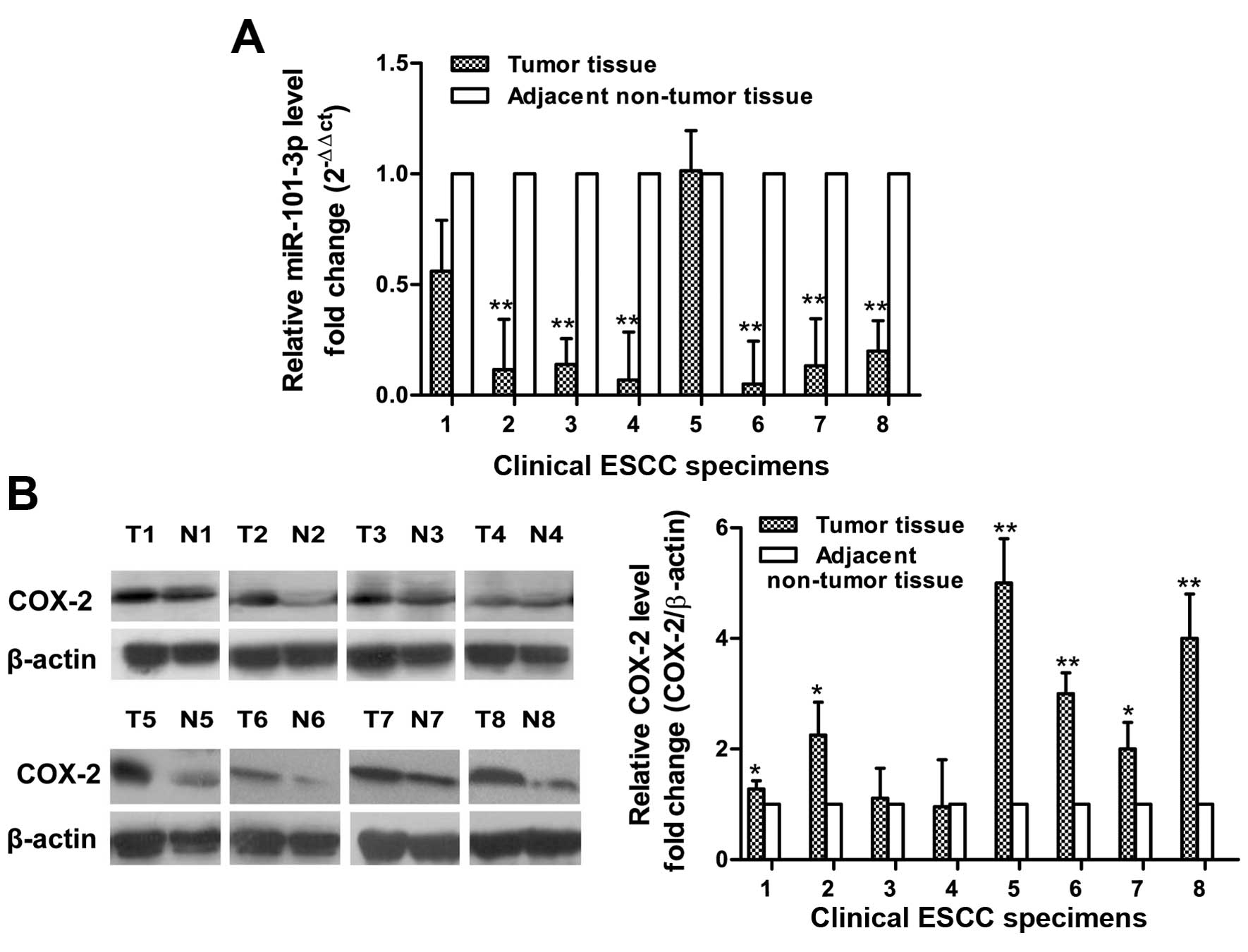
Downregulation of miR-101-3p and
upregulation of COX-2 in ESCC tissue samples
We studied 8 patients who had long history of
smoking but almost no drinking. We detected the level of miR-101-3p
in the 8 paired ESCC patients (tumor and adjacent non-tumor tissue)
by qRT-PCR analysis. Generally, in ESCC patients, miR-101-3p
expression was significantly lower in 7/8 of cancer tissues
compared to adjacent non-tumor tissue (Fig. 5A). The t-test showed that miR-101-3p
was significantly reduced in ESCC tissues than their adjacent
non-cancerous tissues. Then we used western blotting to examine
COX-2 expression in tissue samples collected from those ESCC
patients (Fig. 5B). The 6 of 8
samples showed higher expression of COX-2 protein in ESCC tissues
than in the non-cancerous esophageal tissues.
Discussion
Exposure of cells to CSE causes extensive
alterations in miRNA expression, and those changes in microRNA
(miRNA) expression are an early event following exposure to
cigarette smoke (9). To unravel
miRNA and its function may provide better understanding of how
environmental factors contribute to cancer initiation and
development. miR-101-3p has been reported downregulated in many
types of cancers, including ESCC (13), prostate (14) and colorectal cancer (15). Through post-transcriptional
inhibition of targets, such as cyclooxy-genase-2 (COX-2/PTGS2)
(16), EZH2/ENX-1 (13), Mcl-1(17), mTOR (18), miR-101-3p could suppress cancer cell
proliferation, migration and invasion. In the present study, we
reported miR-101-3p was downregulated upon CSE exposure in both
normal epithelia cell Het-1A and cancer cells, expanding the view
of the miRNA changes that contribute to CSE-induced ESCC
development.
A large body of evidence has suggested that the
promoting effect of smoking on ESCC is mediated by the induction of
COX-2 activity. COX-2 has been found transcriptional upregulated in
Eca109 cells upon cell treatment of chloroform or ethanol extract
of CS, and upregulation of COX-2 stimulates Eca109 cells
proliferation (19). In this
present study, the model of generation of CSE is the extraction in
buffered media, but not organic extract, we also confirmed a
correlation of CSE on COX-2 expression when cells were exposed to a
low concentration (0.25%) of CSE. Yet, the mechanisms of how CSE
regulated COX-2 expression are different from published literature,
revealing a new post-transcriptional mechanism by identifying COX-2
targeting microRMAs. Our finding adds an important piece to the
puzzle of how environmental factors regulate COX-2 expression,
thereby contributing to ESCC development.
In our finding, low concentration of CSE (0.25%)
induced a significant decrease in miR-101-3p expression in Eca109
cells but not in the non-tumorigenic esophageal epithelial cell
line Het1A, indicating dysregulation of miR-101-3p may be
exclusively related to ESCC cancer cell. Considering the anatomy of
esophagus, esophageal cancer cells, unlike oral/airway epithelial
cells, may not directly expose to high concentration of smoking
extract, therefore, the effect of low concentration of smoking
extract on ESCC cells may better imitate the real situation of how
environment factor facilitate ESCC development. CSE solutions of
2.5% used in the present study approximately correspond to direct
exposure to cigarette smoking from those who smoke 0.5 pack/day
(10). Yet, whether 0.25% CSE or
which concentration of CSE could mimic the in vivo
concentration of esophageal epithelial cell exposure when passive
or active inhalation of cigarette smoke are unknown but warrant
further investigation. In the present study, we confirmed that the
proliferation of Eca109 cells when exposed to low concentration of
CSE was dependent on suppression of miR-101-3p and upregulation of
its target COX-2 (Figs. 2C and
4), yet this effect was not found
in Het-1A cells, indicating miR-101-3p may function as a tumor
suppressor in the ESCC development but not in the initiation
stage.
In the present study, we noticed that both normal
esophageal epithelial Het-1A and Eca109 cancer cells induced
downregulation of miR-101-3p when exposed to high concentration of
CSE, yet the functions of downregulation of miR-101-3p in this
circumstance are not known. We speculated that identification of
new targets of miR-101-3p may provide clues for unveiling novel
functions for miR-101-3p, and warrant better understanding of
cellular responses to environmental factors, such as high CSE
exposure.
In conclusion, we revealed CSE-miR-101(−)-COX-2(+)
axis, from which low concentration of CSE facilitated cancer cell
proliferation. These findings provide new regulatory mechanism
involved in the smoking-induced excessive proliferation of ESCC,
providing novel clues into blocking this pathological process.
Acknowledgments
The present study was approved by the Ethics
Committee of The Second Xiangya Hospital, Central South University,
file no. 184 (2010).
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kamangar F, Chow WH, Abnet CC and Dawsey
SM: Environmental causes of esophageal cancer. Gastroenterol Clin
North Am. 38:27–57. vii2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tang WR, Fang JY, Wu KS, Shi XJ, Luo JY
and Lin K: Epidemiological characteristics and prediction of
esophageal cancer mortality in China from 1991 to 2012. Asian Pac J
Cancer Prev. 15:6929–6934. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Castellsagué X, Muñoz N, De Stefani E,
Victora CG, Castelletto R, Rolón PA and Quintana MJ: Independent
and joint effects of tobacco smoking and alcohol drinking on the
risk of esophageal cancer in men and women. Int J Cancer.
82:657–664. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sobus SL and Warren GW: The biologic
effects of cigarette smoke on cancer cells. Cancer. 120:3617–3626.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Huang RY and Chen GG: Cigarette smoking,
cyclooxygenase-2 pathway and cancer. Biochim Biophys Acta.
1815:158–169. 2011.
|
|
7
|
Meng XY, Zhu ST, Zhou QZ, Li P, Wang YJ
and Zhang ST: Promoter methylation regulates cigarette
smoke-stimulated cyclooxygenase-2 expression in esophageal squamous
cell carcinoma. J Dig Dis. 13:208–213. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Izzotti A, Calin GA, Arrigo P, Steele VE,
Croce CM and De Flora S: Downregulation of microRNA expression in
the lungs of rats exposed to cigarette smoke. FASEB J. 23:806–812.
2009. View Article : Google Scholar :
|
|
10
|
Su Y, Han W, Giraldo C, De Li Y and Block
ER: Effect of cigarette smoke extract on nitric oxide synthase in
pulmonary artery endothelial cells. Am J Respir Cell Mol Biol.
19:819–825. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zheng F, Liao YJ, Cai MY, Liu TH, Chen SP,
Wu PH, Wu L, Bian XW, Guan XY, Zeng YX, et al: Systemic delivery of
microRNA-101 potently inhibits hepatocellular carcinoma in vivo by
repressing multiple targets. PLoS Genet. 11:e10048732015.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sancho P, Martín-Sanz P and Fabregat I:
Reciprocal regulation of NADPH oxidases and the cyclooxygenase-2
pathway. Free Radic Biol Med. 51:1789–1798. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lin C, Huang F, Li QZ and Zhang YJ:
miR-101 suppresses tumor proliferation and migration, and induces
apoptosis by targeting EZH2 in esophageal cancer cells. Int J Clin
Exp Pathol. 7:6543–6550. 2014.PubMed/NCBI
|
|
14
|
Hao Y, Gu X, Zhao Y, Greene S, Sha W,
Smoot DT, Califano J, Wu TC and Pang X: Enforced expression of
miR-101 inhibits prostate cancer cell growth by modulating the
COX-2 pathway in vivo. Cancer Prev Res. 4:1073–1083. 2011.
View Article : Google Scholar
|
|
15
|
Strillacci A, Griffoni C, Sansone P,
Paterini P, Piazzi G, Lazzarini G, Spisni E, Pantaleo MA, Biasco G
and Tomasi V: MiR-101 downregulation is involved in
cyclooxygenase-2 overexpression in human colon cancer cells. Exp
Cell Res. 315:1439–1447. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chakrabarty A, Tranguch S, Daikoku T,
Jensen K, Furneaux H and Dey SK: MicroRNA regulation of
cyclooxygenase-2 during embryo implantation. Proc Natl Acad Sci
USA. 104:15144–15149. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Su H, Yang JR, Xu T, Huang J, Xu L, Yuan Y
and Zhuang SM: MicroRNA-101, down-regulated in hepatocellular
carcinoma, promotes apoptosis and suppresses tumorigenicity. Cancer
Res. 69:1135–1142. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lin S, Shao NN, Fan L, Ma XC, Pu FF and
Shao ZW: Effect of microRNA-101 on proliferation and apoptosis of
human osteosarcoma cells by targeting mTOR. J Huazhong Univ Sci
Technolog Med Sci. 34:889–895. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li P, Wu WK, Wong HP, Zhang ST, Yu L and
Cho CH: Chloroform extract of cigarette smoke induces proliferation
of human esophageal squamous-cell carcinoma cells: Modulation by
beta-adrenoceptors. Drug Chem Toxicol. 32:175–181. 2009. View Article : Google Scholar : PubMed/NCBI
|