Introduction
Sirtuins are highly conserved NAD-dependent
deacetylases that have been implicated in influencing a wide range
of cellular processes, such as aging, transcription, apoptosis,
inflammation and stress resistance (1–4).
Mammalian sirtuins have seven homologs (Sirt1-7) that share a
conserved catalytic domain as class III histone deacetylases
(HDACs) (2,5). Of the seven mammalian SIRTs, sirtuin 1
(Sirt1) has been shown to mediate diverse cellular functions,
including maintaining genomic stability, suppressing inflammation,
enhancing synaptic plasticity, and neuroprotection in models of
Alzheimer's disease (6,7). Sirt1 catalyzes the deacetylation of a
large number of non-histone substrates, including p53, PGC-1α,
NF-κB, PTEN, E2F1 and FOXO transcription factors (8–13).
Sirt1 downregulation has been linked to cell senescence and various
pathological events such as insulin resistance and severe oxidative
stress (6,9,13,14).
Like Sirt1, Sirt2 is a strong deacetylase, which deacetylates
internal lysines on histone and α-tubulin as well as many other
proteins such as key transcription factors (15–17).
In addition, Sirt2 deacetylates FOXO1 in response to oxidative
stress or serum deprivation, thereby negatively regulating
FOXO1-mediated autophagy (18).
Sirt2 also plays a major role in cell cycle progression and genomic
stability (19,20). A recent study showed that Sirt1
deacetylates HSF1 and potentiates its DNA-binding ability, and the
downregulation of Sirt1 accelerates the release of HSF1 from its
cognate promoter elements and attenuates the heat shock response
(21,22). Another study showed that the Sirt1
activator SRT1720 prevents colitis by reducing HSF1 acetylation and
increasing the expression of BIP, HSP27 and HSP90. Sirt1 activation
thus participates in protecting cells from stresses related to
damaged, misfolded or aggregated proteins (23). However, the mechanism by which Sirt1
and Sirt2 may regulate HSF1 is still unclear.
We were interested in gaining a better understanding
of the role of Sirt1 and Sirt2 as regulators of HSF1. We found that
inhibitors of Sirt1 and Sirt2, EX527 and AGK2, induced the
acetylation and the degradation of heat stress-induced HSF1. In
addition, cellular HSF1 stability, by controlling Sirt1 expression,
modulated cell migration by influencing HSP27 expression.
Materials and methods
Cell culture and reagents
HeLa cells were cultured in Minimal Essential Medium
(MEM; Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% FBS
(Biochrom AG, Berlin, Germany), 100 U/ml penicillin and 100
μg/ml streptomycin (Sigma-Aldrich) and maintained in 5%
CO2 at 37°C. EX527, AGK2 and hemin were purchased from
Sigma-Aldrich. Fugene HD transfection reagent was from Promega
(Madison, WI, USA). Polyclonal anti-HSF1, anti-HSP27,
anti-caspase-3, and anti-PARP antibodies were from Santa Cruz
Biotechnology, Inc. (Dallas, TX, USA). Monoclonal anti-Sirt1 and
anti-Sirt2 antibodies were from Cell Signaling Technology (Danvers,
MA, USA).
Reverse transcription-polymerase chain
reaction
Total RNA of HeLa cells was extracted using TRIzol
reagent (Life Technologies, Gaithersburg, MD, USA) according to the
manufacturer's instructions. A total of 1 μg of total RNA
was reverse-transcribed to cDNA in a reaction mixture of 20
μl with the Reverse Transcription system (Promega, Leiden,
The Netherlands). Amplification was performed for 30 cycles in a
DNA thermal cycler. GAPDH was used as a control for the PCR
reaction. The PCR products were resolved on a 1.5% agarose gel and
were stained with ethidium bromide.
Cell proliferation assay
Cell proliferation was assessed using a
3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide (MTT)
assay. The cells were seeded in a 96-well plate at a density of
1×104 cells/well. The next day, the cells were washed
twice with PBS, and 500 μg/ml MTT (Sigma-Aldrich) was added
to the wells. The MTT solution was removed after 4 h of incubation
at 37°C. A mixture of 0.01 M glycine and DMSO (Sigma-Aldrich) was
added to each well. The absorbance was measured at 540 nm with a
Benchmark microplate reader (Bio-Rad Laboratories, Philadelphia,
PA, USA).
Immunoprecipitation
Total cell extracts were incubated with anti-HSF1 in
NP-40 lysis buffer (0.5% NP-40, 0.5% Triton X-100, 150 mM NaCl, 50
mM Tris HCl pH 7.4, 1 mM EDTA, 50 mM NaF, 1 mM B-glycerophosphate,
1 mM sodium orthovanadate, 0.5 μg/ml leupeptin, 1
μg/ml pepstatin, 0.2 mM PMSF). The extract mixtures were
incubated at 4°C overnight with rotation before the addition of 20
μl of protein A/G beads (Life Technologies) for 3 h at 4°C.
The beads were washed three times with the same buffer and
suspended in 2X SDS sample buffer. The samples were resolved in
SDS-polyacrylamide gels for western blot analysis with specific
antibodies as indicated.
Western blot analysis
Cell lysates (50 μg) were placed in lysis
buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% NP-40, 0.5%
Na-deoxycholate, 0.1% SDS, and a protease inhibitor cocktail
containing 1 μg/ml aprotinin and leupeptin) and separated by
12% SDS-PAGE. The resolved proteins were transferred onto a
nitrocellulose membrane (Amersham Pharmacia Biotech, UK) according
to standard procedures. The membrane was blocked in 5% non-fat dry
milk for 3 h and incubated with the primary antibodies for 3 h at
room temperature (RT). After incubation with a specific
peroxidase-coupled secondary antibody (Sigma-Aldrich) for 1 h, the
blotted bands were detected using an enhanced chemiluminescence
detection kit (Amersham Pharmacia Biotech).
Wound-scratch assays
Cells were allowed to grow in a culture dish
overnight, and a scratch ~3-mm wide was created in the monolayer
using a pipette tip. After washing twice with PBS, the cells were
treated with or without the Sirt1/Sirt2 inhibitors, and images were
captured after 12 or 36 h. Cells were imaged in 5 random
microscopic fields per well using an Olympus IX2-SLP inverted
microscope (Japan) at a ×100 magnification.
Annexin V-FITC/PI double staining
The cells were harvested and fixed with 70% ethanol
for 1 h at 4°C for cell cycle analysis. After washing with cold
PBS, the cells were incubated with DNase-free RNase and propidium
iodide (PI) at 37°C for 30 min. The specific binding of Annexin
V-FITC/PI was performed by incubating the cells for 15 min at RT in
a binding buffer (10 mM HEPES, 140 mM NaCl, 2.5 mM
CaCl2, pH 7.4) containing saturated concentrations of
Annexin V-FITC and PI. After incubation, the cells were pelleted
and analyzed in a FACScan analyzer (Beckman Coulter Inc.,
Fullerton, CA, USA).
Flow cytometry and cell cycle
analysis
The cell cycle distribution was analyzed by flow
cytometry. Briefly, 1×106 cells were harvested and
washed in PBS, and then fixed in 70% alcohol for 30 min at 4°C.
After washing three times in cold PBS, the cells were resuspended
in 1 ml of PBS solution containing 50 μl of 1 mg/ml PI and 1
unit of DNase-free RNase for 30 min at 37°C. The samples were then
analyzed for their DNA content by FACS (Beckman Coulter Inc.).
Soft agar colony formation assay
Briefly, the cells (8×103 cells/well)
were exposed to different concentrations of EX527 or AGK2 in 1 ml
of 0.3% basal medium Eagle's agar containing 10% FBS. The cultures
were maintained at 37°C in a 5% CO2 incubator for 10–15
days, and the cell colonies were scored using an Olympus IX2-SLP
inverted microscope (Japan).
Statistical analyses
All experiments were performed at least three times.
The mean values for the experiments are expressed as the mean ± SE.
Significant differences were assessed by analysis of variance.
p<0.05 was considered to indicate a statistically significant
difference.
Results
Cytotoxic effect of the Sirt1/2
inhibitors EX527 and AGK2 on cell proliferation
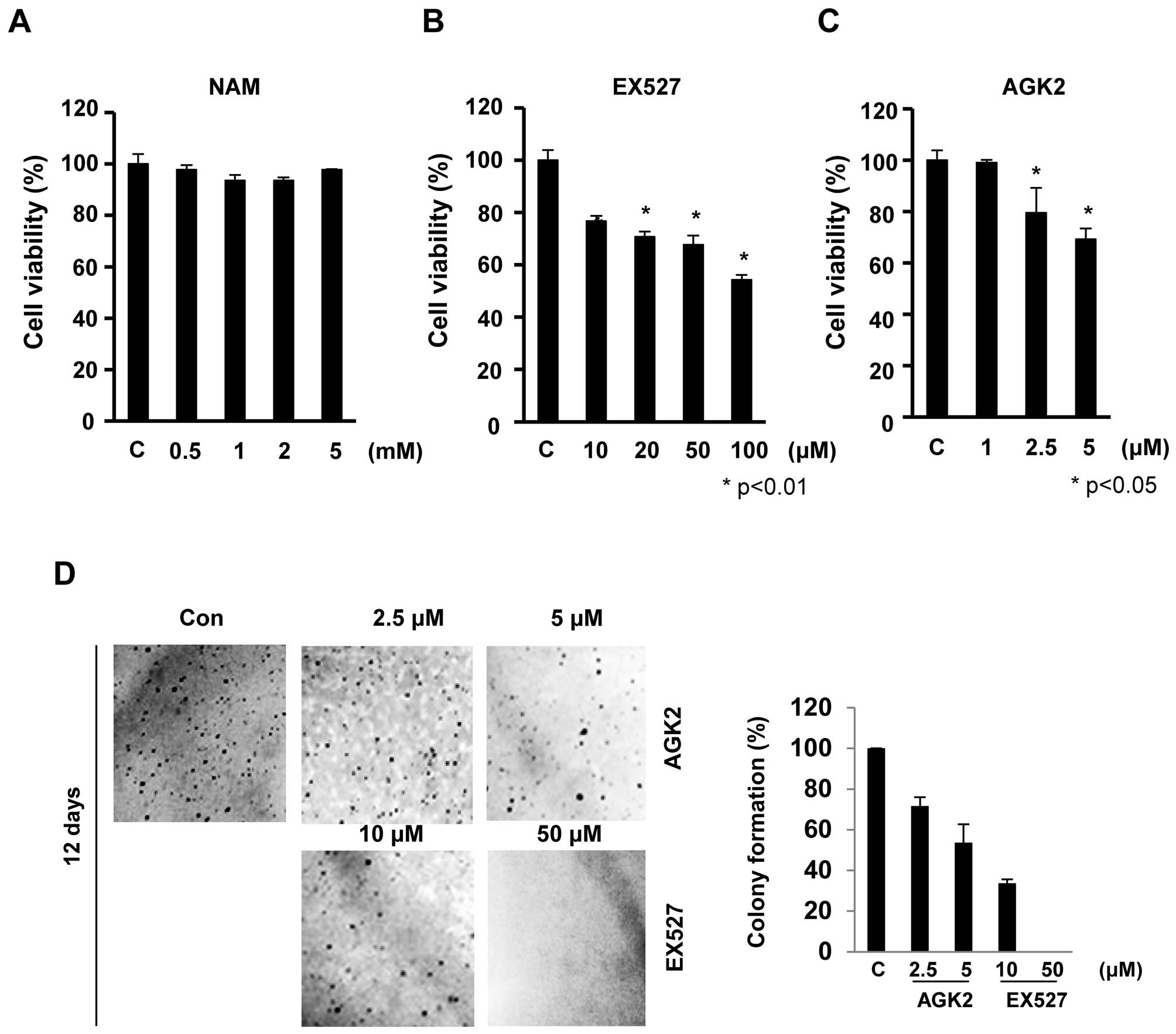
To examine the effect of Sirt1/2 inhibitors, EX527
and AGK2, on the growth of HeLa cells, we performed MTT assays. The
cells were exposed to increasing concentrations of nicotinamide
(NAM), EX527 and AGK2 for 24 h, and the cell viability was
monitored (Fig. 1). EX527 and AGK2
significantly inhibited cell proliferation in a dose-dependent
manner. EX527 at concentrations of 50 and 100 μM resulted in
cell growth inhibition of 68 and 54%, respectively, at 24 h
(Fig. 1B). Similarly, AGK2 also
significantly inhibited cell growth in a dose-dependent manner
without inducing cytotoxicity at low doses (≥1 μM; Fig. 1C). However, these cells exhibited no
significant decrease in cell proliferation after NAM treatment for
24 h (survival rate >93%; Fig.
1A).
To further assess the effect of EX527 and AGK2 on
tumorigenicity, we compared the anchorage-independent growth rates.
Twelve days after EX527 (50 μM) and AGK2 (5 μM)
treatment, HeLa cells showed a significantly reduced colony forming
ability in soft agar to ~95 and 46% of the control cells,
respectively (Fig. 1D). Together,
our observations showed that EX527 and AGK2 suppressed the
malignant phenotype such as cell proliferation and colony
formation.
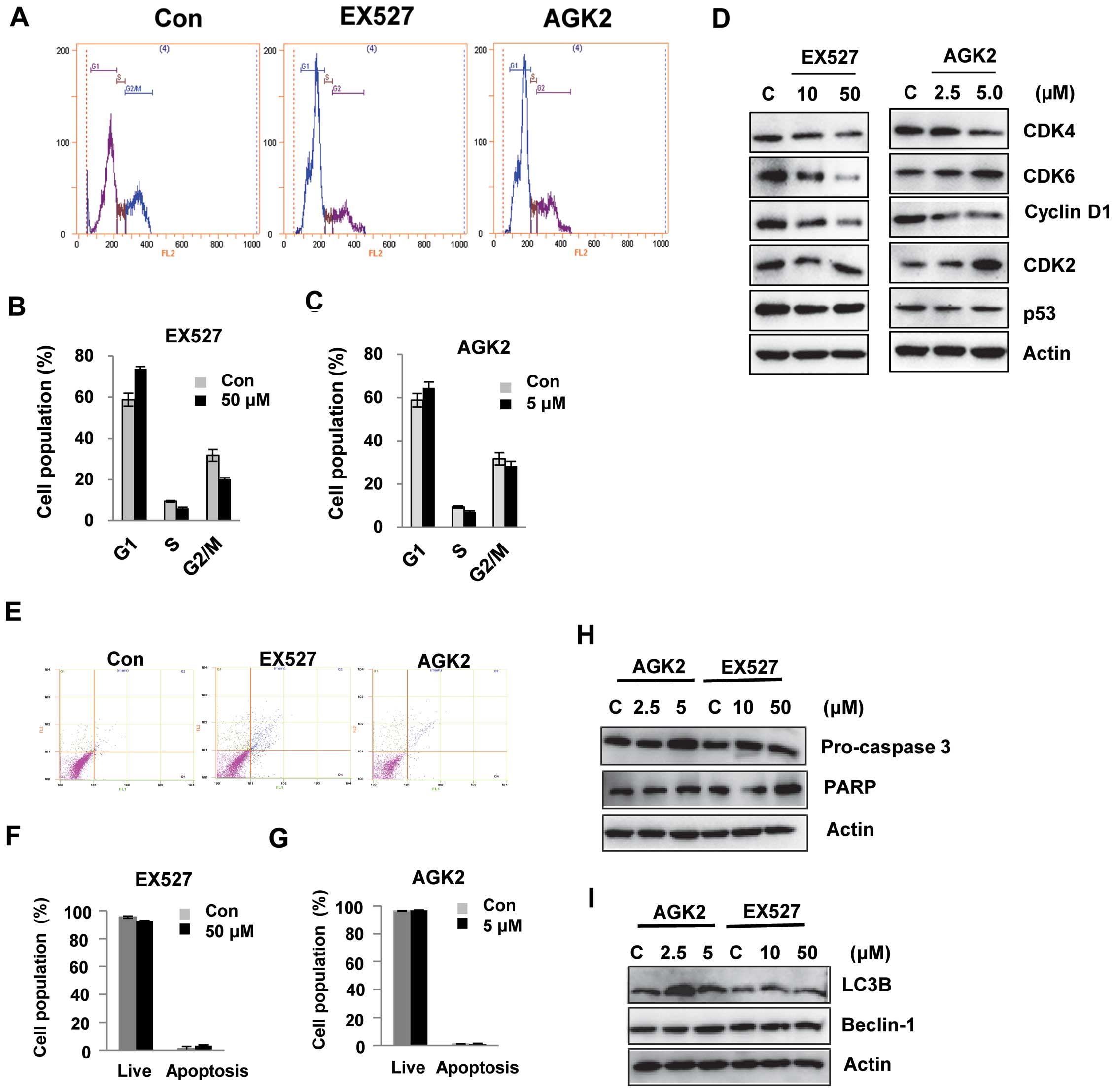
EX527 and AGK2 induce G1 cell cycle
arrest in HeLa cells
It was hypothesized that EX527 or AGK2 induce
alterations in cell cycle regulation. Using flow cytometry, we
analyzed the effect of EX527 or AGK2 on cell cycle progression. As
shown in Fig. 2A–C, EX527 and AGK2
induced cell cycle arrest at the G1 phase. Western blot analysis
showed that the levels of CDK4 or CDK6 and cyclin D1 were decreased
after EX527 or AGK2 treatment in a dose-dependent manner. In
addition, EX527 and AGK2 inhibited the expression of p53 protein
(Fig. 2D). These results suggest
that the effects of EX527 and AGK2 on G1 cell cycle progression
were associated with the inhibition of cell growth.
To determine whether EX527 and AGK2 induce death in
HeLa cells, we quantified apoptosis by flow cytometry using the
Annexin V-FITC/PI double staining assay. As shown in Fig. 2E–H, after 24 h of incubation, there
was no significant decrease in living cells upon EX527 treatment;
93% of the control cells were alive, while only 3.3% of the
EX527-treated cells underwent cell death. Similarly, apoptotic
cells were not markedly observed after AGK2 treatment (Fig. 2G). Consistent with this observation,
caspase-3 and PARP were not cleaved in cells treated with EX527 or
AGK2 compared with the control (Fig.
2H). In addition, key autophagy proteins, LC3B and beclin-1,
were not activated after treatment with EX527 or AGK2 (Fig. 2I).
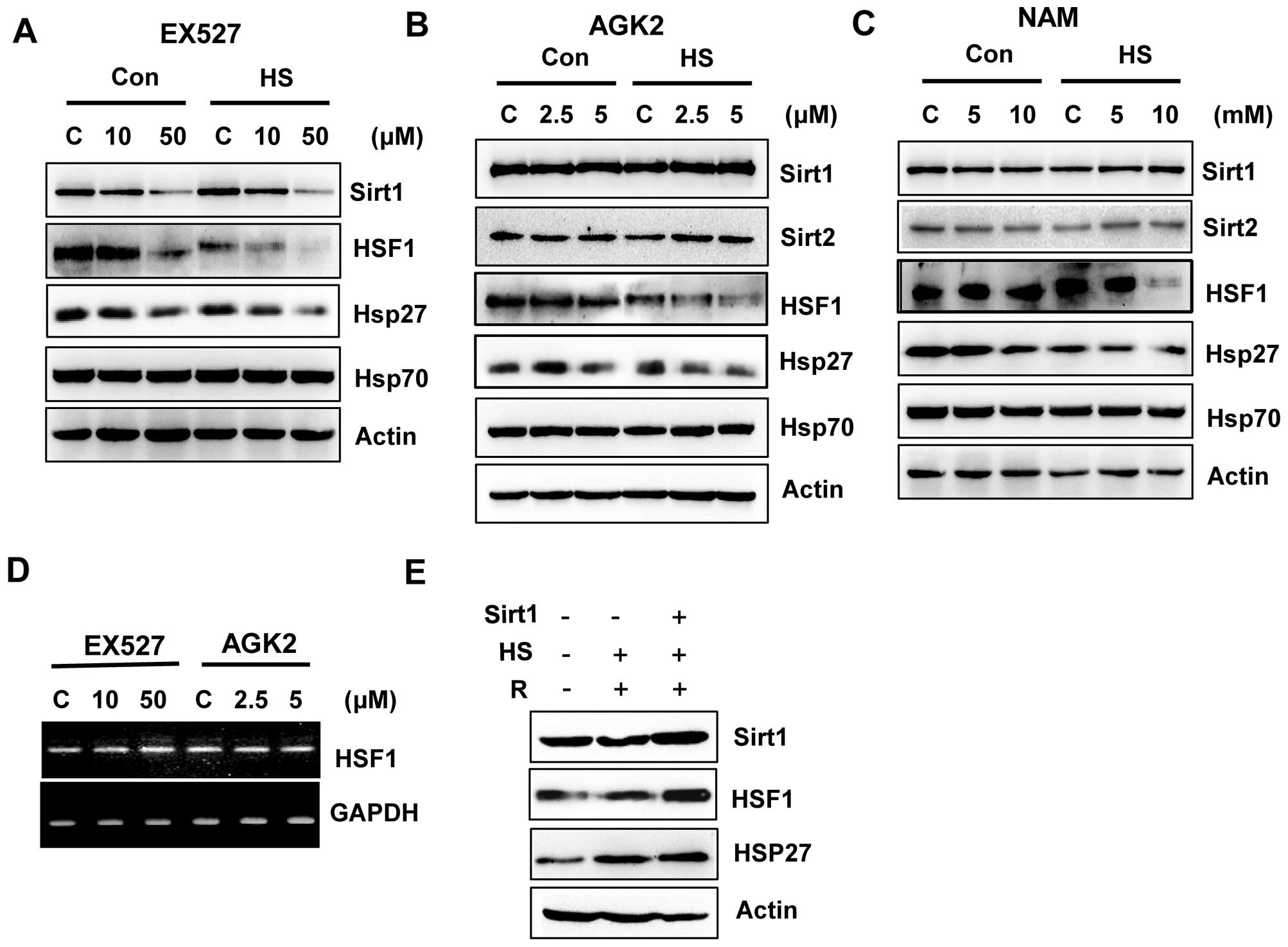
EX527 and AGK2 inhibit the HSF1/HSP27
pathway
A previous study suggested that Sirt1 deacetylates
HSF1 and activates heat shock proteins (HSPs) under a heat stress
condition (21). We investigated
the possibility that Sirt1 and Sirt2 inhibition by EX527 and AGK2
may be responsible for HSF1 inactivation. Therefore, we subjected
EX527- or AGK2-treated cells to heat shock (1 h at 42°C) and
analyzed the expression of endogenous Sirt1, Sirt2 and HSF1, as
well as HSPs such as Hsp27 and Hsp70. EX527 or AGK2 treatment
decreased the expression of HSF1 and HSP27 in non-stress or heat
stress condition (Fig. 3A and B),
which indicated that Sirt1 and Sirt2 are required for the
regulation of the HSF1 pathway. Furthermore, EX527 and AGK2 also
decreased HSF1 phosphorylation after heat shock. However, NAM
decreased HSF1 expression/phosphorylation and HSP27 expression
under heat shock conditions (Fig.
3C).
To determine whether EX527 and AGK2 influence HSF1
transcription in HeLa cells, RT-PCR was performed using specific
oligonucleotides against the HSF1 gene. As shown in Fig. 3D, the HSF1 mRNA level was not
altered by EX527 or AGK2.
To verify the significance of the regulation of HSF1
by Sirt1, cells were transfected with or without Sirt1, exposed to
a 42°C heat shock for 1 h, allowed to recover at 37°C for 24 h, and
analyzed for HSF1 expression. As expected, the heat shock induced
HSP27 expression, which increased with recovery time. Importantly,
at the same time-points, we observed that the cells overexpressing
Sirt1 showed increased HSF1 expression compared with the heat
shock-treated cells (Fig. 3E).
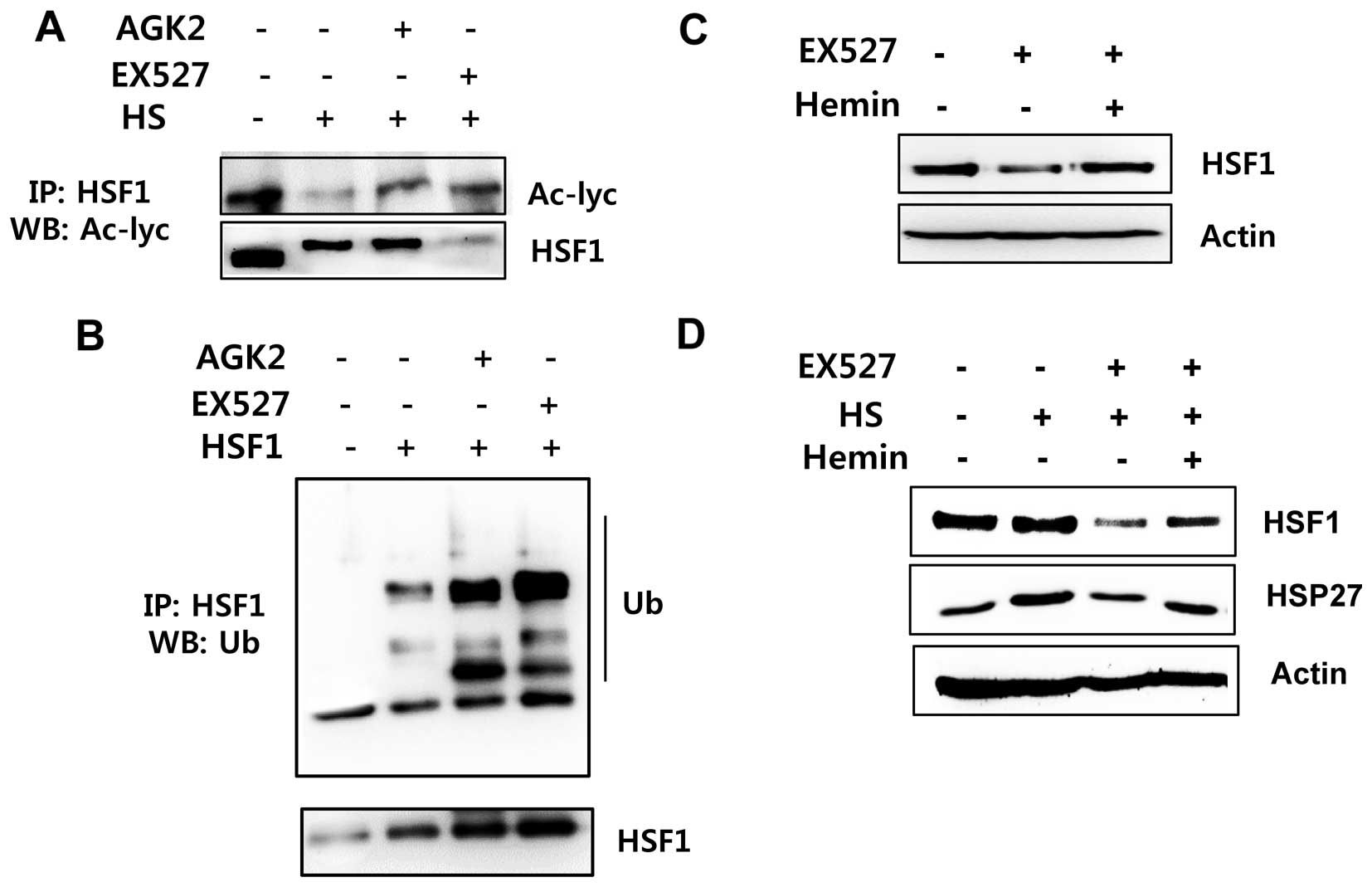
Sirt1/Sirt2 inhibition is linked to
increased HSF1 ubiquitination
To determine whether HSF1 deacetylation by Sirt1 is
involved in HSF1 ubiquitination/stabilization, we examined the
acetylation and ubiquitination status of HSF1 after EX527 and AGK2
treatment. HeLa cells were treated with or without EX527 and AGK2
and exposed to a 42°C heat shock for 1 h. Then, the samples were
immunoprecipitated with an anti-HSF1 antibody and analyzed for HSF1
acetylation. Acetylated HSF1 was detected in the untreated cells
but was decreased in the cells exposed to heat shock stress
conditions. However, acetylated HSF1 levels were induced in the
EX527- or AGK2-treated cells before heat shock (Fig. 4A). In addition, EX527 and AGK2
induced HSF1 ubiquitination (Fig.
4B).
To investigate whether proteasome-dependent protein
degradation is involved in Sirt1 inhibitor-mediated downregulation
of HSF1 protein, we treated cells with the proteasome inhibitor
hemin and/or EX527. As shown in Fig. 4C
and D, the HSF1 protein level in the hemin-treated cells was
substantially higher than that in the EX526-treated cells. Our
above data indicate that EX527 or AGK2 decreases the
expression/phosphorylation of HSF1 protein. However, hemin
inhibited the EX527-mediated reduction of the HSF1 protein. From
the above data, we conclude that Sirt1 and/or Sirt2 inhibition
results in a diminution of HSF1 protein stabilization.
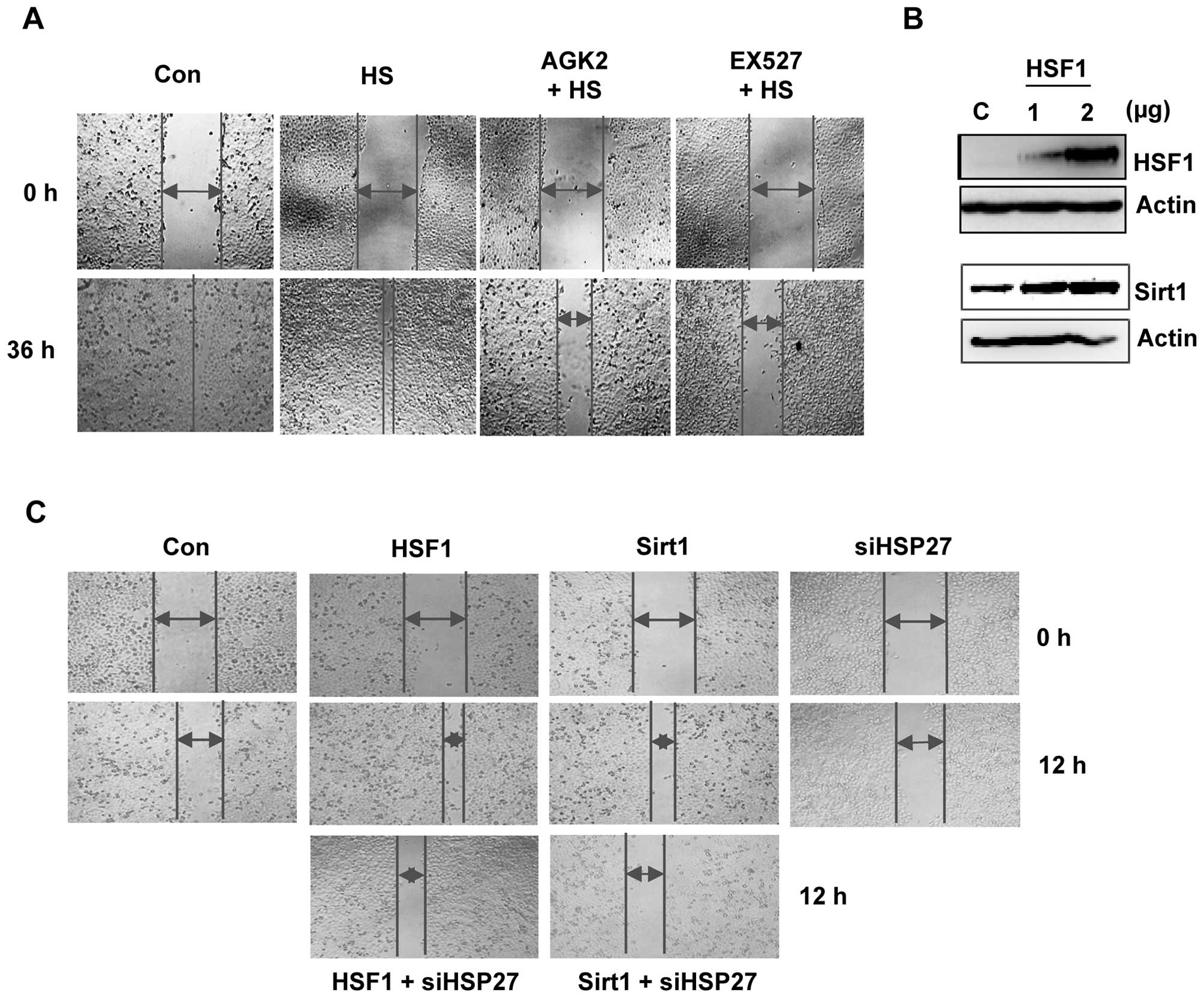
EX527 and AGK2 inhibit cell
migration
To investigate whether Sirt1/HSF1 modulate cell
motility, we treated HeLa cells with EX527 and AGK2 for 36 h and
performed a wound-healing experiment. EX527- or AGK2-treated cells
showed reduced migration ability when compared with the control or
heat shock-treated cells (Fig.
5A).
To further assess the effect of the Sirt1/HSF1/HSP27
pathway on cell migration, we compared the migration capabilities
of Sirt1 or HSF1 with or without siHSP27 transfection. First, the
levels of the HSF1 protein increased in the HSF1-transfected cells
compared to the control cells (Fig.
5B). As expected, HSF1-overexpression led to induced Sirt1
protein levels. A wound-healing assay showed that overexpression of
HSF1 or Sirt1 led to increased cell migration. However, the
increase in gap closure was delayed in cells transfected with
siHSP27; the migration of siHSP27-transfected cells was similar to
the control cells (Fig. 5C). These
data indicate that Sirt1/Sirt2-mediated HSF1/HSP27 regulation is
involved in cell migration.
Discussion
In the present study, the pharmacological inhibition
of Sirt1 or Sirt2 by the specific inhibitor EX527 or AGK2 caused G1
phase arrest, which was mediated by the inhibition of Cdk4, Cdk6,
and cyclin D1 and reduced the cell growth and colony formation of
HeLa cells. Although EX527- or AGK2-treated HeLa cells exhibited
decreased cell growth due to cell cycle arrest at the G1 phase,
they did not exhibit higher levels of apoptosis or autophagy.
Previous studies have suggested that other Sirt
inhibitors, such as AC-93253 and AEM1/2, reduce cell proliferation
by inducing apoptosis (23–26). Other reports have linked Sirt1 to
the regulation of autophagy, such as the report that showed that
Sirt1 (−/−) mouse embryonic fibroblasts fail to activate autophagy
in response to nutrient deprivation (27). The mechanisms underlying the effect
of Sirt1/2 on cell death remain to be determined but may be
dependent on cell type and the duration of Sirt1/2 inhibitor
treatment.
Our study revealed that EX527 and AGK2 negatively
regulated the expression and/or phosphorylation of HSF1, but the
mRNA level of HSF1 did not change. In addition, reduced HSF1 by
EX527 or AGK2 was recovered in the presence of hemin, a proteasome
inhibitor, suggesting that HSF1 may be degraded by proteasomal
machinery following EX527 or AGK2 treatment. To confirm this
hypothesis, we observed that the inhibition of HSF1 deacetylation
by EX527 and AGK2 reduced HSF1 protein abundance through HSF1
ubiquitination, indicating that the ubiquitin-proteasome system
exerts an important role in the acetylation/deacetylation status of
the HSF1 protein. The additional effect of HSF1 acetylation could
induce HSF1 degradation in the nucleus and inhibit target genes
involved in heat shock response, such as HSP27.
Previous studies have shown that Sirt1 is recruited
to the hsp70 promoter and activates HSF1 through deacetylation at a
key lysine 80 residue within the DNA binding domain. In contrast,
Sirt1 downregulation accelerates the release of HSF1 from its
cognate promoter elements and attenuates the heat shock response
(HSR) (21–23). Additionally, Sirt1 was shown to
deacetylate HSF1 and increase HSP70 levels in the brains of A57T
mice, leading to α-synuclein aggregate suppression (28).
Consistently, Sirt1/2 inhibition with EX527 and AGK2
inhibited HSF1-mediated HSP27 induction under heat shock
conditions. These findings could be explained as follows. First,
Sirt1/2 deacetylates HSF1 by binding to HSF1 and promotes the heat
shock response. Second, in contrast, inhibition of Sirt1/2
accelerates HSF1 ubiquitination likely by acetylating the lysine
residue and ubiquitinated HSF1 is recognized and degraded by the
proteasome system.
During elevated stress, HSF1 is subject to
additional enzymatic post-translational modifications including
phosphorylation and SUMOylation as well as acetylation (21,29,30).
These modifications are thought to promote HSF1 expression/activity
to respond to various stresses. The acetylation/deacetylation
status of HSF1 may affect several molecular pathways involved in
the regulation of cell protection, migration/movement, and stress
resistance including MAP kinase signaling, vinculin, and heat shock
protein 27 (Hsp27) (31–33).
As described above, our findings suggest that the
expression/activation of HSF1/HSP27 depend at least in part on the
Sirt1/Sirt2 pathway. This finding prompted us to investigate
whether the Sirt1/2-regulated HSF1/HSP27 pathway may affect
motility such as migration. We demonstrated that the inhibition of
Sirt1/2 by EX527 or AGK2 decreased cell motility in a scratch
assay. Our data indicate that Sirt1 and HSF1/HSP27 play an
important role in cell migration, as specifically demonstrated by
the findings that overexpression of Sirt1 and HSF1 increased cell
migration, whereas downregulation of HSP27 by siRNA decreased
Sirt1- and HSF1-induced cell migration. The direct causal
association between the HSF1/HSP27 pathway and Sirt1/2 was apparent
by the finding that modulation of Sirt1/2 activity by EX527 and
AGK2 strongly affected HSF1 expression/activation and modulated
cell migration. The molecular basis for the activity of HSF1/HSP27
in affecting cell motility and in modulating the response to Sirt1
and Sirt2 still needs to be clarified. It should also be considered
that, although Sirt1 is involved in HSF1 deacetylation, additional
deacetylation sites by Sirt2 on HSF1 activation are present, which
could be differentially regulated by Sirt1 and Sirt2 and may
modulate other cellular functions in these cells.
In conclusion, the inhibition of Sirt1/2 by EX527 or
AGK2 inhibited cell growth and colony formation. Our study is the
first to demonstrate that blocking Sirt1 and Sirt2 can induce HSF1
ubiquitination and degradation in vitro, suggesting that
Sirt1/2 is involved in the activation of heat shock response
signaling pathways. Furthermore, we observed a consistent reduction
in cell migration after EX527 and AGK2 treatment. Our results
suggest that Sirt1 and Sirt2 regulate cell migration via
HSF1/Hsp27-mediated signaling.
Acknowledgments
This study was supported by a National Research
Foundation of Korea (NRF) grant funded by the Korean government
MSIP (no. 2008-0062283).
References
|
1
|
Alcendor RR, Gao S, Zhai P, Zablocki D,
Holle E, Yu X, Tian B, Wagner T, Vatner SF and Sadoshima J: Sirt1
regulates aging and resistance to oxidative stress in the heart.
Circ Res. 100:1512–1521. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Michan S and Sinclair D: Sirtuins in
mammals: Insights into their biological function. Biochem J.
404:1–13. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Finkel T, Deng CX and Mostoslavsky R:
Recent progress in the biology and physiology of sirtuins. Nature.
460:587–591. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yang T and Sauve AA: NAD metabolism and
sirtuins: Metabolic regulation of protein deacetylation in stress
and toxicity. AAPS J. 8:E632–E643. 2006. View Article : Google Scholar
|
|
5
|
Pang M and Zhuang S: Histone deacetylase:
A potential therapeutic target for fibrotic disorders. J Pharmacol
Exp Ther. 335:266–272. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kwon HS and Ott M: The ups and downs of
SIRT1. Trends Biochem Sci. 33:517–525. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lavu S, Boss O, Elliott PJ and Lambert PD:
Sirtuins - novel therapeutic targets to treat age-associated
diseases. Nat Rev Drug Discov. 7:841–853. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Luo J, Nikolaev AY, Imai S, Chen D, Su F,
Shiloh A, Guarente L and Gu W: Negative control of p53 by Sir2alpha
promotes cell survival under stress. Cell. 107:137–148. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Garcia MM, Guéant-Rodriguez RM, Pooya S,
Brachet P, Alberto JM, Jeannesson E, Maskali F, Gueguen N, Marie
PY, Lacolley P, et al: Methyl donor deficiency induces
cardiomyopathy through altered methylation/acetylation of PGC-1α by
PRMT1 and SIRT1. J Pathol. 225:324–335. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yeung F, Hoberg JE, Ramsey CS, Keller MD,
Jones DR, Frye RA and Mayo MW: Modulation of NF-kappaB-dependent
transcription and cell survival by the SIRT1 deacetylase. EMBO J.
23:2369–2380. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ikenoue T, Inoki K, Zhao B and Guan KL:
PTEN acetylation modulates its interaction with PDZ domain. Cancer
Res. 68:6908–6912. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang C, Chen L, Hou X, Li Z, Kabra N, Ma
Y, Nemoto S, Finkel T, Gu W, Cress WD, et al: Interactions between
E2F1 and SirT1 regulate apoptotic response to DNA damage. Nat Cell
Biol. 8:1025–1031. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Brunet A, Sweeney LB, Sturgill JF, Chua
KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, et
al: Stress-dependent regulation of FOXO transcription factors by
the SIRT1 deacetylase. Science. 303:2011–2015. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhao G, Cui J, Zhang JG, Qin Q, Chen Q,
Yin T, Deng SC, Liu Y, Liu L, Wang B, et al: SIRT1 RNAi knockdown
induces apoptosis and senescence, inhibits invasion and enhances
chemosensitivity in pancreatic cancer cells. Gene Ther. 18:920–928.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Skoge RH, Dölle C and Ziegler M:
Regulation of SIRT2-dependent α-tubulin deacetylation by cellular
NAD levels. DNA Repair (Amst). 23:33–38. 2014. View Article : Google Scholar
|
|
16
|
Patel VP and Chu CT: Decreased SIRT2
activity leads to altered microtubule dynamics in
oxidatively-stressed neuronal cells: Implications for Parkinson's
disease. Exp Neurol. 257:170–181. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Donmez G and Outeiro TF: SIRT1 and SIRT2:
Emerging targets in neurodegeneration. EMBO Mol Med. 5:344–352.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang F, Nguyen M, Qin FX and Tong Q: SIRT2
deacetylates FOXO3a in response to oxidative stress and caloric
restriction. Aging Cell. 6:505–514. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Serrano L, Martínez-Redondo P,
Marazuela-Duque A, Vazquez BN, Dooley SJ, Voigt P, Beck DB,
Kane-Goldsmith N, Tong Q, Rabanal RM, et al: The tumor suppressor
SirT2 regulates cell cycle progression and genome stability by
modulating the mitotic deposition of H4K20 methylation. Genes Dev.
27:639–653. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kim HS, Vassilopoulos A, Wang RH, Lahusen
T, Xiao Z, Xu X, Li C, Veenstra TD, Li B, Yu H, et al: SIRT2
maintains genome integrity and suppresses tumorigenesis through
regulating APC/C activity. Cancer Cell. 20:487–499. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Westerheide SD, Anckar J, Stevens SM Jr,
Sistonen L and Morimoto RI: Stress-inducible regulation of heat
shock factor 1 by the deacetylase SIRT1. Science. 323:1063–1066.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu DJ, Hammer D, Komlos D, Chen KY,
Firestein BL and Liu AY: SIRT1 knockdown promotes neural
differentiation and attenuates the heat shock response. J Cell
Physiol. 229:1224–1235. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Melhem H, Hansmannel F, Bressenot A,
Battaglia-Hsu SF, Billioud V, Alberto JM, Gueant JL and
Peyrin-Biroulet L: Methyl-deficient diet promotes colitis and
SIRT1-mediated endoplasmic reticulum stress. Gut. Jan 20–2015.Epub
ahead of print. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Peck B, Chen CY, Ho KK, Di Fruscia P,
Myatt SS, Coombes RC, Fuchter MJ, Hsiao CD and Lam EW: SIRT
inhibitors induce cell death and p53 acetylation through targeting
both SIRT1 and SIRT2. Mol Cancer Ther. 9:844–855. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Karwaciak I, Gorzkiewicz M, Ryba K,
Dastych J, Pulaski L and Ratajewski M: AC-93253 triggers the
downregulation of melanoma progression markers and the inhibition
of melanoma cell proliferation. Chem Biol Interact. 236:9–18. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hoffmann G, Breitenbücher F, Schuler M and
Ehrenhofer-Murray AE: A novel sirtuin 2 (SIRT2) inhibitor with
p53-dependent pro-apoptotic activity in non-small cell lung cancer.
J Biol Chem. 21:5208–5216. 2014. View Article : Google Scholar
|
|
27
|
Lee IH, Cao L, Mostoslavsky R, Lombard DB,
Liu J, Bruns NE, Tsokos M, Alt FW and Finkel T: A role for the
NAD-dependent deacetylase Sirt1 in the regulation of autophagy.
Proc Natl Acad Sci USA. 105:3374–3379. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Donmez G, Arun A, Chung CY, McLean PJ,
Lindquist S and Guarente L: SIRT1 protects against α-synuclein
aggregation by activating molecular chaperones. J Neurosci.
32:124–132. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Björk JK and Sistonen L: Regulation of the
members of the mammalian heat shock factor family. FEBS J.
277:4126–4139. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hietakangas V, Ahlskog JK, Jakobsson AM,
Hellesuo M, Sahlberg NM, Holmberg CI, Mikhailov A, Palvimo JJ,
Pirkkala L and Sistonen L: Phosphorylation of serine 303 is a
prerequisite for the stress-inducible SUMO modification of heat
shock factor 1. Mol Cell Biol. 23:2953–2968. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
O'Callaghan-Sunol C and Sherman MY: Heat
shock transcription factor (HSF1) plays a critical role in cell
migration via maintaining MAP kinase signaling. Cell Cycle.
5:1431–1437. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Toma-Jonik A, Widlak W, Korfanty J, Cichon
T, Smolarczyk R, Gogler-Piglowska A, Widlak P and Vydra N: Active
heat shock transcription factor 1 supports migration of the
melanoma cells via vinculin down-regulation. Cell Signal.
27:394–401. 2015. View Article : Google Scholar
|
|
33
|
Kwon SM, Kim SA, Yoon JH and Ahn SG:
Transforming growth factor beta1-induced heat shock protein 27
activation promotes migration of mouse dental papilla-derived
MDPC-23 cells. J Endod. 36:1332–1335. 2010. View Article : Google Scholar : PubMed/NCBI
|