Introduction
Human hepatocellular carcinoma (HCC) is one of the
most common malignant tumors worldwide, particularly prevalent in
Africa and Asia. In spite of recent advances in surgical techniques
and medical management, the long-term prognosis of patients with
HCC remains unsatisfactory. For diagnosis and adequate treatment of
HCC, understanding of molecules that are responsible for
hepatocarcinogenesis and progression is critical (1).
MicroRNAs (miRNAs) are short (~22 nucleotide),
single-stranded, genome-encoded RNA molecules, which are generated
by the cleavage of precursor hairpins in two sequential processing
reactions. Initially, miRNAs are transcribed as long primary-miRNA
(pri-miRNA) transcripts which are cleaved in the nucleus by the
enzyme DROSHA (Drosha) to liberate the precursor-miRNA (pre-miRNA)
hairpin. The pre-miRNA is subsequently exported from the nucleus
and further processed by the enzyme DICER1 (Dicer) in the cytoplasm
to produce mature miRNAs (2,3). These
tiny molecules have diverse biological functions. In cancer, the
loss of tumor-suppressive miRNAs enhances the expression of target
oncogenes, whereas increased expression of oncogenic miRNAs (known
as oncomirs) can repress target tumor suppressor genes (4,5).
Recent studies have shown that miRNA expression can
be affected by chromosomal abnormalities, mutations, polymorphisms
(SNPs), transcriptional deregulation, defects in the miRNA
biogenesis machinery and epigenetic changes (6). Enhancer of zeste homolog 2
methyltransferase (EZH2), which is a catalytic subunit of the
polycomb repressive complex 2 (PRC2), is responsible for
trimethylation of histone H3 on lysine 27 (H3K27me3), and directly
controls DNA methylation (7). It
has been shown that EZH2 can be regulated by miR-101 in a plethora
of cancers (8–12). Recently, however, several chip-based
studies have shown that EZH2 not only regulates protein-encoding
genes but also miRNAs (13,14).
miR-101 is encoded by two separate genes in human
(miR-101-1 and miR-101-2). miR-101-1 is
located in intergenic region and miR-101-2 in the eighth
intron of RCL1 gene (15).
miR-101 is frequently downregulated in human HCC tissues, and
ectopic overexpression of miR-101 markedly induces apoptosis and
inhibits proliferation, migration, EMT and angiogenesis in HCC by
targeting multiple target genes such as EZH2, COX-2,
STMN1, ROCK2, MCL-1 and FOS (16). Although the antitumor role of
miR-101 is well-documented, the transcriptional regulation and the
regulatory network of miR-101 remain obscure. In the present study,
we found that miR-101 was regulated by EZH2 and they formed a
reciprocal negative feedback loop that kept miR-101 in depleted
state in HCC.
Materials and methods
Cell line and cell culture
A normal hepatic cell line Lo2, and two HCC cell
lines, HepG2 and SMMC-7721 were maintained in Dulbecco's modified
Eagle's medium (DMEM) supplemented with 10% fetal bovine serum at
37°C with 5% CO2.
RT-qPCR
Pre-miRNA and mature miRNA was extracted with RNAiso
for Small RNA (Takara, Dalian, China). The concentration and purity
of RNA were controlled using NanoDrop 2000C (Thermo Scientific,
Waltham, MA, USA). Mature miR-101 was quantified using the
Hairpin-it™ miRNAs qPCR Quantitation kit (GenePharma, Shanghai,
China) according to the manufacturer's instructions. miScript
Precursor Assays (Qiagen, Hilden, Germany) were used for pre-miRNA
quantification according to the manufacturer's instructions. U6 was
used for normalization. EZH2 transcript levels were quantified as
described by Guo et al (17). PrimeScript™ RT reagent kit (Takara)
was used for reverse transcription reaction according to the
manufacturer's instructions. SYBR-Green RT-qPCR was carried out on
StepOnePlus system (Applied Biosystems, Foster City, CA, USA). All
reactions were run in triplicate. The relative quantification in
gene expression was determined using the 2−ΔΔCq
method.
Construction of plasmids
A 292-bp fragment of EZH2 untranslated region
(3′UTR) was amplified by PCR with the following primers: (forward)
5′-ATAGAATTCCATCTGCTACCTCCTCC-3′ and (reverse)
5′-CGCAAGCTTGATTCAACAAGGAC-3′; and cloned downstream of the firefly
lucif-erase gene in pCMV-Tag2A vector (EZH2-3′UTR) (18). The predicted miR-101 binding sites
(Fig. 3C) were mutated by base pair
changes using DpnI-mediated site-directed mutagenesis with
the following primers: (M1S),
CTTCAGGAACCTCGACTGCAGTGGGCAATTTAGAAAA and (M1A),
TTTCTAAATTGCCCACTGCAGTCGAGGTTCCTGAAGC; (M2S),
TTCTGAATTTGCAAAAGATCTTAAGAATAATTTATAG and (M2A),
TATAAATTATTCTTAAGATCTTTTGCAAATTCAGAAT. To explore the
transcriptional regulation of miR-101, a 2118-bp fragment of the
miR-101-1 gene promoter (−1904 to 213 relative to the transcription
start site of pri-miR-101-1) was amplified by PCR using the
following primers: (forward),
5′-GTCGGTACCCACAAAACCAATCCCCATTGAAGACCACA-3′ and (reverse),
5′-ACCAAGCTTGACCAGCACAAATTACAGCAAAGCACCCC-3′. The amplified
fragment was inserted into the pGL3-basic vector (Promega, Madison,
WI, USA) between the KpnI and HindIII sites, named
pGL3-101. For EZH2 overexpression, the coding sequence of EZH2 was
cloned into the pcDNA3.1, named pcDNA-EZH2, using the following
primers: (forward), 5′-ACGGGTACCATCATGGGCCAGACTG-3′ and (reverse),
5′-CCGCTCGAGTCAAGGGATTTCCATTTC-3′. Lentivirus-mediated mature
miR-101 vectors (lenti-miR-101) and control miRNA were constructed
and validated by GenePharma.
Cell transfection and luciferase
assays
For lenti-miR-101 transfection, HepG2 and SMMC-7721
cells were seeded into 6-well plates at 3.5×105
cells/well. After propagation for 24 h, virus particles
(3×107) were added. For the reporter assays, HepG2 and
SMMC-7721 cells were transfected by Lipofectamine 2000 (Invitrogen)
with 1 µg of each constructed vector. For reporter gene
assays, 200 ng of the expression or control vectors (pcDNA3.1) was
co-transfected with 800 ng of the constructed reporter vector. In
each transfection, 50 ng of pRL-TK (Promega) was used to correct
for the transfection efficiency. Luciferase activity was measured
with the Dual-Luciferase Reporter Assay system (Promega). Promoter
activities were expressed as the ratio of Firefly to
Renilla luciferase activity. Chemically synthesized RNAs,
including negative control (NC), EZH2-siRNA were obtained from
GenePharma. For transfection, the cells were transfected with 1
µg of the chemically synthesized RNA.
Quantitative genomic PCR for miR-101
DNA was extracted using Takara MiniBEST Universal
Genomic DNA Extraction kit (Takara). miScript Precursor Assays
(Qiagen) were used for DNA quantification. GAPDH was used for
normalization.
5-Aza-2′-deoxycytidine (5-aza-dC)
treatment
Cells were treated with 5-aza-2′-deoxycytidine (1
µM) (Sigma) for 48 h.
Culture plate colony formation assay
Cells (200) were plated in a fresh 6-well plate and
were maintained in complete medium for 15 days. Colonies were fixed
with methanol and stained with 0.1% crystal violet in 20% methanol
for 15 min. Data were obtained from three independent
experiments.
Migration and invasion assays
For invasion assays, 1.0×105 cells were
seeded in a Matrigel-coated chamber with 8.0-µm pores (BD
Biosciences); for motility assays, 5.0×104 cells were
plated on top of uncoated membranes with 8.0-µm pores. Cells
were seeded in serum-free media and translocated toward complete
growth media for 24 h. All the experiments were repeated at least
three times.
Wound healing assay
Cells grown in a 6-well plate with 90% confluence
were starved in low serum medium (0.5–0.1% serum) overnight. A line
was drawn with a sterile 200 µl pipette tip on the bottom of
the well; the cells were rinsed with phosphate-buffered saline
(PBS) and cultured in the same medium for 48 h before photography.
Percent migration was calculated by measuring the length and width
of the cell-free area. The width was measured at five points along
the scratch area and then averaged to get an accurate
representation of the entire scratch. Percent migration was
determined using the following formula: [Δ area/area (day 0)] ×
100.
Western blot analysis
Total protein was extracted using RIPA buffer
(Beyotime, Haimen, China). Equal amounts of protein was separated
by 10% SDS-PAGE and blotted to polyvinylidene fluoride (PVDF)
membranes (Millipore, Billerica, MA, USA). Membranes were blocked
with 5% skimmed milk at room temperature for 2 h and incubated
overnight with primary antibodies: anti-EZH2 antibody (1:1,000
dilution; Cell Signaling Technology, Beverly, MA, USA) or
anti-GAPDH antibody (1:5,000 dilution; Santa Cruz Biotechnology,
Santa Cruz, CA, USA). After three 5 min washes, membranes were
incubated with horseradish peroxidase-conjugated secondary antibody
(1:5,000 dilution; Cell Signaling Technology).
Statistical analysis
Data are presented as mean ± SD from at least three
separate experiments. Unless otherwise noted, one-way ANOVA was
used for comparisons between groups. All statistical analyses were
carried out with SPSS 12.0 computer software (SPSS, Inc., Chicago,
IL, USA). P-values <0.05 were considered to indicate a
statistically significant result.
Results
EZH2 negatively regulates miR-101-1 in
HCC cell lines
To determine whether EZH2 regulates miR-101 in HCC,
we transfected Lo2 cells with EZH2 expression vector (pcDNA-EZH2).
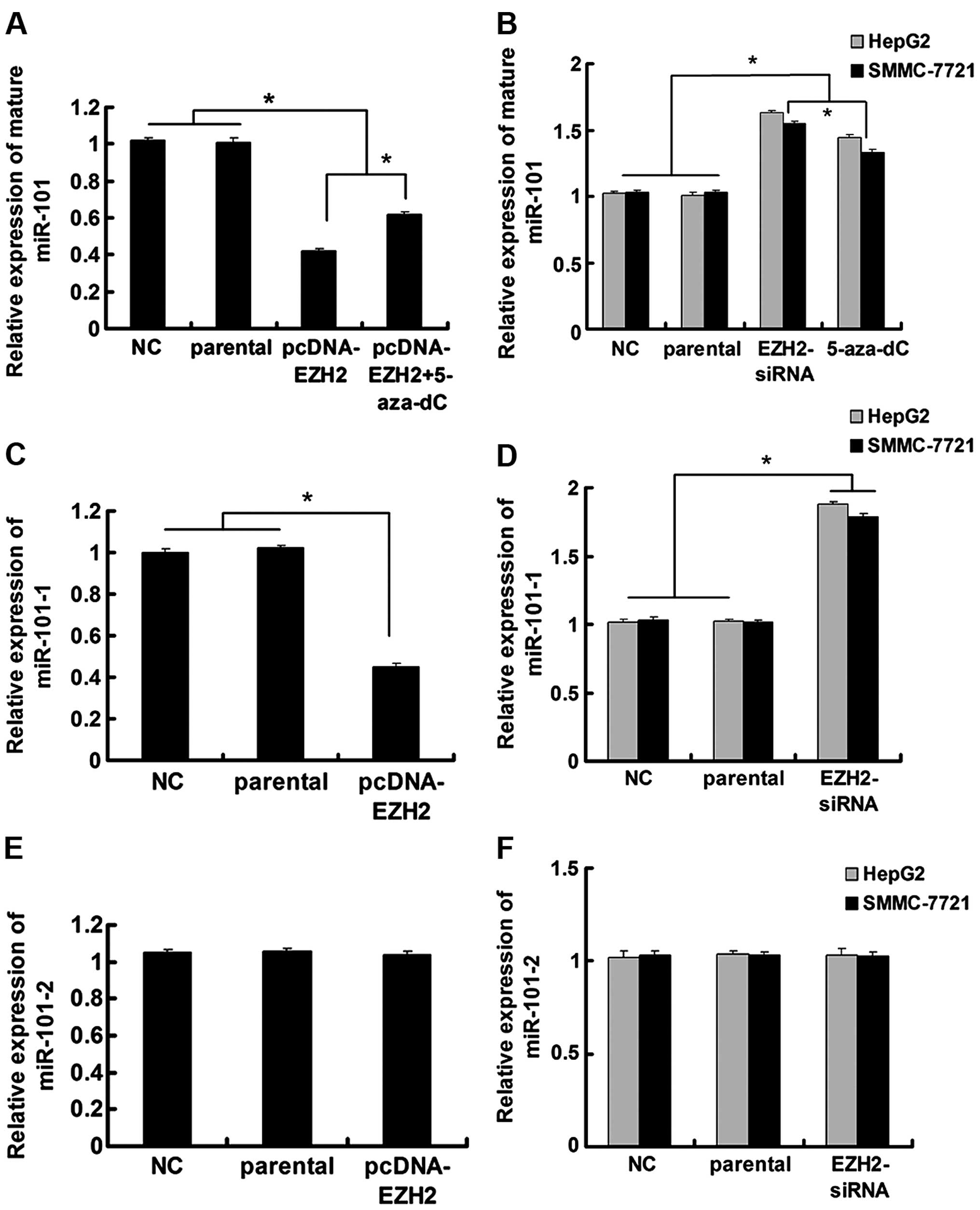
RT-qPCR demonstrated a decrease in miR-101 levels (Fig. 1A). Treatment with the DNA
methylation inhibitor 5-aza-2′-deoxycytidine (5-aza-dC) partially
inhibited this EZH2-mediated miR-101 downregulation in Lo2 cells
(Fig. 1A). In HepG2 and SMMC-7721
cells, treatment with either 5-aza-dC or EZH2-siRNA increased
mature miR-101 levels (Fig.
1B).
Since miR-101 has two genomic loci (miR-101-1, on
chromosome 1; miR-101-2, on chromosome 9), to pinpoint the locus
that is regulated by EZH2, we proceeded to detect miR-101 precursor
levels after EZH2 modulation. Our results showed that even though
ectopic EZH2 could reduce miR-101-1 expression in Lo2 cells and
EZH2 knockdown increased miR-101-1 levels in HepG2 and SMMC-7721
cells, miR-101-2 was resistant to EZH2 modulation (Fig. 1C–F).
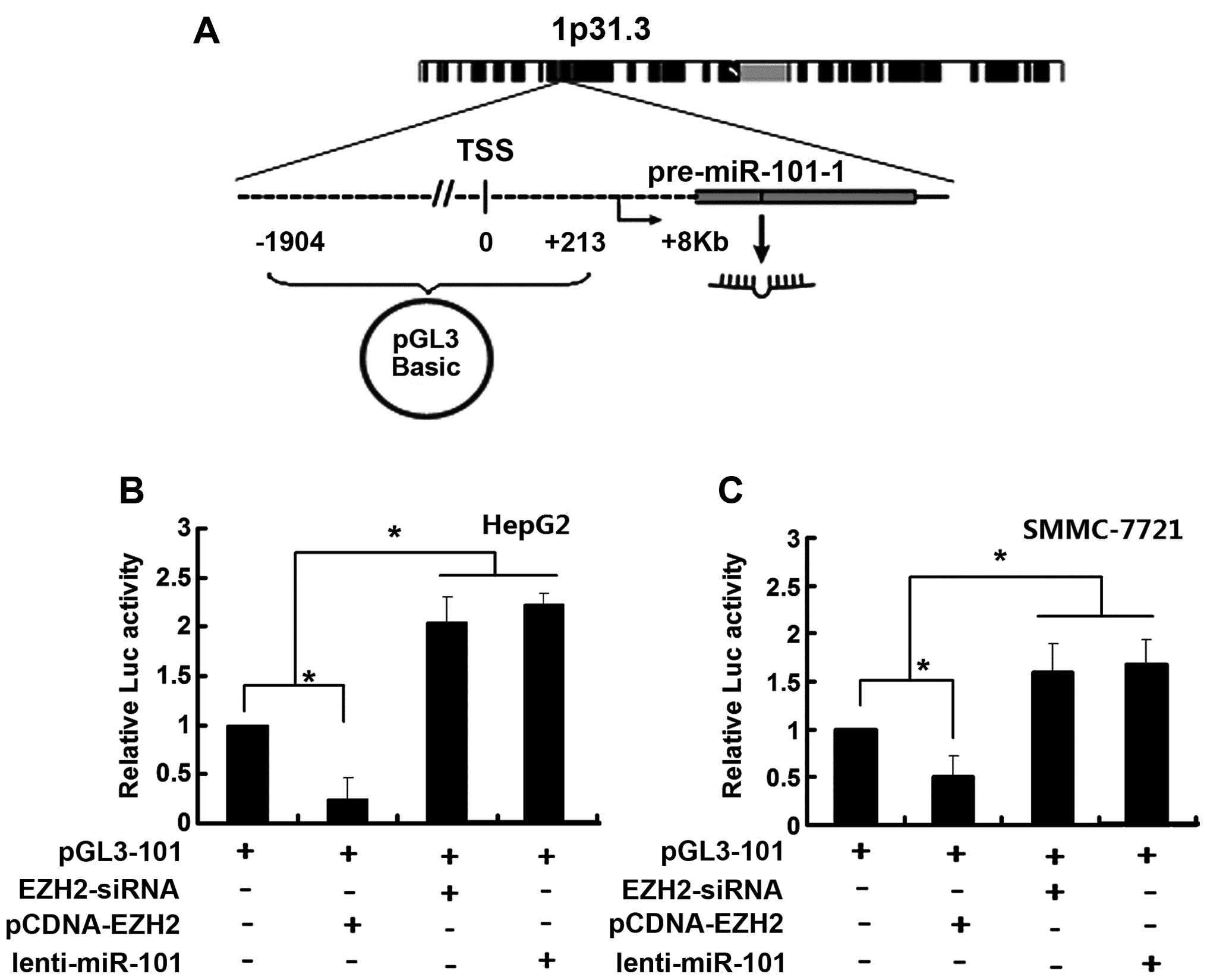
To further corroborate these findings, fragments
from position-1904 to position-213 in the miR-101-1 promoter
(relative to TSS) were cloned into a luciferase reporter plasmid
(pGL3-101) and their transcriptional activity was assessed in HepG2
and SMMC-7721 cells (Fig. 2A). We
found that luciferase activity was enhanced by EZH2-siRNA while
diminished luciferase activity was observed in the cells
transfected with pCDNA-EZH2 (Fig. 2B
and C), suggesting that miR-101-1 promoter region was involved
in EZH2-dependent miR-101-1 regulation.
miR-101 and EZH2 form a reciprocal
negative feedback loop in HCC
EZH2 is a quintessential target of miR-101. If EZH2
is the direct target of miR-101 in HCC, it seems to follow that
miR-101 and EZH2 may form a reciprocal negative feedback loop in
HCC. To test the hypothesis, we transfected HepG2 and SMMC-7721
cells with lentivirus-mediated mature miR-101 vectors
(lenti-miR-101) and a control miRNA. Our results showed ectopic
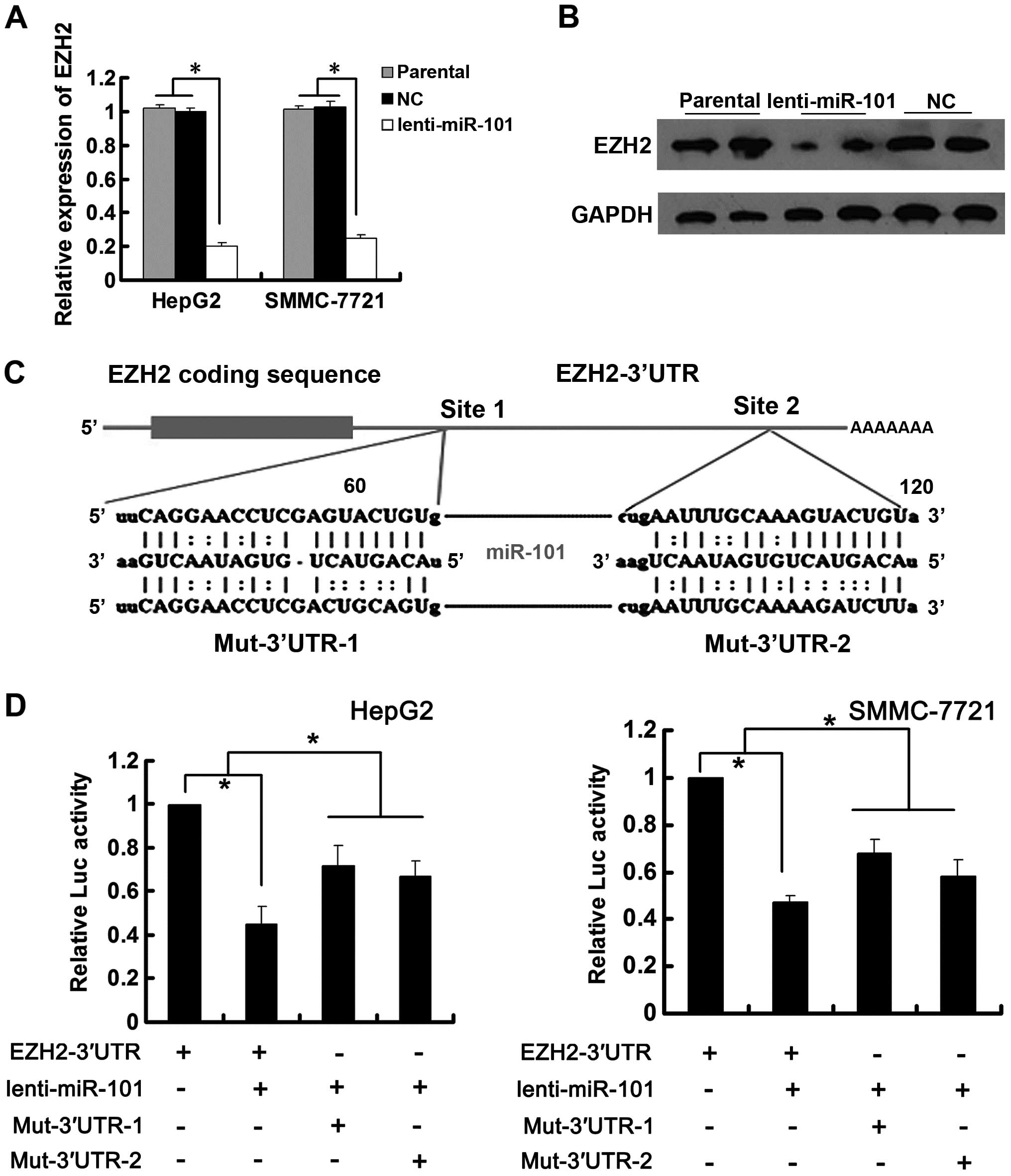
miR-101 decreased EZH2 expression at both mRNA and protein level
(Fig. 3A and B). To test whether
the 3′UTR of EZH2 was responsible for its regulation, we cloned
EZH2 3′UTR downstream of pCMV-Tag2A-luciferase reporter gene and
constructed corresponding mutated reporter vectors (Fig. 3C). Renilla luciferase vector
(pRL-TK) was used as reference control. Reporter gene assays showed
that the luciferase activity was significantly decreased by ectopic
miR-101 as compared with negative control while the mutation in the
predicted target sites abolished the repressive effects of miR-101
on luciferase activity (Fig. 3D),
suggesting that miR-101 regulated EZH2 expression by binding to its
target sites at the 3′UTR of EZH2.
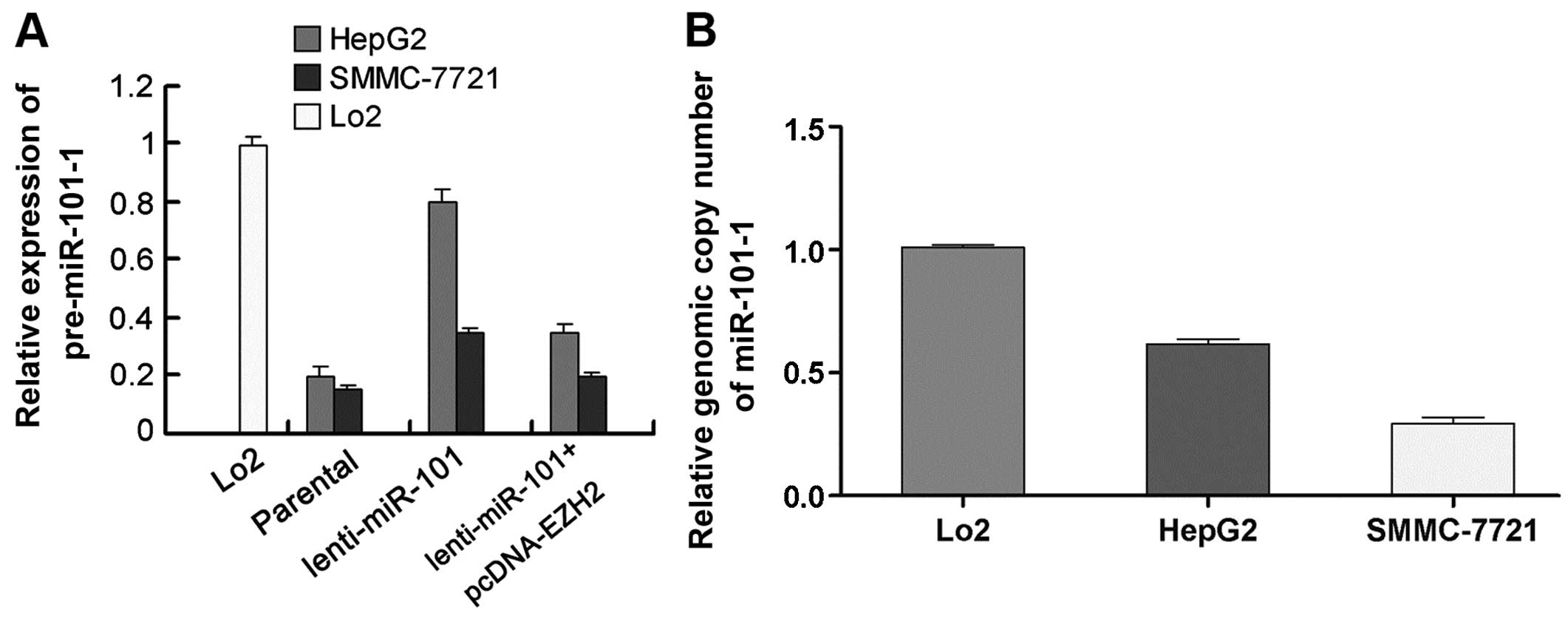
To prove the existence of EZH2/miR-101-1 feedback
loop, we tested the inducibility of precursor miR-101s by mature
miR-101. Compared with parental cell lines, the pre-miR-101-1
levels of HepG2 and SMMC-7721 cells witnessed a 2- to 4-fold
increase after ectopic expression of mature miR-101, while
co-transfection of lenti-miR-101 with pcDNA-EZH2 could reduce the
pre-miR-101-1-inducing effects of ectopic mature miR-101 (Fig. 4A). To further corroborate this
gain-of-function study, we co-transfected pGL3-101 with
lenti-miR-101 in HepG2 and SMMC-7721 cells. Our results indicated
that ectopic miR-101 could enhance the transcriptional activity of
miR-101-1 promoter (Fig. 2B and
C).
To address the issue of why HepG2 and SMMC-7721
cells had different induction efficiencies, we performed
quantitative genomic PCR for miR-101-1, as the inducibility of
pre-miRNA by ectopic mature miRNA depends on the integrity of miRNA
loci. Compared with Lo2 cells, Both HepG2 and SMMC-7721 cells
exhibited miR-101-1 locus deletion. However, SMMC-7721 cells showed
much heavier miR-101-1 locus deletion than HepG2 cells (Fig. 4B). These results suggest that the
lower inducibility of pre-miR-101-1 in SMMC-7721 cells may be
correlated with heavier miR-101-1 locus deletion. Taken together,
our results suggest that EZH2 and miR-101 negatively regulates each
other and form a feedback loop in HCC.
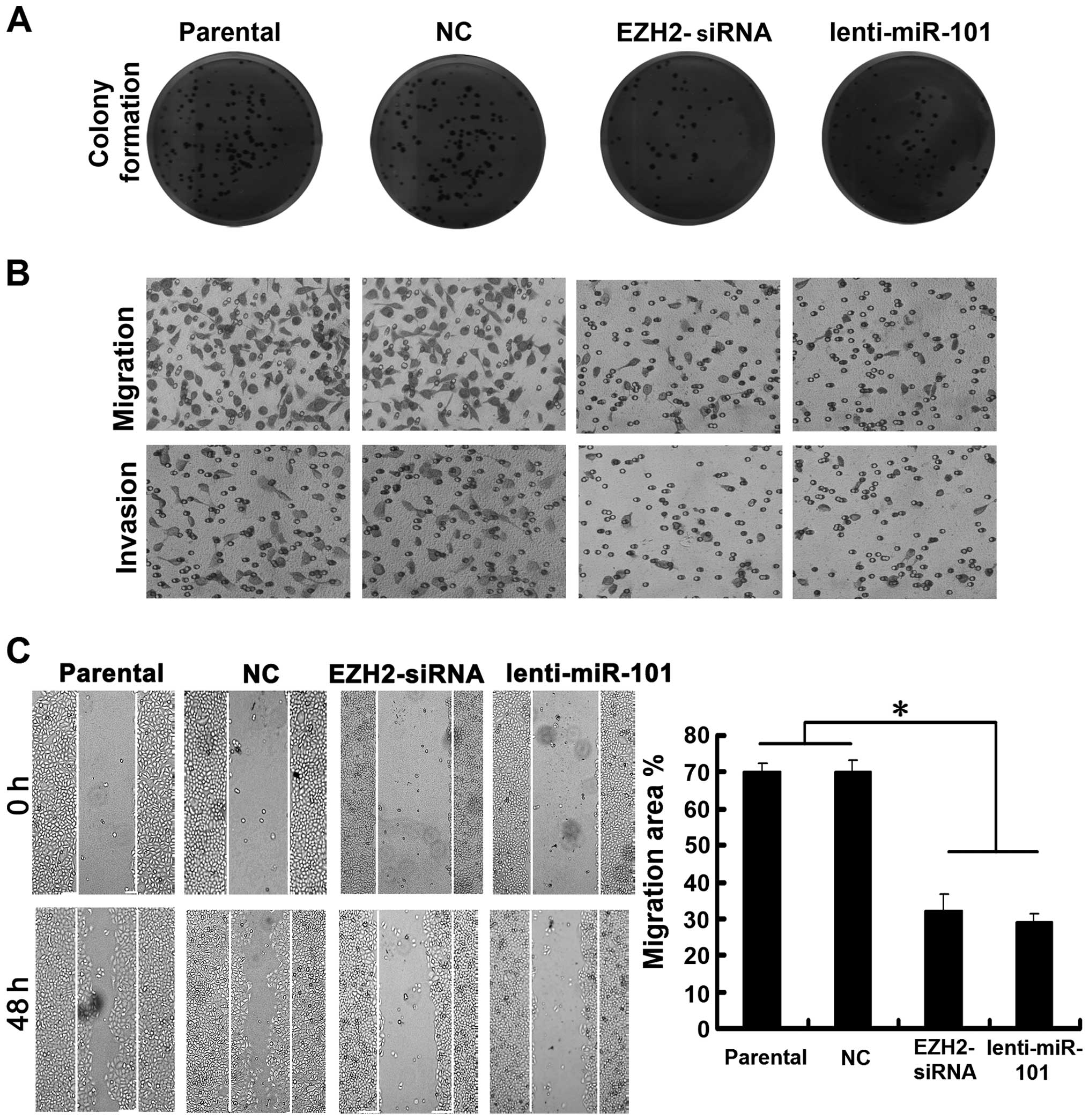
Manipulation of EZH2/miR-101 feedback
loop exerts antitumor effects
To demonstrate the efficacy of targeting
EZH2/miR-101-1 feedback loop in impeding HCC, we transfected HepG2
cells with lenti-miR-101 or EZH2-siRNA. Our results showed that
cells displayed much fewer and smaller colonies compared with NC
transfectants and parental cells (Fig.
5A). Moreover, Transwell migration and invasion assays revealed
that cells transfected with lenti-miR-101 or EZH2-siRNA
demonstrated a decreased ability to pass through 8-µm pore
size polycarbonate membrane compared with NC transfectants and
parental cells (Fig. 5B). The
effects of ectopic miR-101 and EZH2 silencing on cell migration was
further supported by wound healing assays (Fig. 5C). Taken together, our results
suggest that similar antitumor effects can be achieved both by
ectopic miR-101or EZH2 silencing in HCC cells and manipulation of
this miRNA feedback loop may have the therapeutic potential for
treating liver cancer.
Discussion
miRNA genes constitute one of the most abundant gene
families, and are widely distributed in animals, plants and
viruses. The latest release of the miRNA database (miRBase) has
catalogued 2,588 miRNAs in humans. Targeting most protein-coding
transcripts, miRNAs are involved in nearly all developmental and
pathological processes in animals. The biogenesis of miRNAs is
under tight temporal and spatial control, and their dysregulation
is associated with many human diseases, particularly cancer. miRNA
regulation takes place at multiple steps, including their
transcription, their processing by Drosha and Dicer, their loading
onto AGO proteins and miRNA turnover (19,20).
Downregulation of miR-101 is a common event in
cancers and has been implicated in the development and progression
of different malignancies. Previous studies show genomic loss of
miR-101 is one of the main mechanisms of decreased miR-101
expression in prostate, breast and gastric cancers, and
glioblastoma (15). Our genomic PCR
results indicate that this mechanism also plays a role in HCC,
since allele losses were observed in HepG2 and SMMC-7721 cells.
However, cancer cells are a heterogeneous population and no more
than half of the cases of HCC were found to have miR-101 locus
losses in previous comparative genome hybridization studies
(21–23). How cancer cells that have intact
miR-101 loci also exhibit miR-101 depletion is largely unsolved. A
previous study shows that activator protein-1 (AP-1) directly binds
to the −17.4 to −16.4 k region upstream of pre-miR-101-2 and
activated the expression of miR-101 (15). In the present study, we report that
miR-101 expression can be regulated by the key epigenetic regulator
EZH2 in miR-101-1-intact HCC cells and miR-101-1
promoter region is involved in this process. Moreover, our results
show that EZH2-meditated regulation of miR-101 is not simply a
'one-way relationship'. Instead, they form a reciprocal negative
feedback loop: high levels of EZH2 contribute to the depletion of
miR-101, which, in turn, helps HCC cells keep EZH2 at high levels,
hence sustained miR-101 silencing. Several recent studies have
reported similar feedback networks that play critical roles in
cancer (24–26). These studies, along with the present
one, strongly suggest that feedback networks involving miRNAs and
their targets may represent a common mechanism in cancer
development.
Theoretically, EZH2 may regulate miR-101-1 at the
transcription or processing steps. Our evidence, however, indicates
that the regulation takes place at the transcription step rather
than the processing step. First, miRNA-101-2 and
miRNA-101-1 share a common processing pathway except that
miRNA-101-2, being an intragenic miRNA (in the eighth intron
of RCL1 gene), does not require Drosha for cleavage once it
is co-transcribed with the host gene. In this regard, the
resistance of pre-miR-101-2 to induction by EZH2 suggests the
regulation of miR-101-1 by EZH2 should take place prior to Dicer
cleavage. Second, our reporter gene assays showed that ectopic
expression of EZH2 inhibited the transcriptional activities of
miR-101-1 promoter. miR-101-1 promoter region has
multiple CpG islands with high GC content (>90%). This GC-rich
region had defied all our attempts to amplify it using routine PCR
methods such as adding organic additives and jointly using highly
effective DNA polymerase or even slowdown PCR (27,28).
We finally managed to amplify this DNA fragment using long primers
with high T(m) and low ΔT(m) (29).
miRNA transcription is carried out by RNA Pol II and
is controlled by RNA Pol II-associated transcription factors and
epigenetic regulators. Transcription factors, such as p53, MYC,
ZEB1 and ZEB2, and myoblast determination protein 1 (MYOD1)
positively or negatively regulate miRNA expression (30). Epigenetic control, such as DNA
methylation and histone modifications also contribute to miRNA gene
regulation (31). In this regard,
EZH2, which is responsible for trimethylation of histone H3 on
lysine 27 (H3K27me3) and directly controls DNA methylation, may
regulate miR-101-1 expression in two possible ways. That is, it can
directly regulate miR-101-1 promoter by DNA methylation and
histone modification or indirectly via regulating transcription
factor(s). However, which mode of regulation is the case in HCC
should be further studied.
Another property of reciprocal negative feedback
loop is that it allows the system to remain reversible. This
property means targeting the feedback circuit at any level (miR-101
overexpression or EZH2 silencing) would trigger auto-amplification
reactions and bring about stable antitumor effects. Consistent with
this concept, we found that ectopic miR-101 and EZH2-siRNA had
similar antitumor effects on HCC in vitro. In summary, we
have deciphered a feedback mechanism that controls miR-101
expression. The finding of this mechanism sheds new light on
hepatocarcinogenesis. Our data also raise the possibility that
manipulation of this microRNA feedback loop has the therapeutic
potential for treating liver cancer.
Acknowledgments
The present study was supported by grants 81001065
from the National Natural Science Foundation of China.
References
|
1
|
El-Serag HB and Rudolph KL: Hepatocellular
carcinoma: Epidemiology and molecular carcinogenesis.
Gastroenterology. 132:2557–2576. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J,
Lee J, Provost P, Rådmark O, Kim S, et al: The nuclear RNase III
Drosha initiates microRNA processing. Nature. 425:415–419. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lund E, Güttinger S, Calado A, Dahlberg JE
and Kutay U: Nuclear export of microRNA precursors. Science.
303:95–98. 2004. View Article : Google Scholar
|
|
4
|
Bracken CP, Khew-Goodall Y and Goodall GJ:
Network-Based Approaches to Understand the Roles of miR-200 and
Other microRNAs in Cancer. Cancer Res. 75:2594–2599. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lujambio A and Lowe SW: The microcosmos of
cancer. Nature. 482:347–355. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Iorio MV, Piovan C and Croce CM: Interplay
between microRNAs and the epigenetic machinery: An intricate
network. Biochim Biophys Acta. 1799:694–701. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Viré E, Brenner C, Deplus R, Blanchon L,
Fraga M, Didelot C, Morey L, Van Eynde A, Bernard D, Vanderwinden
JM, et al: The Polycomb group protein EZH2 directly controls DNA
methylation. Nature. 439:871–874. 2006. View Article : Google Scholar
|
|
8
|
Sakurai T, Bilim VN, Ugolkov AV, Yuuki K,
Tsukigi M, Motoyama T and Tomita Y: The enhancer of zeste homolog 2
(EZH2), a potential therapeutic target, is regulated by miR-101 in
renal cancer cells. Biochem Biophys Res Commun. 422:607–614. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Alajez NM, Shi W, Hui AB, Bruce J,
Lenarduzzi M, Ito E, Yue S, O'Sullivan B and Liu FF: Enhancer of
Zeste homolog 2 (EZH2) is overexpressed in recurrent nasopharyngeal
carcinoma and is regulated by miR-26a, miR-101, and miR-98. Cell
Death Dis. 1:e852010. View Article : Google Scholar
|
|
10
|
Zhang JG, Guo JF, Liu DL, Liu Q and Wang
JJ: MicroRNA-101 exerts tumor-suppressive functions in non-small
cell lung cancer through directly targeting enhancer of zeste
homolog 2. J Thorac Oncol. 6:671–678. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cho HM, Jeon HS, Lee SY, Jeong KJ, Park
SY, Lee HY, Lee JU, Kim JH, Kwon SJ, Choi E, et al: microRNA-101
inhibits lung cancer invasion through the regulation of enhancer of
zeste homolog 2. Exp Ther Med. 2:963–967. 2011.
|
|
12
|
Ren G, Baritaki S, Marathe H, Feng J, Park
S, Beach S, Bazeley PS, Beshir AB, Fenteany G, Mehra R, et al:
Polycomb protein EZH2 regulates tumor invasion via the
transcriptional repression of the metastasis suppressor RKIP in
breast and prostate cancer. Cancer Res. 72:3091–3104. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Au SL, Wong CC, Lee JM, Fan DN, Tsang FH,
Ng IO and Wong CM: Enhancer of zeste homolog 2 epigenetically
silences multiple tumor suppressor microRNAs to promote liver
cancer metastasis. Hepatology. 56:622–631. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cao Q, Mani RS, Ateeq B, Dhanasekaran SM,
Asangani IA, Prensner JR, Kim JH, Brenner JC, Jing X, Cao X, et al:
Coordinated regulation of polycomb group complexes through
microRNAs in cancer. Cancer Cell. 20:187–199. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu JJ, Lin XJ, Yang XJ, Zhou L, He S,
Zhuang SM and Yang J: A novel AP-1/miR-101 regulatory feedback loop
and its implication in the migration and invasion of hepatoma
cells. Nucleic Acids Res. 42:12041–12051. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zheng F, Liao YJ, Cai MY, Liu TH, Chen SP,
Wu PH, Wu L, Bian XW, Guan XY, Zeng YX, et al: Systemic delivery of
microRNA-101 potently inhibits hepatocellular carcinoma in vivo by
repressing multiple targets. PLoS Genet. 11:e10048732015.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Guo J, Cai J, Yu L, Tang H, Chen C and
Wang Z: EZH2 regulates expression of p57 and contributes to
progression of ovarian cancer in vitro and in vivo. Cancer Sci.
102:530–539. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li Y, Xie J, Xu X, Wang J, Ao F, Wan Y and
Zhu Y: MicroRNA-548 down-regulates host antiviral response via
direct targeting of IFN-λ1. Protein Cell. 4:130–141. 2013.
View Article : Google Scholar
|
|
19
|
Ha M and Kim VN: Regulation of microRNA
biogenesis. Nat Rev Mol Cell Biol. 15:509–524. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Krol J, Loedige I and Filipowicz W: The
widespread regulation of microRNA biogenesis, function and decay.
Nat Rev Genet. 11:597–610. 2010.PubMed/NCBI
|
|
21
|
Qin LX, Tang ZY, Sham JS, Ma ZC, Ye SL,
Zhou XD, Wu ZQ, Trent JM and Guan XY: The association of chromosome
8p deletion and tumor metastasis in human hepatocellular carcinoma.
Cancer Res. 59:5662–5665. 1999.PubMed/NCBI
|
|
22
|
Leung TH, Wong N, Lai PB, Chan A, To KF,
Liew CT, Lau WY and Johnson PJ: Identification of four distinct
regions of allelic imbalances on chromosome 1 by the combined
comparative genomic hybridization and microsatellite analysis on
hepatocellular carcinoma. Mod Pathol. 15:1213–1220. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Patil MA, Gütgemann I, Zhang J, Ho C,
Cheung ST, Ginzinger D, Li R, Dykema KJ, So S, Fan ST, et al:
Array-based comparative genomic hybridization reveals recurrent
chromosomal aberrations and Jab1 as a potential target for 8q gain
in hepatocellular carcinoma. Carcinogenesis. 26:2050–2057. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Brabletz T: MiR-34 and SNAIL: Another
double-negative feedback loop controlling cellular plasticity/EMT
governed by p53. Cell Cycle. 11:215–216. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Luzi E, Marini F, Giusti F, Galli G,
Cavalli L and Brandi ML: The negative feedback-loop between the
oncomir Mir-24-1 and menin modulates the Men1 tumorigenesis by
mimicking the 'Knudson's second hit'. PLoS One. 7:e397672012.
View Article : Google Scholar
|
|
26
|
Yamakuchi M and Lowenstein CJ: MiR-34,
SIRT1 and p53: The feedback loop. Cell Cycle. 8:712–715. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bachmann HS, Siffert W and Frey UH:
Successful amplification of extremely GC-rich promoter regions
using a novel 'slowdown PCR' technique. Pharmacogenetics.
13:759–766. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Frey UH, Bachmann HS, Peters J and Siffert
W: PCR-amplification of GC-rich regions: 'Slowdown PCR'. Nat
Protoc. 3:1312–1317. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li LY, Li Q, Yu YH, Zhong M, Yang L, Wu
QH, Qiu YR and Luo SQ: A primer design strategy for PCR
amplification of GC-rich DNA sequences. Clin Biochem. 44:692–698.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kim VN, Han J and Siomi MC: Biogenesis of
small RNAs in animals. Nat Rev Mol Cell Biol. 10:126–139. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Davis-Dusenbery BN and Hata A: Mechanisms
of control of microRNA biogenesis. J Biochem. 148:381–392.
2010.PubMed/NCBI
|