Introduction
Colorectal cancer (CRC) is one of the most prevalent
malignancies in China and many other parts of the world. The
American Cancer Society recently reported that the rate of
incidence of and death due to CRC have undergone a rapid decline in
the past 30 years, owing to the improvement in early detection
and/or treatment (1). However,
personalized therapeutic strategies targeting different
pro-oncogenic factors and overcoming drug resistance are still
issues in CRC treatment.
Previous clinical research indicated that Smad4 is a
predictive biomarker for 5-fluorouracil (5-FU)-based chemotherapy
in patients with CRC (2–6). In addition, a recent in vitro
study provided evidence that Smad4 inactivation promotes the
resistance of CRC to 5-FU treatment and hypoxia-induced cell death
(7). Low Smad4 expression in human
CRC predicts early recurrence after curative therapy (4), less response to 5-FU (2,3) and
shorter overall and disease-free survival time (2,3,8).
However, the mechanism concerning the promotive effect of Smad4 on
drug sensitivity remains to be elucidated. The PI3K/Akt pathway has
been shown to play an important role in promoting cell survival and
inhibiting apoptosis. Owing to these effects, the PI3K/Akt pathway
may affect cellular responses to DNA damage and cell cycle
checkpoints induced by DNA damage. As crosstalk between Smad and
the non-Smad pathway exists, modification of the non-Smad pathway
by Smad4, including the PI3K/Akt pathway, may effect drug
sensitivity.
The present study suggests, for the first time, that
Smad4 has an effect on the induction of chemosensitivity both in
murine CT26 cells and human SW620 cells, through G1 or G2 cell
cycle arrest by inhibiting the PI3K/Akt/CDC2/survivin pathway.
LY294002 reversed the chemosensitivity of CRC with low Smad4
expression by inhibiting the PI3K/Akt/CDC2/survivin cascade. These
findings imply that Smad4 may be a candidate biomarker for the
combined use of LY294002 and 5-FU-based chemotherapy for patients
with CRC.
Materials and methods
Cell lines, animals, reagents and
antibodies
CT26 cells, an undifferentiated murine
adenocarcinoma-derived cell line from the BALB/c mouse, and human
colon adenocarcinoma cell line SW620 were maintained in RPMI-1640
containing 10% fetal bovine serum (FBS). Smad4 knockdown CT26 cells
and SW620 cells expressing Smad4 were established as previously
described (9). Female BALB/c and
athymic nude mice (6 weeks of age) were used for the experiments.
All animal studies were approved by the Ethics Committee of
Huazhong University of Science and Technology. 5-FU was obtained
from Sigma. The PI3K inhibitor, LY294002, and the MEK1/2 inhibitor,
PD98059, were obtained from Calbiochem (San Diego, CA, USA).
Antibodies were purchased as follows: anti-Smad4, anti-cyclin B1,
anti-survivin, anti-CDC2, anti-ERK (Santa Cruz Biotechnology, Santa
Cruz, CA, USA); anti-cleaved-caspase 3, anti-p-ERK, ant-p-Akt and
anti-Akt (Cell Signaling Technology, Inc., Danvers, MA, USA).
Cell counting assay
CT26 cells (2,000/well) or SW620 cells (3,000/well)
were seeded into 12-well plates and were cultured in media with 10%
FBS. Media were replaced every other day. Cells were counted each
day, and the average cell numbers from triplicate measurements were
plotted.
Cell isolation from tumor tissues
The liver metastasis model was established by
splenic injection of CT26 cells in BALB/c mice as previously
described (10). Liver metastases
were removed 4 weeks after cell inoculation and cut into small
pieces in phosphate-buffered saline (PBS). Tissues were then
incubated with collagenase (Sigma) medium for 3–4 h at 37°C, under
shaking. Cells were centrifuged and washed with Dulbecco's modified
Eagle's medium (DMEM) 4–5 times, and then suspended in complete
DMEM in 10 ml for 2 min. Eight milliliters was taken from the top
for culture, and the liver metastasis cell line (CT26 LM1) was
established. LM1 cells were splenic injected into the mice, and LM2
cells were established by repeating the experiment.
Soft agarose tumorigenicity assay
Semisolid (1 ml) 0.8% sea plaque agarose was plated
into 6-well plates, allowed to solidify for at least 1 h. Then,
CT26 (1.0×104) and SW620 (2.5×104) cells were
suspended in 1 ml of 0.4% agarose containing 7% FBS and plated on
the top of the agarose. After 48 h, the cells were treated with
media containing 20 µM 5-FU. The media were replaced with or
without 5-FU every other day. Images of the colonies were captured
by phase contrast microscope 10 days after cell splitting.
Western blotting
Lysates from the CT26 or SW620 cells were treated as
indicated, and tumors from the animal models were used for western
blot analysis as previously described (11).
Flow cytometric analysis for cell cycle
analysis
Cell cycle distribution analysis was performed by
flow cytometry as previously described (12). Cells (1×105/well) were
split into 6-well plates and were incubated in serum-free media for
48 h to synchronize the cell cycle. Then cells were treated with
kinase inhibitors and/or different concentrations of 5-FU for 48 h.
Cells were harvested, washed twice with PBS and were fixed in 70%
cold ethanol, and stained with propidium iodide (PI) in PBS
containing RNase (both from Sigma). Stained cells were
analyzed.
Tumorigenicity assay
CT26 (1×105) or SW620 cells
(2×106) were injected subcutaneously into BALB/c or
athymic nude mice (n=5), respectively. 5-FU treatment was started
as soon as the tumor had reached palpable size. For the SW620 tumor
xenografts, 5-FU (50 mg/kg) was administered i.p. on a 3 times/week
basis for three consecutive weeks, and for CT26 allografts, 5-FU
(100 mg/kg) was administered once a week during the first two weeks
and 50 mg/kg once a week thereafter as previously described
(13,14). Tumor volume was calculated as
previously described (10). Mice
were sacrificed according to IACUC guidelines.
MTT cell proliferation assay and
IC50 calculation
CT26 and SW620 cells (5×103) were plated
into 96-well flat-bottomed microtiter plates and were incubated
overnight. Cells were treated with the indicated concentrations of
5-FU for 72 h. The number of viable cells was determined by MTT
assay following the kit protocol (CT01; Millipore). The percent
viability was calculated as: [Absorbance of
drug-treatment)/(control absorbance)] × 100%. IC50
values were calculated using linear or logarithmic regression
(R2>0.9).
Cell migration assay
Cells (2×104) in 100 µl serum-free
media were seeded to the upper chamber of 8-µm pore
Transwells coated with collagen. Cells were allowed to migrate
towards 10% FBS containing medium in the lower chamber for 5 h.
Cells on the upper side of the membrane were removed with a cotton
swab. Cells that passed through the Transwells were fixed with 4%
paraformaldehyde and stained with 1% crystal violet. Cells in five
random fields (magnification, ×200) in each well were counted.
Statistical analysis
The Student's t-test was used to determine the
statistical differences among the analyzed groups. Calculations
were performed using Prism 5.0 software for Macintosh (GraphPad
Software, San Diego, CA, USA). P<0.05 was considered to indicate
a statistically significant result.
Results
Highly malignant CRC cells exhibit lower
Smad4 expression
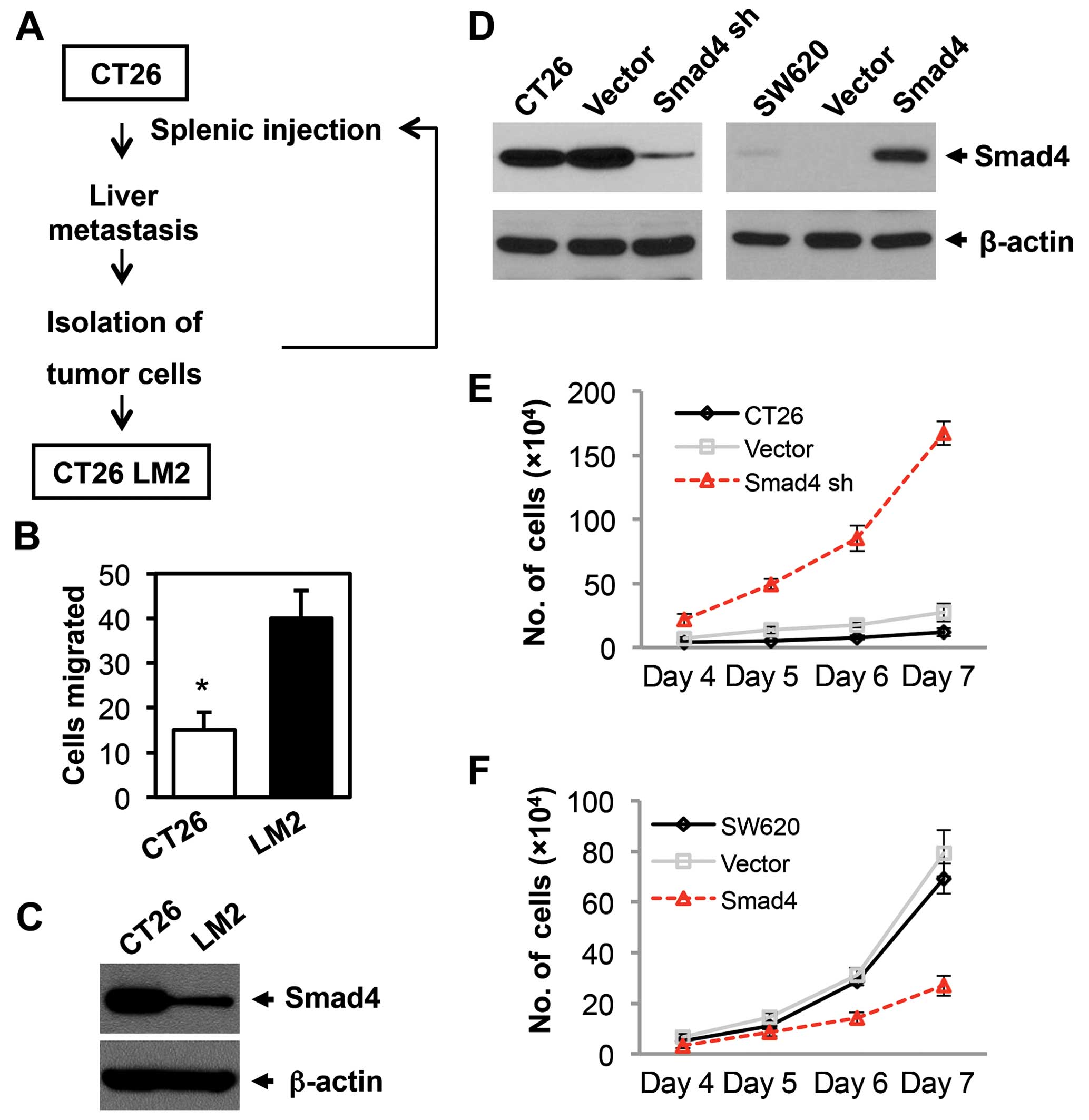
A highly aggressive and highly metastatic mouse cell
line was generated using a splenic injection model of liver
metastasis. CT26 cells have less potential to produce liver
metastasis. To develop this highly metastatic cell line, CT26 cells
were injected into the spleen of BALB/c mice, and 4 weeks later the
mice were sacrificed, liver metastases were harvested and the cell
line was established. Following this procedure, a highly metastatic
CT26-LM2 cell line was established after 2 cycles of stepwise
selection (Fig. 1A). The
aggressiveness of the LM2 cells was tested by in vitro
migration assays using Boyden chambers. We found that the LM2 cells
exhibited a 2.7-fold increase in the level of cell migration as
compared to the CT26 parental cells (Fig. 1B). Therefore, these results
confirmed that the LM2 cells are highly aggressive. We next
examined the Smad4 expression in the CT26 parental and LM2 cells.
Smad4 expression in the LM2 cells was sharply lower when compared
with the parental CT26 cells (Fig.
1C). These results indicated that lower Smad4 expression
predicts higher malignancy in CRC.
Smad4 inhibits the proliferation of
CRC
In order to investigate the role of Smad4 in CRC
progression and drug sensitivity, we established a Smad4-knockdown
mouse CRC cell line (CT26 Smad4 sh) and a Smad4-overexpressed human
CRC cell line (SW620 Smad4). Smad4 expression was examined by
western blotting (Fig. 1D). Cell
proliferation was assessed by a cell counting assay. Smad4
deficiency significantly promoted cell proliferation in the CT26
cells (Fig. 1E), while Smad4
overexpression reduced the cell proliferation in the SW620 cells
(Fig. 1F). In summary, Smad4
reduces cell proliferation in CRC, which is consistent with our
previous study (9).
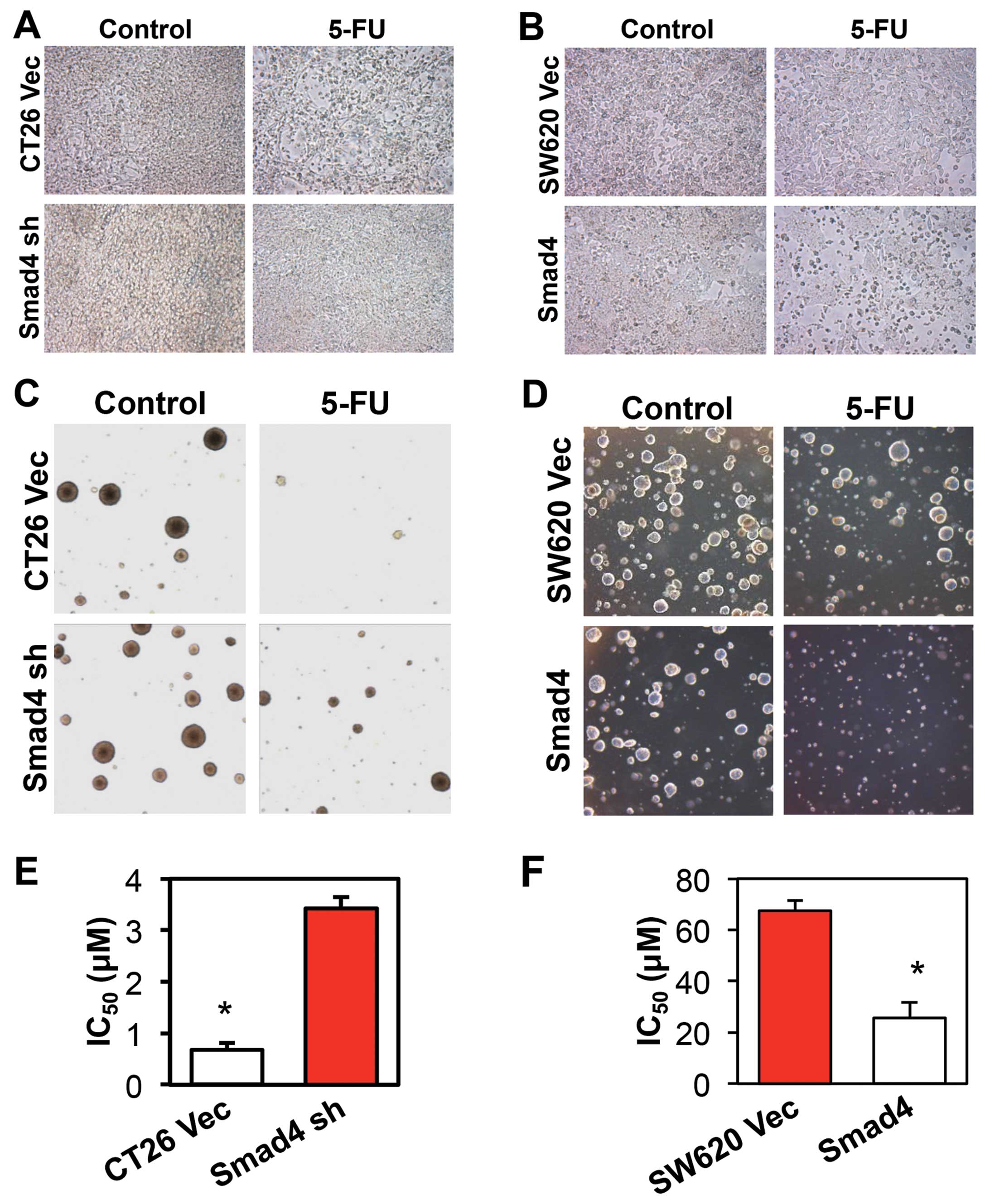
Smad4 promotes the chemosensitivity of
CRC to 5-FU in vitro and in vivo
To determine whether Smad4 affects the
chemosensitivity of CRC cells, we first treated the CRC cells with
5-FU. After a 48-h treatment with 5-FU, the CT26 empty vector group
presented a large number of floating cells, while the
Smad4-knockdown group showed few floating cells (Fig. 2A). Consistent with this result, 5-FU
killed more cells in the Smad4-overexpressing SW620 cells when
compared with that in the empty vector group (Fig. 2B). We next treated the CRC colonies
in soft argarose with 5-FU, and achieved the confirmed result
(Fig. 2C and D). MTT cell
proliferation assay was applied to check the IC50 value
to 5-FU in the different cells. The IC50 value to 5-FU
in the CT26 Smad4-knockdown group was >5 times higher when
compared with the CT26 vector control group (Fig. 2E). In contrast, in the
Smad4-overexpressing SW620 cells, the IC50 value of 5-FU
was >2.6 times lower when compared with the IC50
value in the vector control group (Fig.
2F).
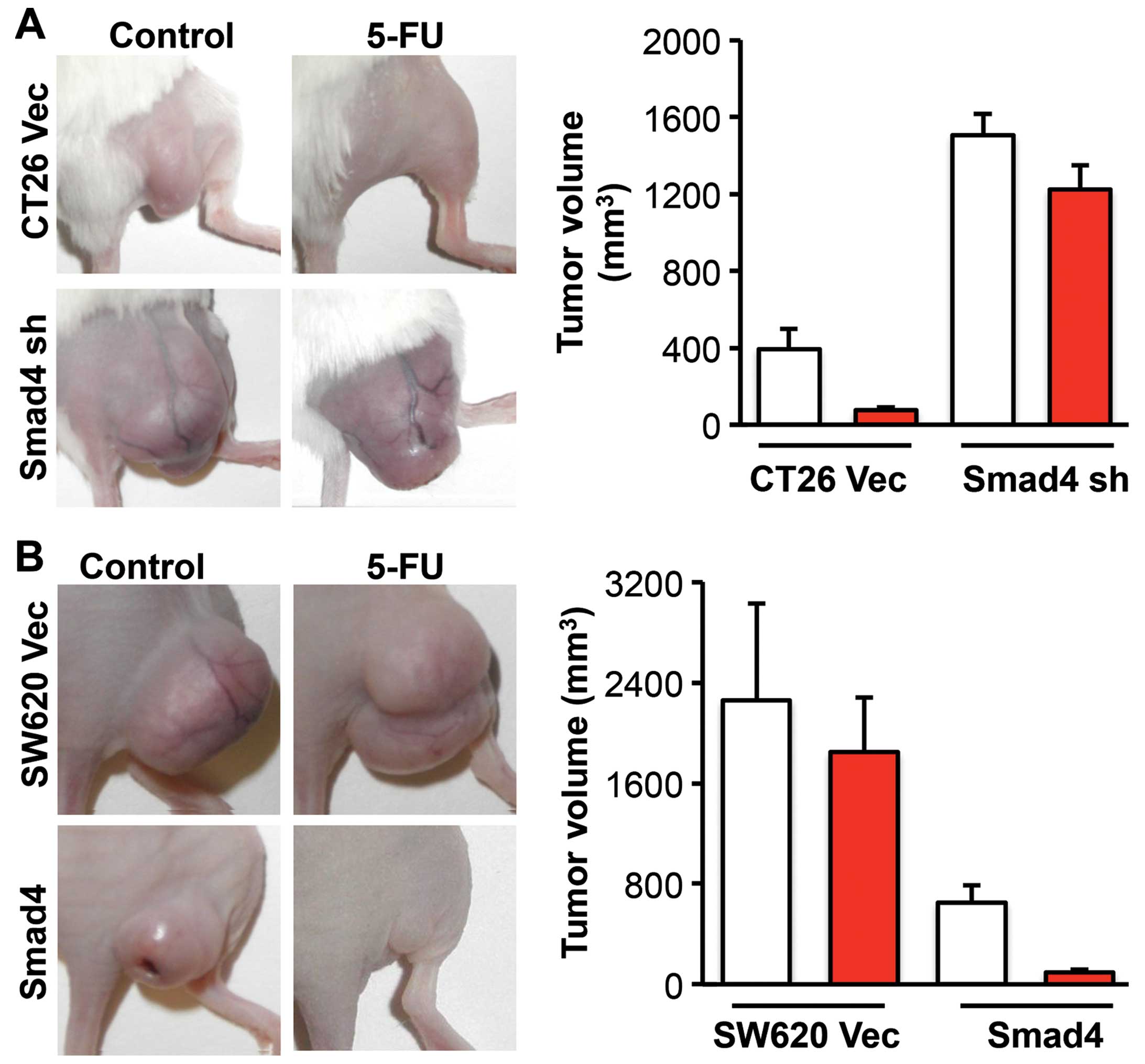
We next treated the mice bearing CRC tumors with
5-FU to investigate the effect of Smad4 on 5-FU drug sensitivity
in vivo. At the end of the experiment, the mice were
sacrificed according to the IACUC standard and tumors were removed
and measured. 5-FU reduced the tumor volume by 4.2 times in the
CT26 empty vector group, while it reduced the tumor volume by only
0.2 times in the CT26 Smad4-knockdown group (Fig. 3A). In the SW620 empty vector control
group, the tumor volume was reduced only by 0.2 times following
5-FU treatment. In the SW620 Smad4-overexpressing group, the tumor
volume was reduced by 5.9 times following 5-FU treatment (Fig. 3B). These results provide evidence
that Smad4 promotes chemosensitivity to 5-FU in CRC.
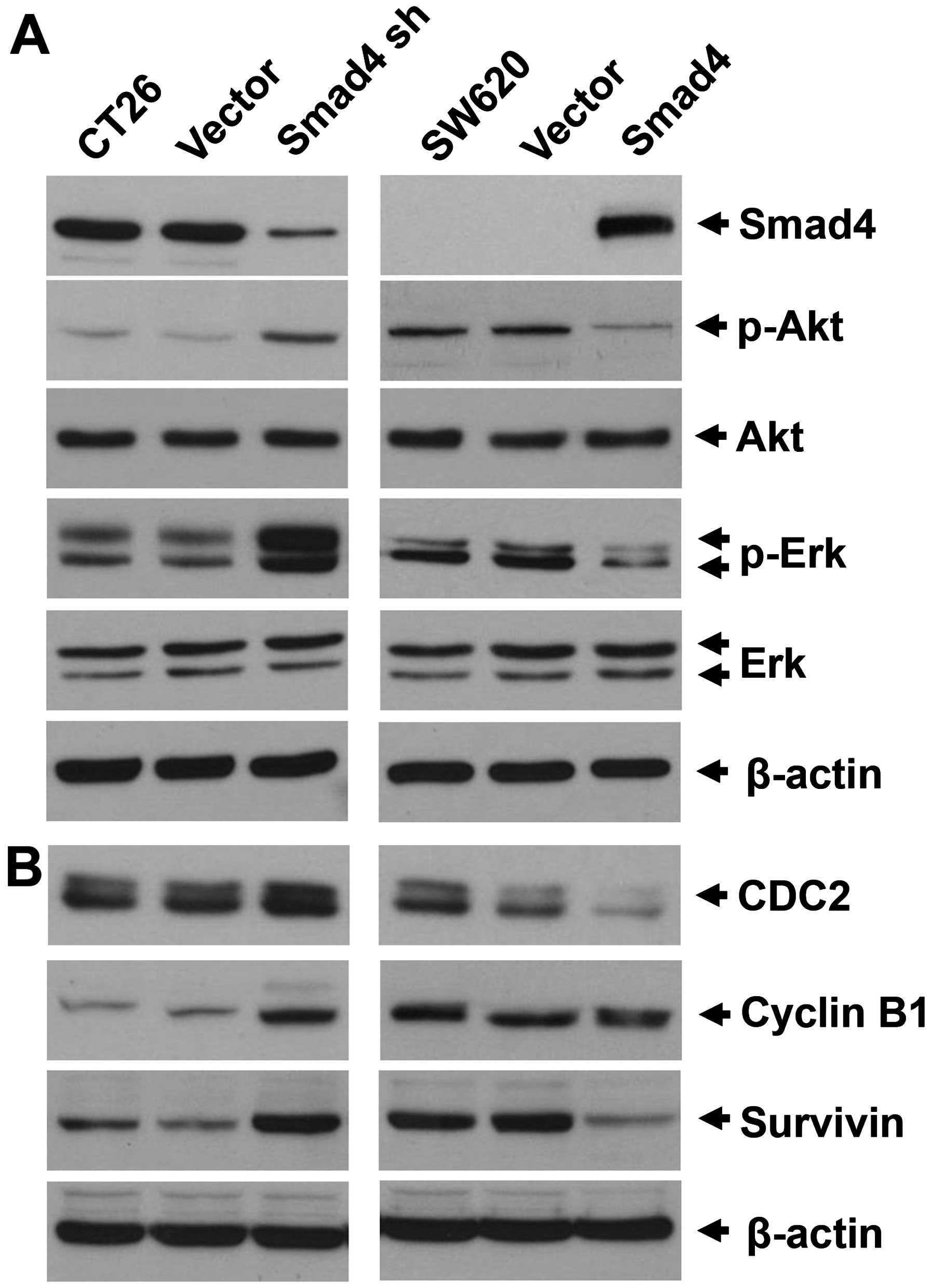
Smad4 deficiency in CRC activates Akt and
ERK, and attenuates cell cycle arrest in the G1 and G2 phases
As crosstalk between Smad and the non-Smad pathway
exists, and several non-Smad pathways were reported to affect
chemosensitivity, we examined the activation of Akt and ERK in the
Smad4-knockdown or overexpressing cells. Consistent with our
previous study, Akt was activated in the Smad4 knockdown CT26 and
Smad4-null SW620 cells. Moreover, ERK was also activated in the
Smad4-deficient cells (Fig. 4A). As
5-FU is a cell cycle-specific drug, we next tried to investigate
whether the Akt or ERK pathway modified by Smad4 affects cell
cycle-related proteins.
CDC2 (CDK1) is a cyclin-dependent protein kinase,
which regulates the G2-M mitotic cell cycle checkpoint through the
CDC2/cyclin B complex (15). In
addition, the CDC2/cyclin B complex regulates survivin, which
inhibits apoptosis and also regulates the cell cycle (16,17).
In the present study, we observed that CDC2 and cyclin B were
amplified in the CT26 cells when Smad4 was knockdown, while they
were reduced in the SW620 Smad4-overexpressing cells (Fig. 4B).
Cell cycle distribution was next examined by flow
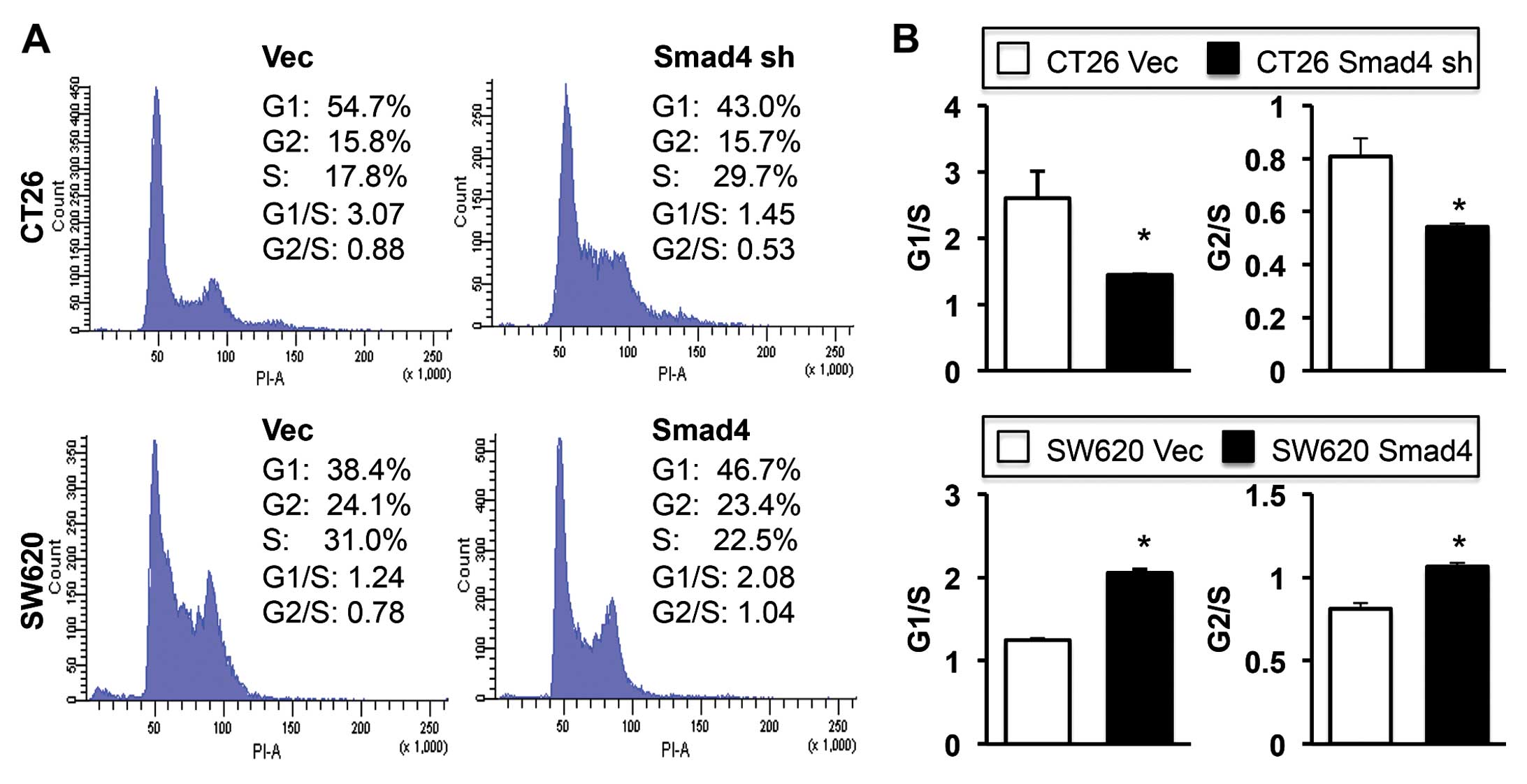
cytometric analysis. We observed that Smad4 in the CRC cells
triggered cell cycle arrest in both the G1 and G2 phase (Fig. 5A). G1/S and G2/S ratios indicated
that the cell cycle arrest reached significance at the G1 and G2
checkpoint in both the CT26 vector control and SW620
Smad4-overexpressing cells when compared with the Smad4-deficient
cells (Fig. 5B).
Smad4 promotes 5-FU sensitivity through
G2/M cell cycle arrest and apoptosis by inhibition of the
PI3K/Akt/CDC2/survivin pathway
As both cell cycle arrest and cell apoptosis altered
by CDC2 expression may affect drug sensitivity, this result led us
to hypothesis that CDC2 amplification in Smad4 deficient/null cells
may contribute to 5-FU resistance.
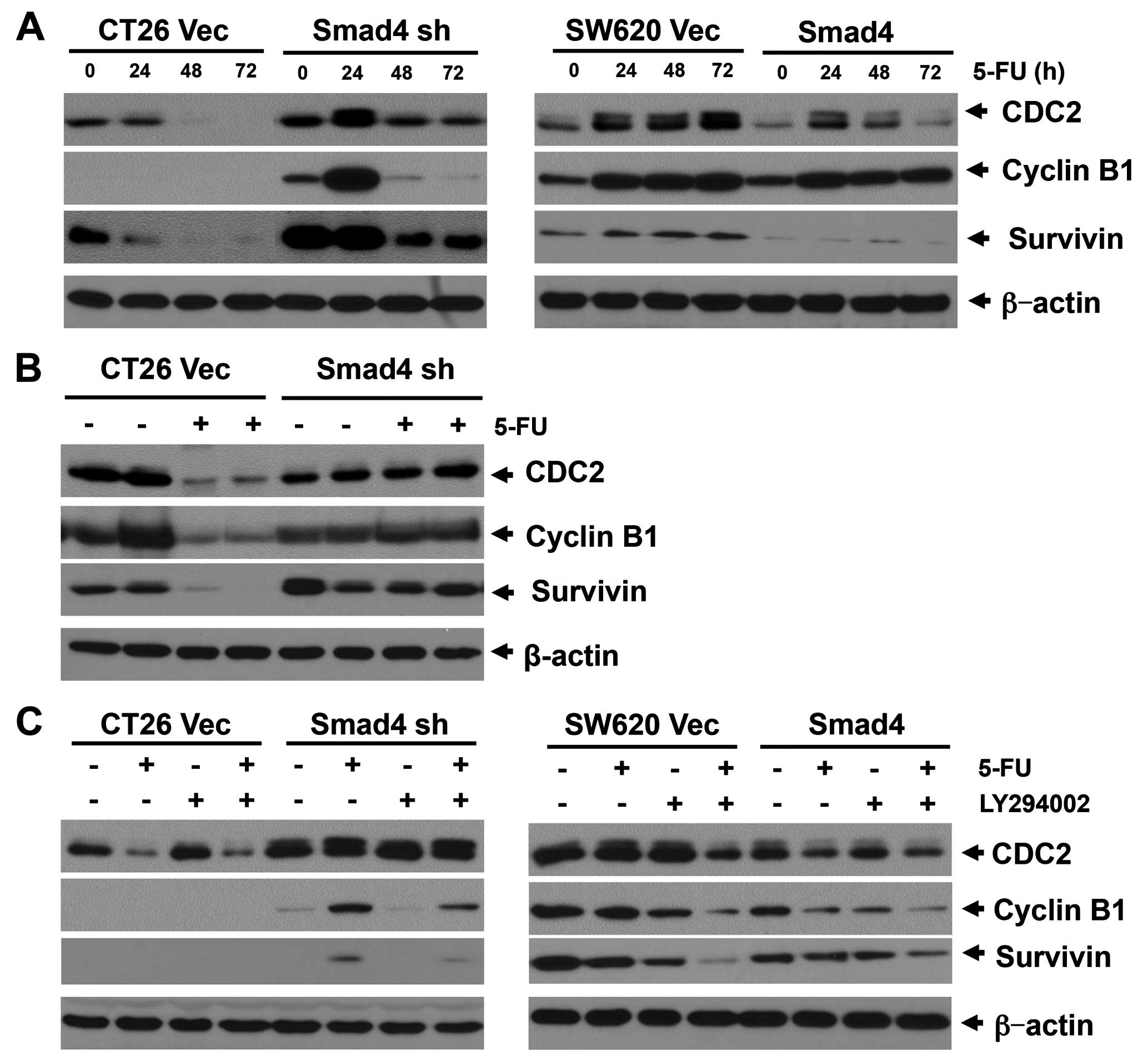
We then examined the CDC2/cyclin B/survivin cascade
with 5-FU treatment in the two model cell lines. In the CT26
Smad4-deficient cells, CDC2 expression was significantly increased
24 h after 5-FU treatment, and thereafter was reversed to a lower
level after a 48-h treatment. In contrast, CDC2 expression in the
CT26 vector control cells was continuously downregulated after a
24-h treatment with 5-FU. In this cell line, the cyclin B1 and
survivin expression levels were strictly correlated with CDC2
expression (Fig. 6A). Consistent
with this result, 5-FU treatment downregulated the CDC2/cyclin
B1/survivin cascade in the CT26 vector tumors, but not in the CT26
Smad4-deficient tumors in the mouse model (Fig. 6B). In the SW620 vector control
cells, 5-FU treatment continuously amplified CDC2 and survivin
expression. CDC2 and survivin were also increased in the SW620
Smad4-transfected cells at 24 and 48 h following 5-FU treatment,
but was decreased following a 72-h treatment with 5-FU (Fig. 6A). However, cyclin B1 which was
promoted by 5-FU treatment showed no change between the vector
control and the Smad4-overexpressing cells in this cell line.
Upregulation of CDC2/cyclin B and survivin plays a role in
promoting cell survival and G2-M cell cycle transition, both of
which may contribute to 5-FU resistance. These data indicated that
Smad4 inhibited the CDC2/cyclin B/survivin cascade, and thus
promoted 5-FU sensitivity through promotion of cell apoptosis and
G2-M cell cycle arrest.
To elucidate the mechanism of Smad4 in regulating
the CDC2/cyclin B/survivin cascade, we next focused on the PI3K/Akt
pathway that plays a pivotal role in cell survival and cell cycle
regulation. Akt was reported to regulate CDC2 in different types of
malignancies (18,19). In the present study, the expression
levels of CDC2 and survivin were consistently correlated with p-Akt
expression in both of the two model cell lines (Fig. 4), which imply that CDC2/survivin is
regulated by the Akt pathway. We next used a low dose of the PI3K
inhibitor (LY294002) in combination with 5-FU to treat the CT26 and
SW620 cells. In the CT26 Smad4-knockdown and SW620 Smad4-null
cells, LY294002 significantly inhibited CDC2 and cyclin B
expression under 5-FU treatment, while a lower effect was noted in
these cells without 5-FU treatment (Fig. 6C).
In contrast, LY294002 showed a slight effect on CDC2
and cyclin B expression in the Smad4 expressed CT26 cells or Smad4
overexpressed SW620 cells. In addition, we previously reported that
LY294002 had a stronger effect on inhibiting survivin expression in
Smad4-deficient cells under 5-FU treatment, when compared with CT26
or SW620 cells with higher Smad4 expression (9). This evidence indicates that LY294002
has a synergistic effect on promoting 5-FU-induced apoptosis,
through inhibition of the CDC2/cyclin B/survivin cell survival
pathway.
Taken together, these results indicated that the
inhibition of Smad4 by the PI3K/Akt/CDC2/survivin pathway
contributes to cell cycle arrest, thereby restoring 5-FU
sensitivity in CRC.
MEK/ERK pathway contributes little to
Smad4-induced chemosensitivity in CRC
The ERK pathway is a controversial factor that may
effect 5-FU sensitivity. In the different cell lines, the ERK
pathway was reported either required or resistant to 5-FU-induced
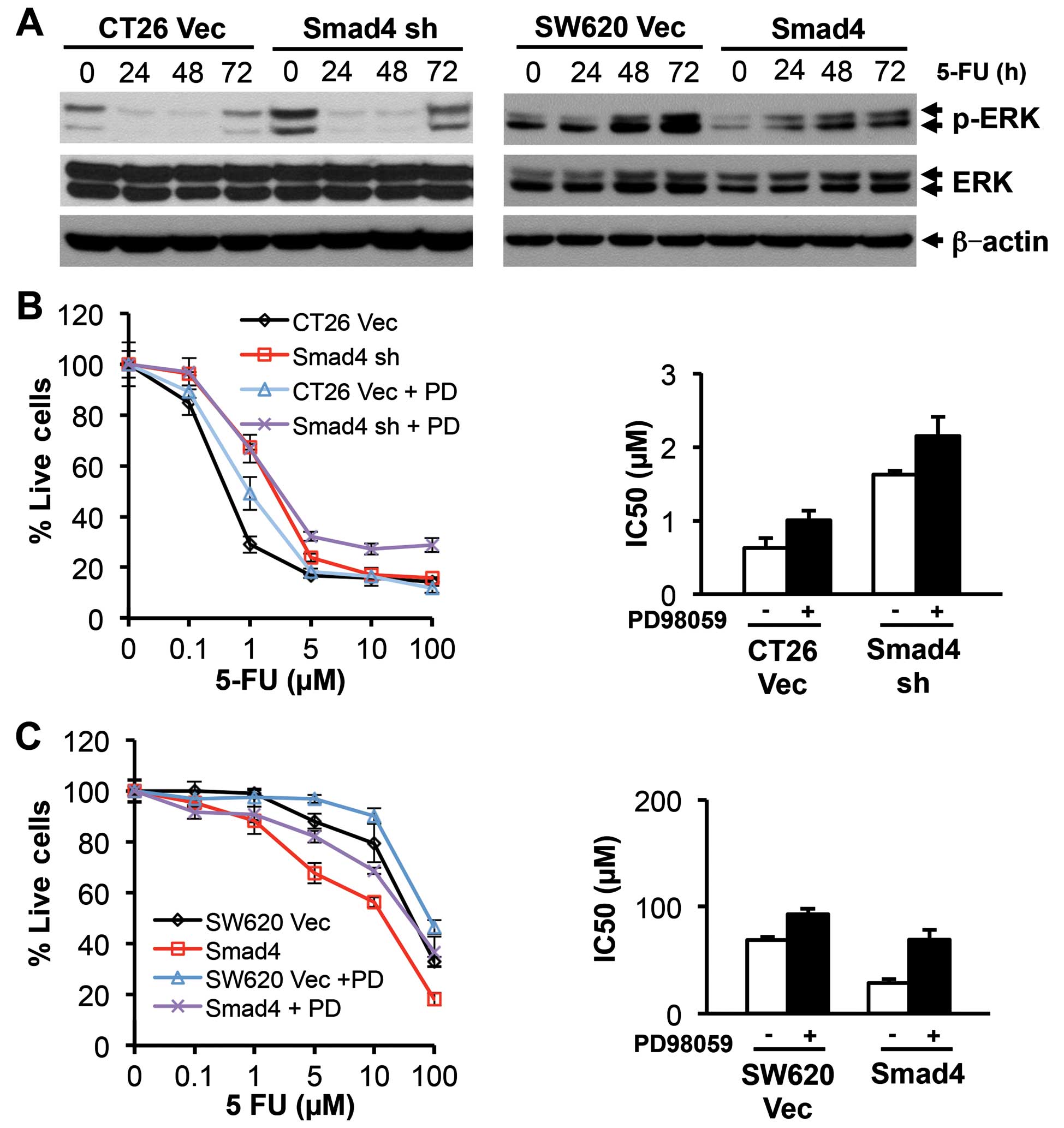
apoptosis (20). In CT26 cells,
p-ERK was activated in the Smad4-deficient clones compared with the
vector control, and was depressed following 5-FU treatment in both
vector control and Smad4-knockdown clones (Fig. 7A). In the SW620 cells, p-ERK was
downregulated in the Smad4-overexpressing clones, and was increased
in a time-dependent manner following 5-FU treatment in both the
control and Smad4-transfected clones (Fig. 7A). We next used MEK inhibitor,
PD98059, to elucidate the role of ERK on 5-FU sensitivity in these
two cell lines. PD98059 treatment made both CT26 and SW620 cells
less sensitive to 5-FU treatment (Fig.
7B and C), but this effect was not in a Smad4-dependent manner.
In addition, inhibition of the MEK/ERK pathway did not affect
protein expression of either the anti-apoptotic Bcl2 family or
pro-apoptotic Bim and Bad (data not shown). These results indicate
that the ERK pathway contributes little to Smad4-induced
chemosensitivity in either the CT26 or SW620 cells.
Discussion
TGF-β signaling plays a bilateral role in CRC
progression and prognosis, depending on the condition of the
intracellular Smad and non-Smad pathway. Smad4, a pivotal signal of
the TGF-β Smad pathway, acts as a tumor-suppressor gene in CRC via
interruption of the TGF-β signaling pathway (21). This gene was reported to predict the
sensitivity to chemotherapy and prognosis in CRC in several
clinical studies (2–6,8). Our
present research provides evidence that Smad4 expression was
reduced with the advance of tumor stages (Fig. 1), which was consistent with previous
clinical studies. In the present study, we used two CRC cell lines:
a murine CT26 cell line with Smad4 expression sensitive to 5-FU and
a human Smad4-null SW620 cell line resistant to 5-FU. Smad4
deficiency in CRC cells promoted tumor progression. This evidence
is consistent with our previous results (10) and a recent study (22). In addition, we showed for the first
time in the present study that Smad4 sensitizes CRC to chemotherapy
through downregulation of the PI3K/Akt/CDC2/survivin cascade.
It is believed that inactivation of the Smad pathway
coupled with activation of the non-Smad pathway is essential to
reverse TGF-β from a tumor suppressor to a tumor promoter.
Therefore, crosstalk between Smad and the non-Smad pathway plays a
critical role in regulating tumor progression, although the exact
mechanism of this crosstalk remains elusive. We previously showed
that Smad4 overexpression reverses TGF-β from a tumor promoter to a
tumor suppressor (10). In the
present study, we identified that Smad4 deficiency in CT26 cells
induced tumor progression in vivo and in vitro. In
both cell line models, TGF-β inhibited cell growth through the Smad
pathway while promoted cell growth through the non-Smad pathway. In
this sense, knockdown or loss of Smad4 in these two cell lines
promoted tumor progression through inhibition of the
tumor-suppressive activity of TGF-β coupled with activation of the
tumor-promotor function of the non-Smad pathway.
5-FU is the most commonly used chemotherapeutic
agent for CRC. However, resistance to 5-FU-based chemotherapy is
still an obstacle owing to great polymorphism in drug metabolizing
enzymes, as well as several signaling pathways including TGF-β Smad
and non-Smad pathway. The combination of agents affecting these
pathways with 5-FU-based chemotherapy may improve the effects of
therapeutic strategies. In previous clinical studies, low Smad4
expression in CRC predicted less sensitivity to 5-FU-based
chemotherapy and poor prognosis (2–6,8). Our
animal studies showed a consistent observation (Fig. 3). The present study also showed that
Smad4 expression was reduced with tumor progression. In the
experimental study, we showed that Smad4 had an effect on inducing
5-FU chemosensitivity both in vitro and in vivo,
which was consistent with the clinical results.
Although increasing evidence shows the role of Smad4
in 5-FU chemoresistance, the mechanism remains unknown. To the best
of our knowledge, there is little research regarding the mechanism
of Smad4 in inducing chemosensitivity. To reveal the mechanism, we
focused on the non-Smad signals which were regulated by Smad4
expression, as several non-Smad signals contribute to
chemosensitivity, with the MEK/ERK and PI3K/Akt pathway being well
elucidated (23–29). There is much controversy regarding
the effect of the MEK/ERK pathway on chemotherapeutic drug-induced
apoptosis. This pathway was reported to be either required
(30–32) or resistant (28) to chemotherapy, depending on the cell
types and genetic background. In the present study, the MEK/ERK
pathway was required for 5-FU-induced apoptosis in the CT26 and
SW620 cells, but it did not contribute to Smad4-induced
chemosensitivity. We finally observed that Smad4 promoted
chemosensitivity through the inhibition of the PI3K/Akt pathway
(Fig. 4).
Akt activation upregulates CDC2, which in
combination with cyclin B is necessary for G2-M cell cycle
transition. CDC2 upregulates the anti-apoptotic factor survivin,
which inhibits caspase-dependent apoptosis, therefore promoting
cell survival (16,17). Inhibition of the CDC2 and cyclin B
complex led to G2-M cell cycle arrest, and genetic knockout of CDC2
or survivin was reported to result in embryo lethality (15). Survivin has been reported to be a
highly differentially expressed gene in different types of human
cancers including CRC (33), when
compared with corresponding normal tissues. The role of the
Akt/CDC2/survivin pathway in regulating cell cycle checkpoint and
apoptosis makes it crucial for cell sensitivity to drug treatment.
Our results obtained by evaluation of cell cycle kinetics revealed
that cells with high Smad4 expression were arrested in the G2-M
phase under 5-FU treatment, owing to inhibition of the CDC2/cyclin
B/survivin cascade. Accumulation of cells in the G1/S phase may
result in resistance to 5-FU treatment, which is consistent with
previous studies (34-36). Smad4 reduced the CDC2/cyclin B
complex and survivin in both the CT26 and SW620 cells, through the
downregulation of the PI3K/Akt pathway, resulting in cell cycle
arrest and cell apoptosis. Thus, we believe that Smad4 sensitizes
CT26 and SW620 cells to 5-FU treatment, at least partially, through
inhibition of the Akt/CDC2/survivin pathway. Either the
anti-apoptotic effect or cell cycle checkpoint regulation of this
pathway may contribute to 5-FU sensitivity.
In summary, the present study suggests that Smad4
induces chemosensitivity both in murine CT26 and human SW620 cells,
through G1 or G2 cell cycle arrest by inhibiting the
PI3K/Akt/CDC2/survivin cascade. LY294002 reversed the
chemosensitivity of CRC with low Smad4 expression by inhibiting the
PI3K/Akt/CDC2/survivin cascade. Smad4 may be a candidate biomarker
for the combined use of LY294002 and 5-FU-based chemotherapy for
patients with CRC.
Acknowledgments
The present study was supported by the State Key
Project on Infectional Disease of China (grant no.
2012ZX10002016-004 and 2012ZX10002010-001-004), the Chinese
Ministry of Public Health for Key Clinical Projects (no. 439, 2010)
to Dr Xiaoping Chen and the National Nature Science Foundation of
China (no. 81502524) to Dr Binhao Zhang.
Abbreviations:
|
5-FU
|
5-fluorouracil
|
|
CRC
|
colorectal cancer
|
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Boulay JL, Mild G, Lowy A, Reuter J,
Lagrange M, Terracciano L, Laffer U, Herrmann R and Rochlitz C:
SMAD4 is a predictive marker for 5-fluorouracil-based chemotherapy
in patients with colorectal cancer. Br J Cancer. 87:630–634. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Alhopuro P, Alazzouzi H, Sammalkorpi H,
Dávalos V, Salovaara R, Hemminki A, Järvinen H, Mecklin JP,
Schwartz S Jr, Aaltonen LA, et al: SMAD4 levels and response to
5-fluorouracil in colorectal cancer. Clin Cancer Res. 11:6311–6316.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ahn BK, Jang SH, Paik SS and Lee KH: Smad4
may help to identify a subset of colorectal cancer patients with
early recurrence after curative therapy. Hepatogastroenterology.
58:1933–1936. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kawakami M, Yamaguchi T, Takahashi K,
Matsumoto H, Yasutome M, Horiguchi S, Hayashi Y, Funata N and Mori
T: Assessment of SMAD4, p53, and Ki-67 alterations as a predictor
of liver metastasis in human colorectal cancer. Surg Today.
40:245–250. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Baraniskin A, Munding J, Schulmann K,
Meier D, Porschen R, Arkenau HT, Graeven U, Schmiegel W, Tannapfel
A and Reinacher-Schick A: Prognostic value of reduced SMAD4
expression in patients with metastatic colorectal cancer under
oxaliplatin-containing chemotherapy: A translational study of the
AIO Colorectal Study Group. Clin Colorectal Cancer. 10:24–29. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Papageorgis P, Cheng K, Ozturk S, Gong Y,
Lambert AW, Abdolmaleky HM, Zhou JR and Thiagalingam S: Smad4
inactivation promotes malignancy and drug resistance of colon
cancer. Cancer Res. 71:998–1008. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Roth AD, Delorenzi M, Tejpar S, Yan P,
Klingbiel D, Fiocca R, d'Ario G, Cisar L, Labianca R, Cunningham D,
et al: Integrated analysis of molecular and clinical prognostic
factors in stage II/III colon cancer. J Natl Cancer Inst.
104:1635–1646. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang B, Zhang B, Chen X, Bae S, Singh K,
Washington MK and Datta PK: Loss of Smad4 in colorectal cancer
induces resistance to 5-fluorouracil through activating Akt
pathway. Br J Cancer. 110:946–957. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang B, Halder SK, Kashikar ND, Cho YJ,
Datta A, Gorden DL and Datta PK: Antimetastatic role of Smad4
signaling in colorectal cancer. Gastroenterology.
138:969.e1-3–980.e1-3. 2010. View Article : Google Scholar
|
|
11
|
Halder SK, Beauchamp RD and Datta PK:
Smad7 induces tumorigenicity by blocking TGF-beta-induced growth
inhibition and apoptosis. Exp Cell Res. 307:231–246. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Halder SK, Beauchamp RD and Datta PK: A
specific inhibitor of TGF-beta receptor kinase, SB-431542, as a
potent antitumor agent for human cancers. Neoplasia. 7:509–521.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fanciullino R, Giacometti S, Mercier C,
Aubert C, Blanquicett C, Piccerelle P and Ciccolini J: In vitro and
in vivo reversal of resistance to 5-fluorouracil in colorectal
cancer cells with a novel stealth double-liposomal formulation. Br
J Cancer. 97:919–926. 2007.PubMed/NCBI
|
|
14
|
Wagner M, Roh V, Strehlen M, Laemmle A,
Stroka D, Egger B, Trochsler M, Hunt KK, Candinas D and Vorburger
SA: Effective treatment of advanced colorectal cancer by rapamycin
and 5-FU/oxaliplatin monitored by TIMP-1. J Gastrointest Surg.
13:1781–1790. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Malumbres M and Barbacid M: Cell cycle,
CDKs and cancer: A changing paradigm. Nat Rev Cancer. 9:153–166.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
O'Connor DS, Wall NR, Porter AC and
Altieri DC: A p34cdc2 survival checkpoint in cancer.
Cancer Cell. 2:43–54. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
O'Connor DS, Grossman D, Plescia J, Li F,
Zhang H, Villa A, Tognin S, Marchisio PC and Altieri DC: Regulation
of apoptosis at cell division by p34cdc2 phosphorylation
of survivin. Proc Natl Acad Sci USA. 97:13103–13107. 2000.
View Article : Google Scholar
|
|
18
|
Chen Q and Li W, Wan Y, Xia X, Wu Q, Chen
Y, Lai Z, Yu C and Li W: Amplified in breast cancer 1 enhances
human cholangiocarcinoma growth and chemoresistance by simultaneous
activation of Akt and Nrf2 pathways. Hepatology. 55:1820–1829.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chao JI, Su WC and Liu HF: Baicalein
induces cancer cell death and proliferation retardation by the
inhibition of CDC2 kinase and survivin associated with opposite
role of p38 mitogen-activated protein kinase and AKT. Mol Cancer
Ther. 6:3039–3048. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
McCubrey JA, Steelman LS, Chappell WH,
Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M,
Tafuri A, et al: Roles of the Raf/MEK/ERK pathway in cell growth,
malignant transformation and drug resistance. Biochim Biophys Acta.
1773:1263–1284. 2007. View Article : Google Scholar
|
|
21
|
Cancer Genome Atlas N; Cancer Genome Atlas
Network: Comprehensive molecular characterization of human colon
and rectal cancer. Nature. 487:330–337. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Freeman TJ, Smith JJ, Chen X, Washington
MK, Roland JT, Means AL, Eschrich SA, Yeatman TJ, Deane NG and
Beauchamp RD: Smad4-mediated signaling inhibits intestinal
neoplasia by inhibiting expression of β-catenin. Gastroenterology.
142:562–571. e5622012. View Article : Google Scholar
|
|
23
|
Kodach LL, Bos CL, Durán N, Peppelenbosch
MP, Ferreira CV and Hardwick JC: Violacein synergistically
increases 5-fluorouracil cytotoxicity, induces apoptosis and
inhibits Akt-mediated signal transduction in human colorectal
cancer cells. Carcinogenesis. 27:508–516. 2006. View Article : Google Scholar
|
|
24
|
Jeung HC, Che XF, Haraguchi M, Zhao HY,
Furukawa T, Gotanda T, Zheng CL, Tsuneyoshi K, Sumizawa T, Roh JK,
et al: Protection against DNA damage-induced apoptosis by the
angiogenic factor thymidine phosphorylase. FEBS Lett.
580:1294–1302. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cantley LC: The phosphoinositide 3-kinase
pathway. Science. 296:1655–1657. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zheng G, Xiong Y, Yi S, Zhang W, Peng B,
Zhang Q and He Z: 14-3-3σ regulation by p53 mediates a chemotherapy
response to 5-fluorouracil in MCF-7 breast cancer cells via Akt
inactivation. FEBS Lett. 586:163–168. 2012. View Article : Google Scholar
|
|
27
|
Knuefermann C, Lu Y, Liu B, Jin W, Liang
K, Wu L, Schmidt M, Mills GB, Mendelsohn J and Fan Z:
HER2/PI-3K/Akt activation leads to a multidrug resistance in human
breast adenocarcinoma cells. Oncogene. 22:3205–3212. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jin W, Wu L, Liang K, Liu B, Lu Y and Fan
Z: Roles of the PI-3K and MEK pathways in Ras-mediated
chemoresistance in breast cancer cells. Br J Cancer. 89:185–191.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Taiyoh H, Kubota T, Fujiwara H, Matsumura
A, Murayama Y, Okamoto K, Ichikawa D, Ochiai T, Nakamura T,
Matsumoto K, et al: NK4 gene expression enhances
5-fluorouracil-induced apoptosis of murine colon cancer cells.
Anticancer Res. 31:2217–2224. 2011.PubMed/NCBI
|
|
30
|
Wang X, Martindale JL and Holbrook NJ:
Requirement for ERK activation in cisplatin-induced apoptosis. J
Biol Chem. 275:39435–39443. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Woessmann W, Chen X and Borkhardt A:
Ras-mediated activation of ERK by cisplatin induces cell death
independently of p53 in osteosarcoma and neuroblastoma cell lines.
Cancer Chemother Pharmacol. 50:397–404. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Basu A and Tu H: Activation of ERK during
DNA damage-induced apoptosis involves protein kinase Cdelta.
Biochem Biophys Res Commun. 334:1068–1073. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hernandez JM, Farma JM, Coppola D, Hakam
A, Fulp WJ, Chen DT, Siegel EM, Yeatman TJ and Shibata D:
Expression of the antiapoptotic protein survivin in colon cancer.
Clin Colorectal Cancer. 10:188–193. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Noda T, Nagano H, Takemasa I, Yoshioka S,
Murakami M, Wada H, Kobayashi S, Marubashi S, Takeda Y, Dono K, et
al: Activation of Wnt/beta-catenin signalling pathway induces
chemoresistance to interferon-alpha/5-fluorouracil combination
therapy for hepatocellular carcinoma. Br J Cancer. 100:1647–1658.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Guo X, Goessl E, Jin G, Collie-Duguid ES,
Cassidy J, Wang W and O'Brien V: Cell cycle perturbation and
acquired 5-fluoro-uracil chemoresistance. Anticancer Res. 28:9–14.
2008.PubMed/NCBI
|
|
36
|
Gavin EJ, Song B, Wang Y, Xi Y and Ju J:
Reduction of Orc6 expression sensitizes human colon cancer cells to
5-fluorouracil and cisplatin. PLoS One. 3:e40542008. View Article : Google Scholar : PubMed/NCBI
|