Introduction
The clinical and biological anticancer effects
produced by tyrosine kinase inhibitors (TKIs) in renal cell cancer
(RCC) are the result of their inhibitory activities on a variety of
cell receptors on cancer cells, endothelial cells, pericytes, and
stromal cells (1). The targeted
effects of TKIs are dependent on the inhibition of downstream
mediators upregulated in response to molecular abnormalities (i.e.,
VHL, c-MET) in RCC. Specifically, in clear cell renal cell cancer
(ccRCC), the mutations and/or epigenetic silencing of the VHL gene
promote subsequent overexpression of growth factors, including the
vascular endothelial growth factor receptor (VEGFR) and
platelet-derived growth factor (PDGF), and multiple other
hypoxia-regulated genes (EPO, NOS, GLUT-1, CA IX), all of which are
co-responsible for tumor angiogenesis and cell proliferation
(2,3). A group of targeted agents selective
against RCC has been developed and introduced in the clinic over
the last decade (4,5). More recently, cancer stem cells (CSCs)
or tumor-initiating cells have come into focus as potential
treatment targets in solid tumors (6,7) and
RCCs (8,9).
Sunitinib (SU11248) is a multi-targeted kinase
inhibitor of VEGFR-1, -2 and -3, PDGFR-β, mast/stem cell growth
factor receptors (SCFR, c-Kit) and FMS-like tyrosine kinase 3
(FLT3) (10). Sunitinib inhibits
cancer growth primarily through an anti-angiogenic mechanism, by
halting endothelial cell proliferation and motility (11). The elucidated mechanisms of action
of sunitinib may also include targeting CSCs. The first study to
show that sunitinib targets CSCs was conducted in pancreatic
cancer. In this study,
CD24+CD44+ESA+ triple-positive
pancreatic CSCs were shown to be responsive to this TKI in
combination with liposome-coated doxorubicin (12). In prostate cancer, in a PC3
cell-based model, sunitinib was shown to reduce the number of
ALDH+ cancer stem-like cells, and to sensitize these
cells to the radiation-mediated loss of clonogenicity (13). In xenograft RCC models, sunitinib
has been shown to generate resistance to its own therapeutic
mechanism due to the induction of hypoxia in perinecrotic areas.
Moreover, CD133/CXCR4 co-expressing cells, also considered CSCs,
were found in these areas at higher numbers. Under hypoxia, the
tumorigenic potential of CD133/CXCR4+ cells increased,
and their sensitivity to sunitinib decreased (14).
Sorafenib (BAY 43-9006) was discovered based on its
ability to inhibit kinases, including C-RAF and B-RAF (wild-type
and V600E mutant), and subsequent MEK and ERK phosphorylation. It
also targets VEGFR-2 and VEGFR-3, PDGFR-β, FLT3, and c-Kit
(15). The complete mechanisms of
action include a reduction in angiogenesis and cancer cell
proliferation. However, the exact mechanisms remain undefined. In
the case of hepatocellular carcinoma (HCC), it was demonstrated
that label-retaining cancer cells (LRCCs), a subpopulation ofCSCs,
were significantly resistant to sorafenib. In addition, the
proportion of LRCCs in the HCC cell lines was increased in the
total culture after sorafenib treatment (16). Moreover, CD44/CD133+ and
CD44/ALDH1A1+ HCC cells (CSCs) were shown to increase in
number after treatment with sorafenib (17,18).
After treatment with sorafenib, starting at a concentration of 1
μM and increased by 10% every 2 weeks until reaching the
maximum tolerated dose (4–7 μM), the number of
CD44/CD133+ stem cells increased in the HCC cell lines
(17). However, another HCC
subpopulation, CD13+/CD166+/ALDH+
CSCs, was determined to be sensitive to sorafenib (19). On the contrary, HCC CSC-defined by
the expression of the Nanog gene also exhibited resistance to
sorafenib (20).
It was later determined that it is the combination
of two drugs that enables sorafenib to target the HCC CSCs.
Sorafenib, in combination with FH535 (an inhibitor of Wnt/β-catenin
signaling and dual antagonist of PPARγ/δ activity), inhibited the
proliferation of liver CSCs (CD133, CD44, CD24, and aldehyde
dehydrogenase 1-positive) (21). A
similar effect was noted for the combination of sorafenib and
gedatolisib (PKI-587), a highly potent dual inhibitor of PI3Kα,
PI3Kγ and mTOR (22). Sorafenib was
also shown to effectively target pancreatic CSCs
(CD24−/CD44+ and ALDH+) when
administered with sulforaphane, a phytochemical belonging to the
family of isothiocyanates (23). In
the case of breast cancer, sorafenib has been shown to be effective
against breast cancer CD44+CD24− stem cells
in combination with radiation (24). Contrary to its low activity in HCC,
sorafenib was shown to be effective in vitro against CSCs in
glioblastomas (tumor-initiating cells) in primary cell cultures by
inhibiting the PI3K/Akt and MAPK pathways. In this case, sorafenib
significantly induced apoptosis by downregulating the survival
factor myeloid cell leukemia 1 (Mcl-1) (25), which was further potentiated by
metformin (26). The combination of
metformin and sorafenib was also shown to be significantly toxic to
radio-iodine refractory anaplastic thyroid carcinoma CSCs (27). In addition, it was shown to be
effective against prostate cancer stem-like cells isolated from
holoclones of the PC3 cell line (28). Finally, RCC CXCR4+ cells,
which express stem cell-associated transcription factors
(NANOG, OCT3/4, and SOX2) at elevated levels,
were reported to be more resistant to sorafenib (and sunitinib)
than the parental cells in adherent cultures (9).
Axitinib (AG-013736) is a potent small-molecule
inhibitor of multiple tyrosine kinases, including VEGFR-1, -2 and
-3 and PDGFRβ. Therefore, axitinib inhibits endothelial cell
survival, new tube formation, and nitric oxide (NO), protein kinase
B (PKB, Akt), and extracellular signal-regulated kinase (ERK)
signaling in endothelial cells (29). It was shown that AG-013736 alone, or
in combination with radiotherapy treatment, induces functional
normalization of the tumor vasculature (30). In glioblastomas, in an S1-M1-80 cell
line xenograft model, axitinib was shown to target a side
population (SP), referred to by the authors as cancer stem-like
cells. It was shown to inhibit the transporter activity of the
adenosine triphosphate (ATP)-binding cassette subfamily G member 2
(ABCG2), reversing ABCG2-mediated drug resistance. In this model,
axitinib (every 4 days x 9, p.o., 25 mg/kg) enhanced the
cytotoxicity of topotecan and mitoxantrone against an SP (31). Subsequently, it was also shown that
axitinib exerts direct cytotoxic activity against patient-derived
glioblastoma CSCs (32) and
potentiates myxoma virus-based treatment directed against brain
tumor-initiating cells (33). In
another study based on the PC3 cell line, holoclone-derived cancer
stem-like cells (called PC3/2G7) in a prostate cancer model were
shown to be sensitive to axitinib (28). Later, it was shown that 1 μM
axitinib in vitro increased the toxic effects of non-small
cell lung cancer (NSCLC) cell irradiation respectability when
applied to spheres derived from CD24/CD44+ NSCLC CSCs
(34).
In terms of the tumor microenvironment-dependent
activity of TKIs, it was first suggested that sunitinib (and
possibly other VEGF inhibitory compounds) increased the population
of CSCs in the tumor by generating intratumoral hypoxia.
Xenograft-based breast cancer studies revealed that hypoxia-driven
cancer stem or progenitor cell enrichment resulted from
hypoxia-inducible factor 1α (HIF1α) signaling. Therefore, it was
concluded that an increase in the number of hypoxia-driven CSCs
limits the effectiveness of anti-angiogenic agents as a result of
CSC drug resistance (35). It was
multitargeted VEGFR inhibition (with sunitinib), not VEGF
sequestration (with bevacizumab), that rapidly created a vascular
gradient, inducing tumor hypoxia, promoting the aggressive
mesenchymal phenotype, and increasing the cancer stem cell number
(36). In a mouse based study, oral
sunitinib administered at 80 mg/kg/2 days for 4 weeks significantly
reduced the tumor volume and angiogenesis, but increased the number
of CSCs in the tumors (37). We
hypothesized that TKIs suppress tumor angiogenesis and tumor growth
and progression via inhibition of the paracrine and autocrine
effects of VEGF; however, TKI-induced tumor hypoxia may promote the
CSC phenotype. Since the data on TKI activity against renal cell
CSCs is not available, we aimed to verify whether renal CSCs are
targeted by TKIs in an RCC model. We aimed to verify the influence
under both normoxic and hypoxic conditions, which would represent
conditions prior to and post-TKI exposure.
Materials and methods
Renal cell cancer-cancer stem cell
isolation
Individual donor samples were selected for the
study, and selected donations were used for analysis. Primary tumor
tissue was obtained. The tumor samples obtained after surgery were
placed immediately in tumor transportation media and shipped at
4–8°C for processing. Tissues were washed with 1X PBS solution,
divided into two halves and aseptically cut into 0.5-mm sections
and cultured in 6-well tissue culture plates pre-coated with
extracellular matrix. First, the control half was processed in
regular media with serum and was referred to as the parental cell
line, while CSCs were selected in Kidney Cancer Stem Cell Complete
Growth Medium (Celprogen, San Pedro, CA, USA) (38,39).
All cell cultures remained viable and maintained their native
architecture for at least 14 days. After 14 days of culture, the
cells were characterized for stem cell phenotype.
As previously described, cancer, kidney cancer and
CSC surface markers, transcription factors and
epithelial-mesenchymal transition (EMT) markers (linked to the
induction of a stem-cell like phenotype) were quantified by qPCR or
immunocytochemistry per cell culture. These markers included: human
kidney injury molecule-1 (hK1M-1), renal cell carcinoma marker
(RCC-Ma), chromophobe renal cell carcinoma (chRCC), calveolin-1
(CAV-1), carbonic anhydrase IX (CA9/CAIX), vascular endothelial
growth factor (VEGF), chemokine (C-X-C motif) ligand 16 (CXCL16), A
disintegrin and metalloproteinase domain-containing protein 10
(ADAM10, CD156c), programmed death-ligand 1 (PD-L1; also known as
cluster of differentiation 274, CD274 or B7 homolog 1, B7-H1),
Ki-67, survivin, P53, glucose transporter 1 (GLUT-1), galctosyl
transferase II (GalT-II), glyceraldehyde 3-phosphate dehydrogenase
(GAPDH), cancer antigen 19 (CA19-9), cancer antigen 72-4 (CA72-4),
carcinoembryonic antigen (CEA), α-fetoprotein (AFP),
β-2-microglobulin (B2M), octamer-binding transcription factor 4
(OCT4), sex-determining region Y-box 2 (SOX-2), stage-specific
embryonic antigen-3 and -4 (SSEA3/4), aldehyde dehydrogenase
(ALDH), alkaline phosphatase (ALP), tissue-non-specific alkaline
phosphatase (TRA1-81 and TRA1-61), telomerase, CD9, CD10, CD24,
CD34, CD40, CD44, CD105 (endoglin), CD133 (prominin-1), CXCR-4, SHH
(sonic hedgehog), epithelial cell adhesion molecule (EpCAM),
epithelial-specific antigen (ESA), CBX7, SNAIL, SLUG, TWIST, Ki-67,
E-cadherin, β-catenin, nestin and vimentin (38,40,41).
SCID nude mice were subcutaneously injected with
1,000 CSCs in the hind limbs. Subcutaneous tumors developed. Cells
were capable of generating tumors in mice within 20 days. The mice
were also evaluated for metastases. Cells tumorigenic at the above
mentioned injection concentration were selected for further
experiments. Stem cell marker expression was re-confirmed in the
developed tumors (38,41,42).
Cells positive for Sox 2, Oct4, SSEA3/4, ALDH, ALP,
telomerase, calveolin-1, CD133 and CD44, that were also capable of
clonal self-renewal and were tumorigenic with <1,000 cells
injected as described above, were used for further investigation.
Cells were characterized and stable for markers for up to seven
passages. Markers were significantly expressed at a higher level
compared with the expression levels in normal kidney tissues and
primary renal cancer cells (38,40,41).
Cell culture
Human kidney cancer stem cells (HKCSCs, RCC-CSCs;
Celprogen, Torrance, CA, USA) were cultured in Human Kidney Cancer
Stem Cell media as per the manufacturer's protocol requirements.
786-0 (CRL-1932), a primary tumor-derived VHL mutated cell line,
was obtained from the American Type Culture Collection (ATCC)
Global Bioresource Center (Manassas, VA, USA). The SK-RT-42
metastatic bone-derived cell line was established in the laboratory
of Dr Lloyd Old, from patients undergoing nephrectomy at Memorial
Hospital, Memorial Sloan Kettering Cancer Center (MSKCC) and was
obtained from the Core Facility, MSKCC (New York, NY, USA)
(43). The 786-0 and SK-RT-42 cell
lines were cultured in RPMI-1640 medium with 10% FBS (Biochrom
GmbH, Cambridge, UK) and GlutaMAX (Life Technologies, Carlsbad, CA,
USA). RCC-CSCs were differentiated with RPMI-FBS under culture
conditions described above. Cells were cultured under normoxic (21%
O2) and hypoxic (1% O2) conditions. Human
Kidney Cancer Stem Cell Extracellular Matrix was used to increase
RCC-CSC viability (Celprogen).
3D cell culture
In the 3D approach, CSCs were grown in media as
above but as aggregates and in hanging drop culture forming
spheroids as previously described (44,45),
with modification of 100 cells/10 μl drop. After formation
of the spheroids (72 h), sunitinib was added in a volume of 5
μl to each drop in order to obtain 0, 1, 2, 4, 8, and 15
μM concentrations. Spheres were photographed under a Nikon
TMS-F phase contrast microscope after 24, 48 and 72 h. The size of
the colonies was analyzed using ImageJ software as previously
published (46,47).
Assessment of drug toxicity
The cells were treated with TKIs, sunitinib malate,
axitinib (Sigma-Aldrich, St. Louis, MO, USA) or sorafenib (Cayman
Chemical Co., Ann Arbor, MI, USA) at different concentrations (0,
1, 2, 4, 6, 8, 10, 12, 15 and 20 μM) with DMSO <0.5%
(control). Subsequently IC50 values of the TKIs were
evaluated after 24, 48 and 72 h. The cells were also treated with
TKIs under normoxic (21% O2) or hypoxic (1%
O2) conditions. HKCSCs were seeded in 96-well 2D plates
and cultured under standard conditions (37°C, 5% CO2).
After 24 h, sunitinib was added and the plates were moved to a
normoxic or hypoxic incubator. Subsequently, inhibition of
proliferation was quantified after 24, 48 and 72 h. Alamar blue
(resazurin) (Life Technologies) assay was performed as per the
manufacturer's protocol to quantitatively measure cell viability
and cytotoxicity (48) which were
read by microplate reader Multiskan GO and analyzed using ScanIt™
software package (Thermo Scientific, Waltham, MA USA). MTT
[3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide]
assay (Life Technologies) was used to assess cell viability under
TKI treatment as previously published (49,50).
Results
Tyrosine kinase inhibitors target renal
cell cancer-cancer stem cells
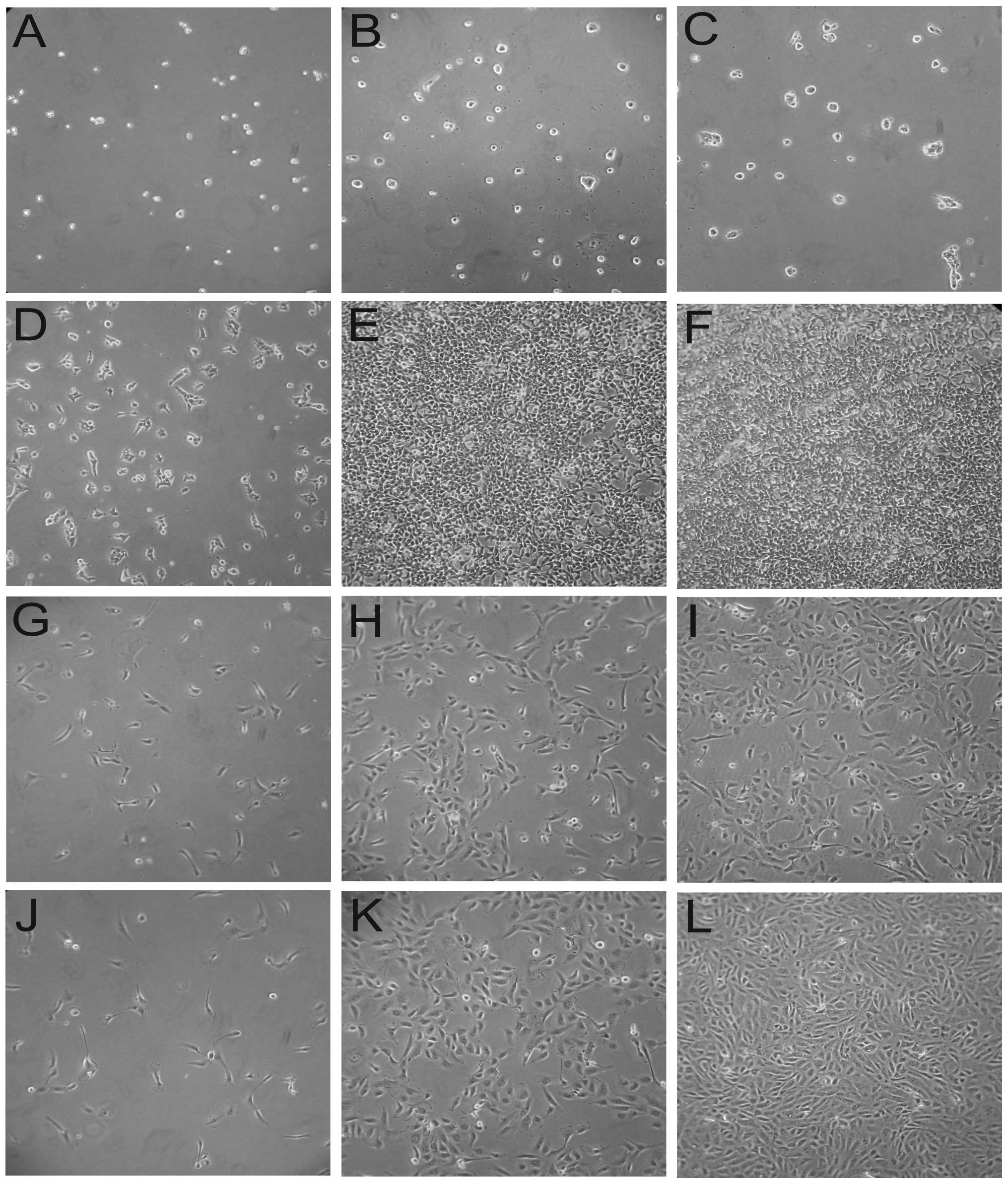
RCC-CSCs were found to be slow proliferators in
comparison to stable (differentiated) renal cell cancer cells, and
all TKIs (sunitinib, sorafenib, axitinib) were found to directly
influence RCC-CSCs. Under TKI treatment, the proliferation rate of
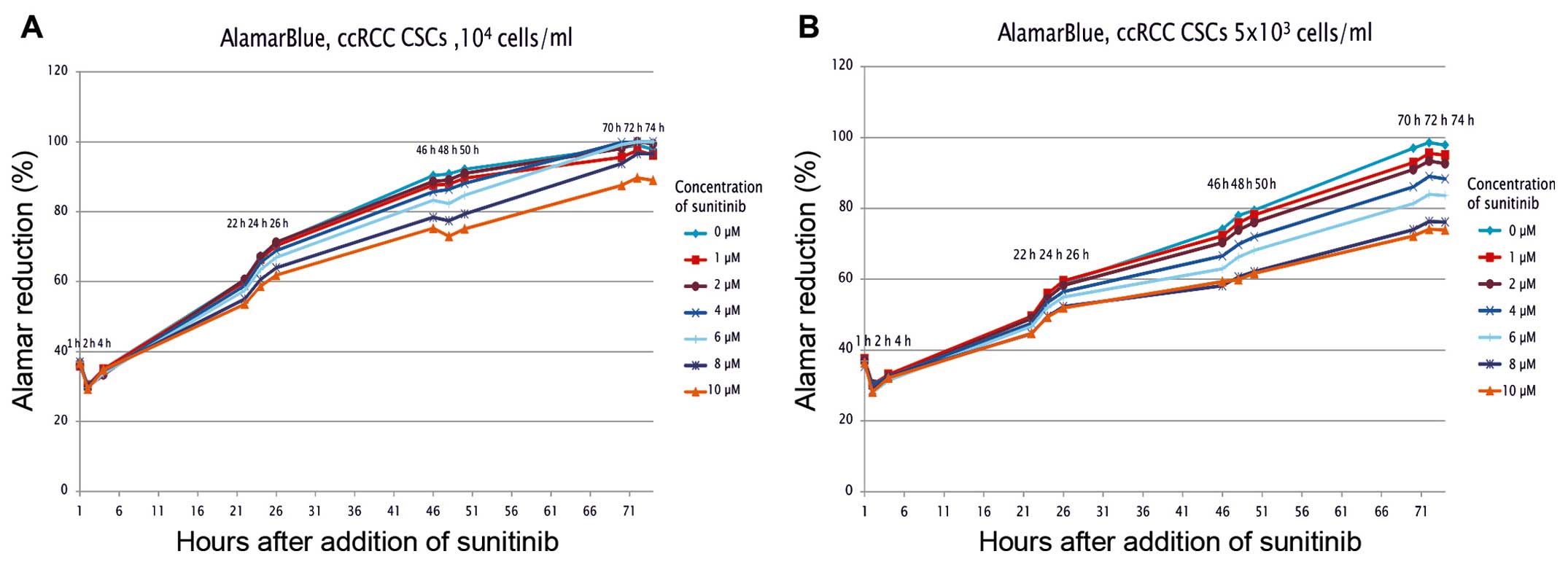
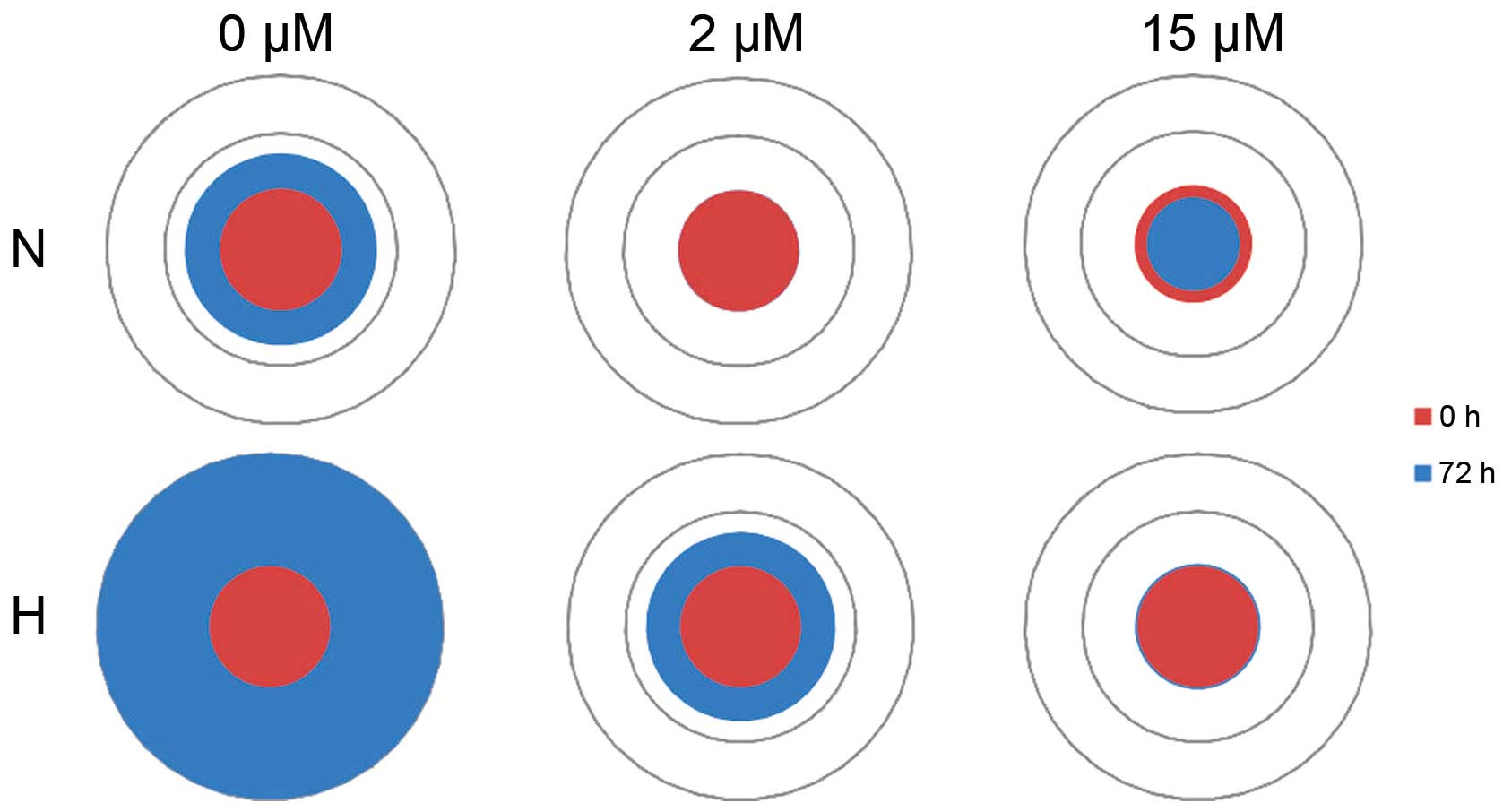
the RCC-CSCs was decreased (Figs. 1
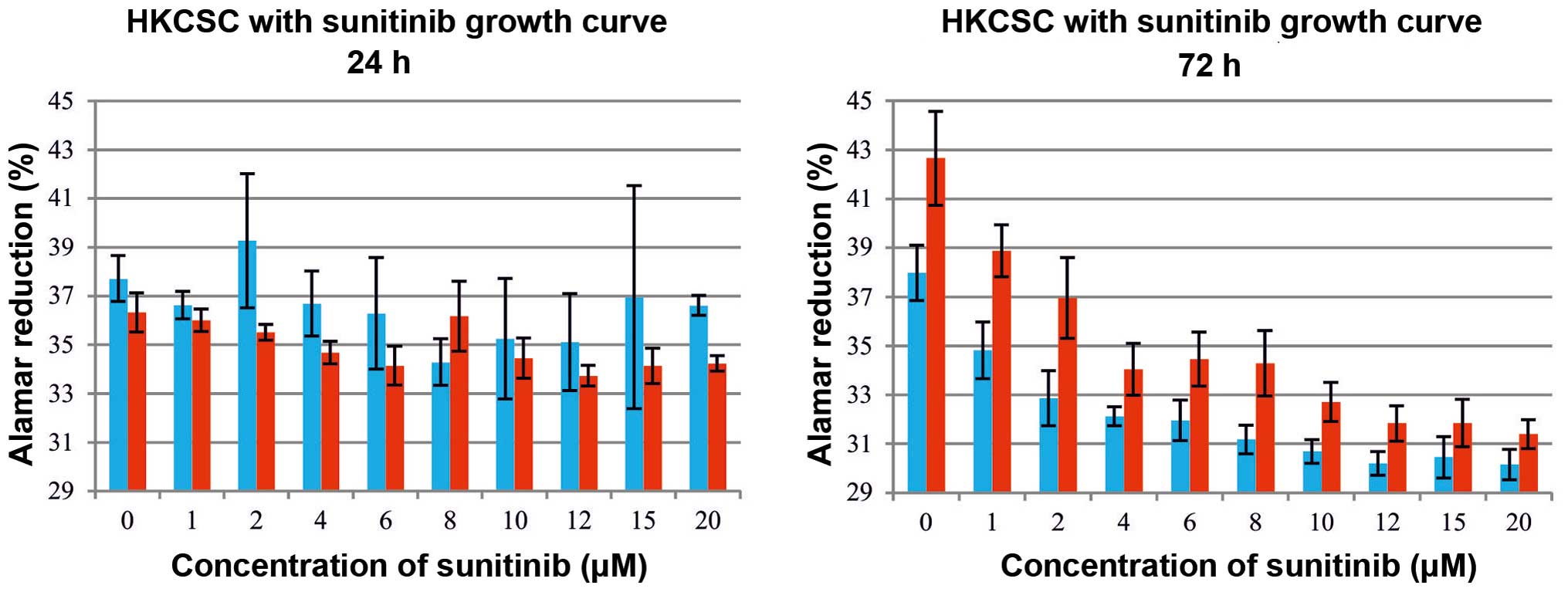
and 2). The inhibition of
proliferation was dose-dependent, with a significant RCC-CSC toxic
minimal dose of 2 μM TKI (Fig.
1); however, this inhibition was evident after 22 h of drug
treatment at high sunitinib doses (>6 μM) (Fig. 1A), and also significant at a low
dose (2 μM) (Fig. 1B) after
72 h of culture. The metabolic activity of the cells (seeded at
least at 1×104 cells/ml) growing under TKIs appeared to
slow down by day 3, suggesting that the surfaces were advancing
toward confluency. At a lower seeding number (starting at
500/well), the cell culture was extended for 6 days of treatment in
order to prolong the observation of the exponential/linear growth
phase, focusing on the effect of TKIs on the cells at their point
of maximum growth (Figs. 1B and
3). The inhibition of the
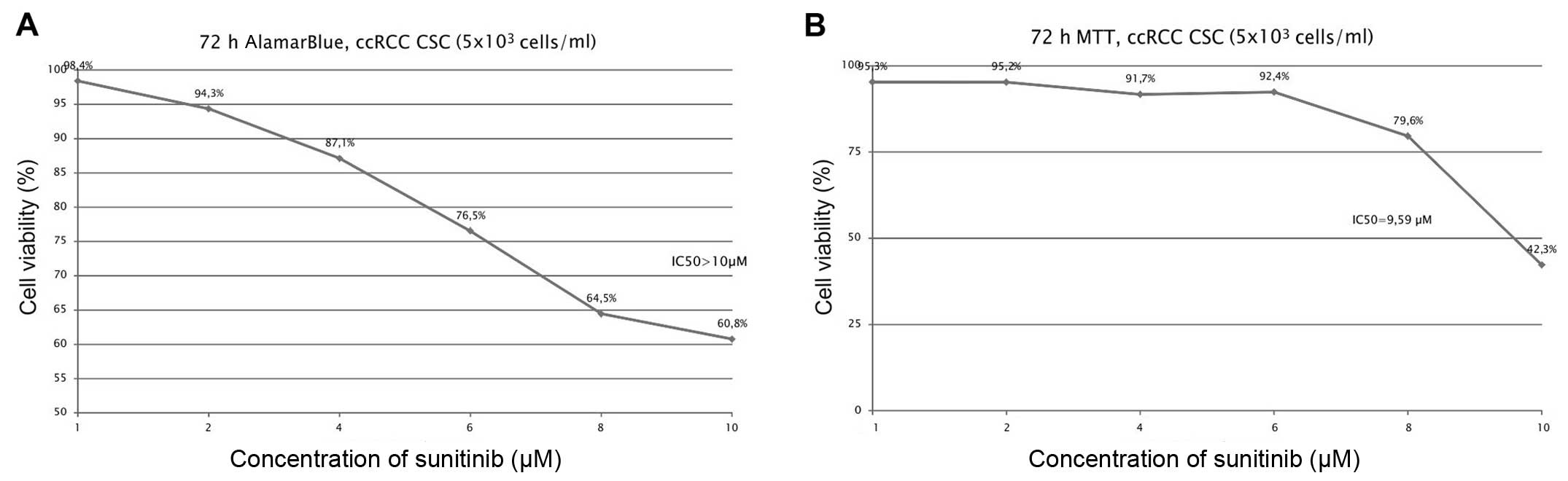
proliferation by sunitinib was shown to be dose-dependent (Figs. 1 and 3A), and it was determined (for RCC-CSCs)
that the half-maximal inhibitory concentration (IC50)
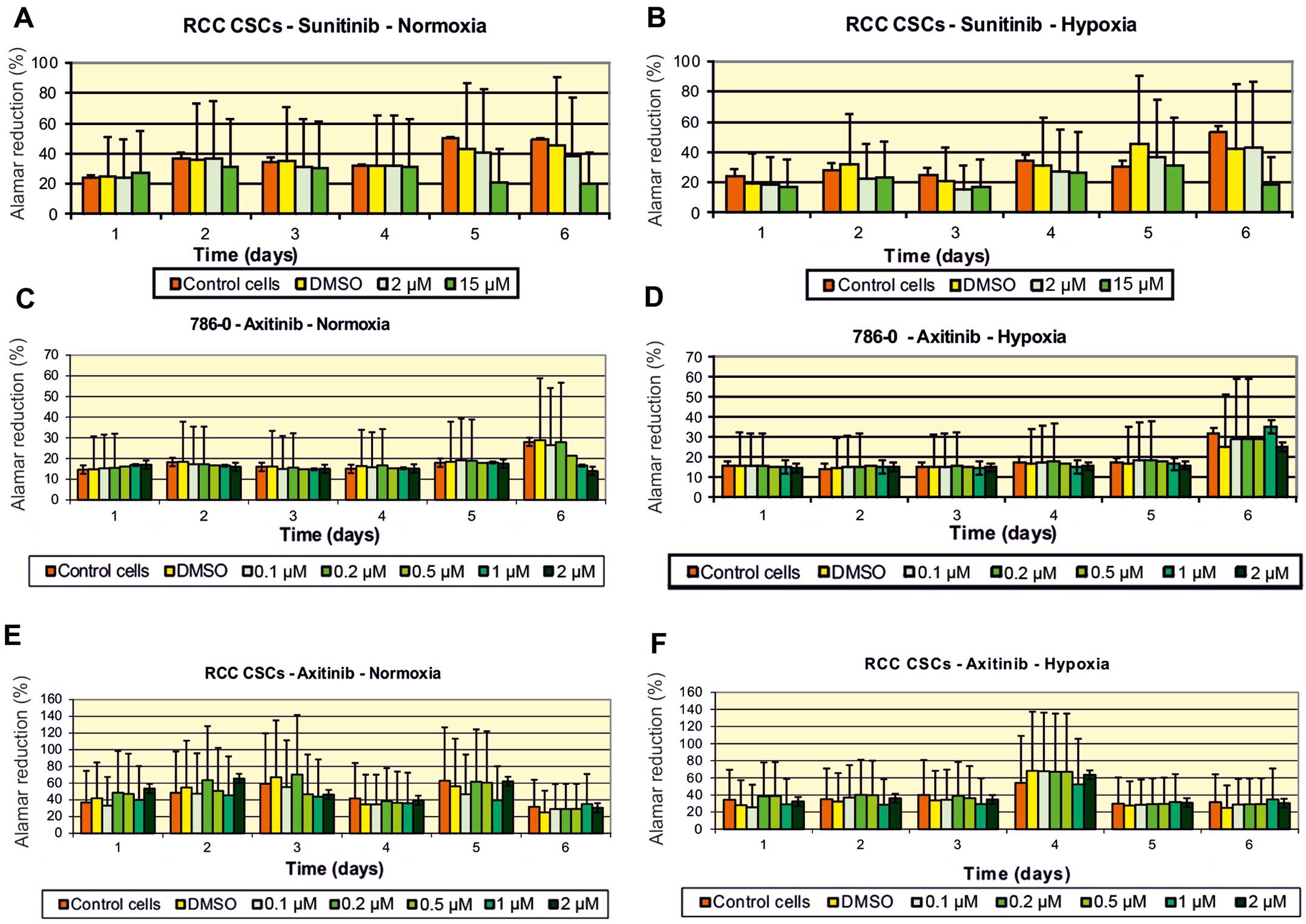
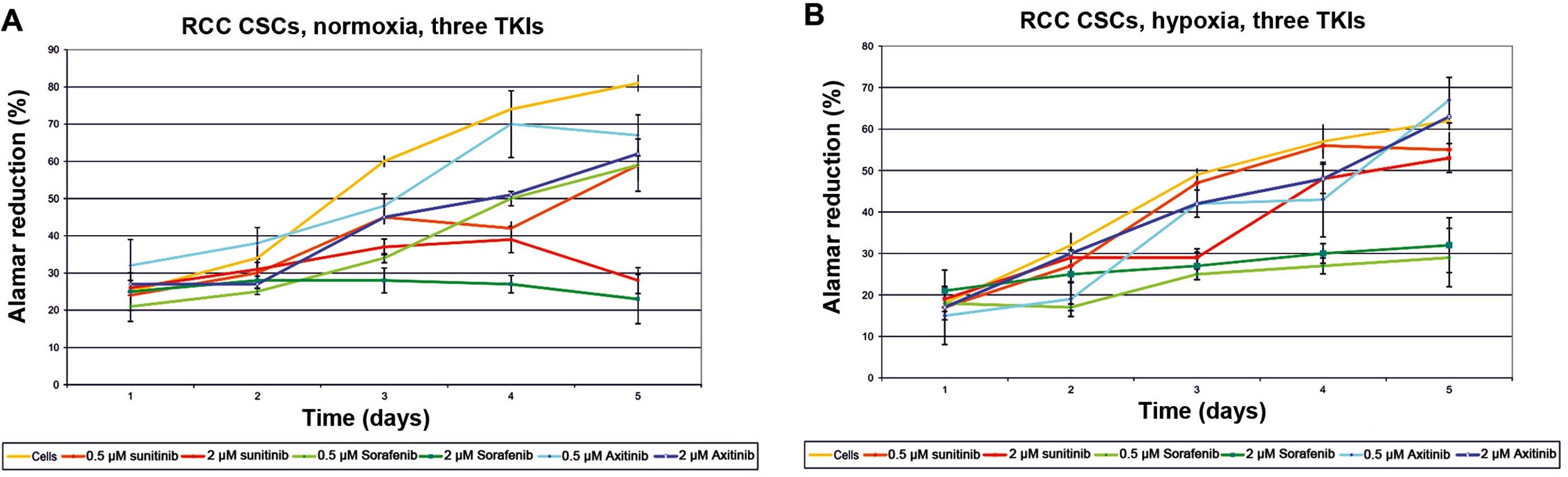
for sunitinib was at the 10 μM level (Fig. 2). The activity of axitinib against
RCC-CSCs was lower than that for sunitinib. In regards to the
regular ccRCC 786-0 cell line, the inhibition of cell proliferation
was significant on day 6 of the axitinib treatment, while the cell
growth of the RCC-CSCs was not inhibited using the same
concentration of the drug (Figs. 3C
vs. E and 4). A similar trend was confirmed for the SK-RC-42
metastatic cell line (data not shown).
Anti-proliferative activity of tyrosine
kinase inhibitors is altered by hypoxia
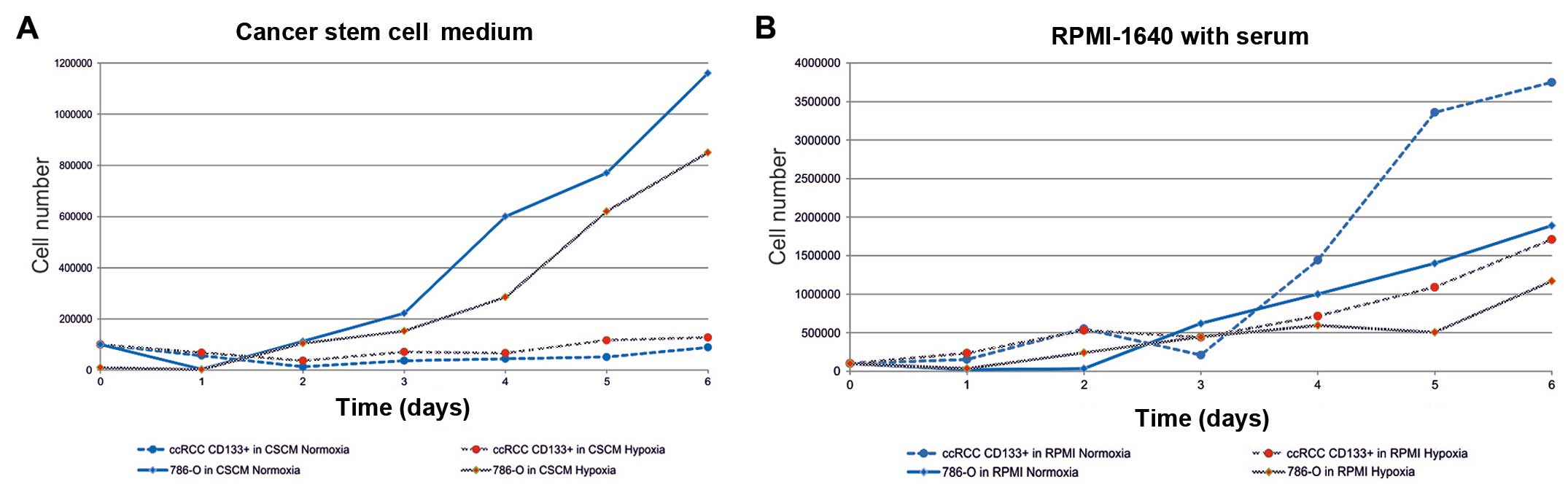
The proliferation of renal cell cancer cells
representing differentiated cancer cells (786-0 cells and
post-RCC-CSCs), as well as RCC-CSCs/HKCSCs are influenced by oxygen
tension in the environment. Normoxic (21% O2) conditions
promoted the growth of the renal cancer cell lines 786-O and
SM-KT-42 under no-TKI treatment (Fig.
5). Moreover, when differentiated by the overload of growth
factors (FBS), RCC-CSCs also proliferated at a significantly higher
rate under high oxygen availability. Differentiated post-RCC-CSCs
showed a high proliferation rate, similar to that of the 786-0 cell
line, and significantly higher than in RCC-CSCs in stem cell
promoting conditions (Fig. 6). At
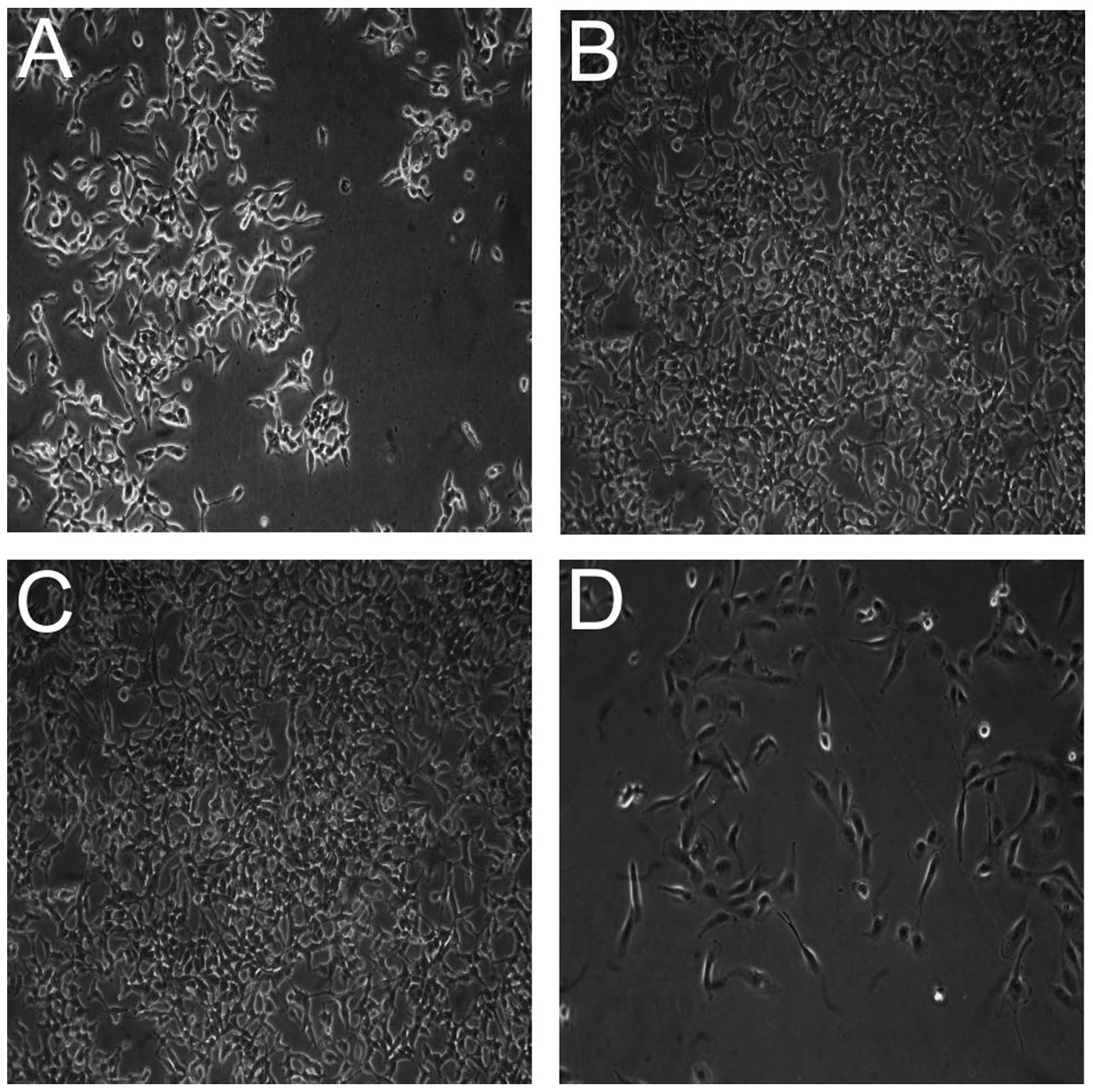
the same time, RCC-CSCs under hypoxia changed their morphology and
developed less invadopodia (Fig.
7D) (51), which is also
typical when RCC-CSCs are cultured in ECM-rich conditions (Fig. 7A). Under a hypoxic condition, the
activity of sunitinib (Fig. 8) and
other TKIs was limited with the highest activity of sorafenib
(Fig. 4B), and the influence of low
oxygen tension was visible after 72 h in a hypoxic culture. During
the first 24 h, cells in normoxia and hypoxia proliferate at
similar rates, but the reduction in sunitinib toxicity and
concurrent induction of RCC-CSC proliferation under hypoxia were
time-dependent (48 h; data not shown), which increased with time at
low cell seeding and were visible after 6 days of culture (Fig. 3B).
The anti-proliferative activity of sunitinib
decreased under a hypoxic condition, which may occur due to tumor
cell-cell interactions between cancer cells, and has been
investigated in tumorispheres. Additionally, the anti-proliferative
activity of sunitinib also decreased in the 3D environment, which
further increased the hypoxic condition, likely to occur in tumors
(Fig. 9). The size of the spheres
that developed in hypoxia under sunitinib treatment was larger than
those developing during normoxia (Fig.
10), which further confirms the pro-proliferatory effect of
hypoxia on RCC-CSCs.
Discussion
Recent achievements in the development of
multi-targeted molecular inhibitors has necessitated a better
understanding of the activity against individual targets with
regard to their efficacy. Sunitinib, sorafenib, and axitinib are
the most recently identified and extensively investigated
anti-angiogenic drugs (52);
nevertheless, it must be kept in mind that, at a cellular level, it
has been shown that TKIs target not only endothelial cells, but
also RCC cancer cells, pericytes and renal stromal cells (1). The first pathological condition that
was shown to be sunitinib-sensitive was acute myelogenous leukemia
(AML), in which sunitinib markedly inhibited cellular
proliferation, including AML-stem cell proliferation, in a
dose-dependent manner with an IC50 of 10–50 nM (53,54).
In this study, we report the activity of TKIs against RCC-CSCs,
which has not been confirmed previously.
It is known that the anti-proliferative activity is
dependent on the presence of constitutively active receptor
tyrosine kinase (RTK) targets. Sunitinib binds 73 kinases in
addition to its main target, VEGFR-2, while sorafenib binds 40
additional kinases. However, axitinib is the most selective, with a
limited number of targets (55).
The activity of sorafenib in our CSC research appeared to be the
highest under the harsh conditions of hypoxia. In addition, the
high activity against a generally drug-resistant target (CSCs)
remains in accordance with a wide spectrum of kinases that are
inhibited by sorafenib, and therefore, a wider panel of anti-stem
cell proliferation inhibitors and hypoxia induced genes. It is
known that sorafenib targets kinases in addition to VEGFR, which
are in order of increasing IC50 value: platelet-derived
growth factor receptor α (PDGFRα) → discoidin domain receptor
tyrosine kinase 2 (DDR2) → rearranged during transfection (RET) →
homeodomain-interacting protein kinase 4 (HIPK4) → fms-like
tyrosine kinase 4 (FLT4, also known as VEGFR3) → FLT1 (also known
as VEGFR1) → kinase insert domain receptor (KDR, also known as
VEGFR2) → PDGFRβ → RAF1 and FLT3. The IC50 values of
sorafenib for these 10 kinases were no greater than 100-fold those
against its top target. At the same time, sunitinib had 30 kinase
targets within similar IC50 ranges, suggesting that
sorafenib might be more selective against the VEGFR family
(Fig. 4) (56).
In vitro, sunitinib inhibits the growth of
cancer cell lines driven by VEGF, SCF and PDGF, and also induces
apoptosis of vein endothelial cells (57); however, a full understanding of the
targets and mechanism of action of sunitinib, as well as the other
TKIs in ccRCC treatment, remains incomplete. To complement this
activity data, we showed direct inhibition of the ccRCC-CSC growth
in a dose-dependent manner (Figs.
1, 2 and 4). At the same time, axitinib that was
found to have lower activity against RCC-CSCs (Fig. 3) was shown to have an 8- to 25-fold
higher IC50 against PDGFR-β, KIT, and PDGFR-α (1.6–2.0
nmol/l) and a significantly lower activity against FGFR-1, Flt-3
and RET (1 μmol/l) (29).
This specificity may be contradictory to the phenotype of RCC-CSCs
and their proliferation triggering pathways.
Our results with regard to the direct activity of
TKIs against RCC cancer cells are confirmed by other in
vitro and animal studies in which sunitinib targeted the tumor
cells themselves, since these cells express one or more target
RTKs, including the human kidney cancer 786-O cell line which we
used as the control (58). In 786-0
cells, the PDGFRβ is highly constitutively activated and VEGFR-2
expression is upregulated (58);
therefore, it is a target of TKIs. Similar gene expression
deregulation has been reported in RCC tumors in clinical reports;
for example, PDGFβ and VEGFR-2 were reported to be overexpressed in
RCCs, relative to normal renal tissues (11). Therefore, we suggest that the
multi-targeted inhibition of tyrosine kinase by
sunitinib/sorafenib/axitinib contributes to its anti-proliferative
effects against ccRCC-CSCs, and may contribute to its clinical
efficacy in RCC (Figs. 4, 8 and 10).
The concentrations that are being described as
inhibiting RCC-CSCs (Figs. 2,
3 and 7) are in the range that is found within
RCC tumors in patients. For example, the intratumoral
concentrations of sunitinib in mice and human patients are 10.9±0.5
and 9.5±2.4 μmol/l, respectively, whereas the serum measured
concentrations are 10-fold lower, and described as 1.0±0.1 and
0.3±0.1 μmol/l, respectively (59). The serum concentration of sunitinib
was similar to that in other healthy organs, including the skin,
where the concentration was measured at a level of 0.1 to 0.4
μM (60). The high
concentration of TKIs in RCC tumors has been investigated, and was
determined to result in multiple codependent mechanisms. The TKIs
extravasate into the tumor's extracellular matrix (ECM), while the
decrease in the tumor interstitial fluid pressure that arises as a
result of anti-angiogenesis slows the leakage of TKIs from the
tumor, leading to longer TKI retention in the tumor. At the same
time, as the blood flow to the tumor decreases in response to
anti-angiogenesis, the blood flow out of the tumor may also
decrease. In particular, axitinib was shown to slow the drug efflux
from tumors (61); as a result,
sunitinib inhibits the tumor cell growth at clinically relevant
concentrations in vitro, with IC50 values of at
least 1.4 to 2.3 μM (59).
In addition, in RCC cell lines, the anti-proliferative effects of
sorafenib are both concentration- and time-dependent, as is the
case in our RCC-CSCs. The calculated IC50 of sorafenib
was in the range of 7.5–10 μM, depending on the RCC cell
line, and sorafenib-induced RCC cell apoptosis was reported after
48–72 h of treatment (62).
In terms of the RCC cell targeted activity of TKIs,
it was shown that the short-term (24 h) application of sunitinib in
renal cell carcinoma Caki-1 and KTC-26 cell lines induced cell
growth inhibition, which was halted in the M and G2
phases. The signs of anti-RCC cell toxicity became apparent when
the cells were exposed to 10 μM of sunitinib; additionally,
sorafenib caused a distinct downregulation of the tumor cell number
at a dosage of ≥5 μM (63).
However, when RCC cells were exposed to 1 μM sunitinib for 8
weeks (equivalent to a 1.3 treatment cycle) cdk1 and cdk2 were
overexpressed, p27 was downregulated, and Akt, Rictor, and Raptor
were activated (63). It was
previously shown that the wild-type pVHL expressing CAKI-1 and
786-0-VHL (VHL-transfected) cells were ~2-fold more resistant to
the anti-proliferative effects of sorafenib (2.5–20 μM)
under hypoxic conditions, when compared with the mutant pVHL
expressing CAKI-2 and 786-0 cells. Such a difference was not
reported under normoxia (62). The
phenomenon of TKI activity dependence on tumor niche oxygen
availability is also true for RCC-CSCs (Figs. 3, 6
and 7). In order to explain this
phenomenon in RCCs, the gene expression in the 786-0-VHL cells (wt
VHL transfected) vs. the 786-0-neo cells (VHL mutant) were
investigated. As many as 40 genes, mostly related to the inhibition
of apoptosis (e.g., BAG1-bcl2-associated athanogene) and
angiogenesis (e.g., PDGFβ), were shown to be >5-fold
overexpressed under hypoxia in the RCC model. These genes,
including BAG-1 and PDGFβ, were downregulated >2-fold under
hypoxia with sorafenib treatment in both cell types. It was finally
concluded that wt-VHL ccRCC under hypoxic (1% 02)
conditions promoted the overexpression of anti-apoptotic and
pro-angiogenic genes that attenuated the anti-proliferative effects
of sorafenib (62). In our study,
hypoxia limited the efficacy of the TKIs in both the 3D model and
RCC-CSC proliferation and influenced their morphology, which
further confirmed the significant role of oxygen in tumor
development (Figs. 7, 8 and 10).
Anti-angiogenic agents generate intratumoral
hypoxia, modulating the metastatic process and stimulating cancer
stem cells (CSCs) (37). Currently,
determining the most effective application of TKIs clinically in
RCC treatment is imperative to investigate the mechanisms
underlying their efficacy. It remains to be determined whether the
in vivo efficacy of compounds such as sunitinib, sorafenib
or axitinib can be explained in terms of its inhibition of one (or
a combination) of its targets in various tumor cells. As an example
of this approach, the primary goal of our study was to describe the
action of these TKIs in vitro, but in a clinically relevant
model or RCC-CSCs. The findings presented here not only broaden our
understanding of the role of hypoxia in RCC-CSC biology, but may
have significant clinical implications, since angiogenesis has been
a long-standing therapeutic target in ccRCC.
Acknowledgments
This project was supported by the Foundation for
Polish Science Team project TEAM/2010-6/8. Scribendi proofreading
service (Scribendi Inc. Chatham, ON, Canada) was covered by
A.M.C.
References
|
1
|
Buczek M, Escudier B, Bartnik E, Szczylik
C and Czarnecka A: Resistance to tyrosine kinase inhibitors in
clear cell renal cell carcinoma: From the patient's bed to
molecular mechanisms. Biochim Biophys Acta. 1845:31–41. 2014.
|
|
2
|
Młot B, Szczylik C and Rzepecki P: Seeking
new prognostic and predictive factors in patients with metastatic
renal cell carcinoma - hypoxia-induced factors. Contemp Oncol
(Pozn). 16:250–253. 2012.
|
|
3
|
Bielecka ZF, Czarnecka AM and Szczylik C:
Genomic analysis as the first step toward personalized treatment in
renal cell carcinoma. Front Oncol. 4:1942014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Porta C, Bellmunt J, Eisen T, Szczylik C
and Mulders P: Treating the individual: The need for a
patient-focused approach to the management of renal cell carcinoma.
Cancer Treat Rev. 36:16–23. 2010. View Article : Google Scholar
|
|
5
|
Escudier B, Szczylik C, Porta C and Gore
M: Treatment selection in metastatic renal cell carcinoma: Expert
consensus. Nat Rev Clin Oncol. 9:327–337. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ning X, Shu J, Du Y, Ben Q and Li Z:
Therapeutic strategies targeting cancer stem cells. Cancer Biol
Ther. 14:295–303. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chow EK: Implication of cancer stem cells
in cancer drug development and drug delivery. J Lab Autom. 18:6–11.
2013. View Article : Google Scholar
|
|
8
|
Matak D, Szymanski L, Szczylik C,
Sledziewski R, Lian F, Bartnik E, Sobocinska A and Czarnecka AM:
Biology of renal tumour cancer stem cells applied in medicine.
Contemp Oncol (Pozn). 19:A44–A51. 2015.
|
|
9
|
Czarnecka AM and Szczylik C: Renal cell
carcinoma cancer stem cells as therapeutic targets. Curr Signal
Transduct Ther. 8:203–209. 2013. View Article : Google Scholar
|
|
10
|
Roskoski R Jr: Sunitinib: A VEGF and PDGF
receptor protein kinase and angiogenesis inhibitor. Biochem Biophys
Res Commun. 356:323–328. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Huang D, Ding Y, Li Y, Luo WM, Zhang ZF,
Snider J, Vandenbeldt K, Qian CN and Teh BT: Sunitinib acts
primarily on tumor endothelium rather than tumor cells to inhibit
the growth of renal cell carcinoma. Cancer Res. 70:1053–1062. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Padhye SS, Guin S, Yao HP, Zhou YQ, Zhang
R and Wang MH: Sustained expression of the RON receptor tyrosine
kinase by pancreatic cancer stem cells as a potential targeting
moiety for antibody-directed chemotherapeutics. Mol Pharm.
8:2310–2319. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Diaz R, Nguewa PA, Redrado M, Manrique I
and Calvo A: Sunitinib reduces tumor hypoxia and angiogenesis, and
radiosensitizes prostate cancer stem-like cells. Prostate.
75:1137–1149. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Varna M, Gapihan G, Feugeas JP, Ratajczak
P, Tan S, Ferreira I, Leboeuf C, Setterblad N, Duval A, Verine J,
et al: Stem cells increase in numbers in perinecrotic areas in
human renal cancer. Clin Cancer Res. 21:916–924. 2015. View Article : Google Scholar
|
|
15
|
Adnane L, Trail PA, Taylor I and Wilhelm
SM: Sorafenib (BAY 43-9006, Nexavar), a dual-action inhibitor that
targets RAF/MEK/ERK pathway in tumor cells and tyrosine kinases
VEGFR/PDGFR in tumor vasculature. Methods Enzymol. 407:597–612.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xin HW, Ambe CM, Hari DM, Wiegand GW,
Miller TC, Chen JQ, Anderson AJ, Ray S, Mullinax JE, Koizumi T, et
al: Label-retaining liver cancer cells are relatively resistant to
sorafenib. Gut. 62:1777–1786. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chow AK, Ng L, Lam CS, Wong SK, Wan TM,
Cheng NS, Yau TC, Poon RT and Pang RW: The enhanced metastatic
potential of hepatocellular carcinoma (HCC) cells with sorafenib
resistance. PLoS One. 8:e786752013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hashimoto N, Tsunedomi R, Yoshimura K,
Watanabe Y, Hazama S and Oka M: Cancer stem-like sphere cells
induced from de-differentiated hepatocellular carcinoma-derived
cell lines possess the resistance to anti-cancer drugs. BMC Cancer.
14:7222014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yamada T, Abei M, Danjoh I, Shirota R,
Yamashita T, Hyodo I and Nakamura Y: Identification of a unique
hepatocellular carcinoma line, Li-7, with CD13(+) cancer stem cells
hierarchy and population change upon its differentiation during
culture and effects of sorafenib. BMC Cancer. 15:2602015.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shan J, Shen J, Liu L, Xia F, Xu C, Duan
G, Xu Y, Ma Q, Yang Z, Zhang Q, et al: Nanog regulates self-renewal
of cancer stem cells through the insulin-like growth factor pathway
in human hepatocellular carcinoma. Hepatology. 56:1004–1014. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Galuppo R, Maynard E, Shah M, Daily MF,
Chen C, Spear BT and Gedaly R: Synergistic inhibition of HCC and
liver cancer stem cell proliferation by targeting RAS/RAF/MAPK and
WNT/β-catenin pathways. Anticancer Res. 34:1709–1713.
2014.PubMed/NCBI
|
|
22
|
Gedaly R, Galuppo R, Musgrave Y, Angulo P,
Hundley J, Shah M, Daily MF, Chen C, Cohen DA, Spear BT, et al:
PKI-587 and sorafenib alone and in combination on inhibition of
liver cancer stem cell proliferation. J Surg Res. 185:225–230.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rausch V, Liu L, Kallifatidis G, Baumann
B, Mattern J, Gladkich J, Wirth T, Schemmer P, Büchler MW, Zöller
M, et al: Synergistic activity of sorafenib and sulforaphane
abolishes pancreatic cancer stem cell characteristics. Cancer Res.
70:5004–5013. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lee JH, Shim JW, Choi YJ, Heo K and Yang
K: The combination of sorafenib and radiation preferentially
inhibits breast cancer stem cells by suppressing HIF-1α expression.
Oncol Rep. 29:917–924. 2013.PubMed/NCBI
|
|
25
|
Carra E, Barbieri F, Marubbi D, Pattarozzi
A, Favoni RE, Florio T and Daga A: Sorafenib selectively depletes
human glioblastoma tumor-initiating cells from primary cultures.
Cell Cycle. 12:491–500. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Aldea MD, Petrushev B, Soritau O,
Tomuleasa CI, Berindan-Neagoe I, Filip AG, Chereches G, Cenariu M,
Craciun L, Tatomir C, et al: Metformin plus sorafenib highly
impacts temozolomide resistant glioblastoma stem-like cells. J
BUON. 19:502–511. 2014.PubMed/NCBI
|
|
27
|
Chen G, Nicula D, Renko K and Derwahl M:
Synergistic anti-proliferative effect of metformin and sorafenib on
growth of anaplastic thyroid cancer cells and their stem cells.
Oncol Rep. 33:1994–2000. 2015.PubMed/NCBI
|
|
28
|
Zhang K and Waxman DJ: Impact of tumor
vascularity on responsiveness to antiangiogenesis in a prostate
cancer stem cell-derived tumor model. Mol Cancer Ther. 12:787–798.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hu-Lowe DD, Zou HY, Grazzini ML, Hallin
ME, Wickman GR, Amundson K, Chen JH, Rewolinski DA, Yamazaki S, Wu
EY, et al: Nonclinical antiangiogenesis and antitumor activities of
axitinib (AG-013736), an oral, potent, and selective inhibitor of
vascular endothelial growth factor receptor tyrosine kinases 1, 2,
3. Clin Cancer Res. 14:7272–7283. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fenton BM and Paoni SF: The addition of
AG-013736 to fractionated radiation improves tumor response without
functionally normalizing the tumor vasculature. Cancer Res.
67:9921–9928. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang F, Mi YJ, Chen XG, Wu XP, Liu Z, Chen
SP, Liang YJ, Cheng C, To KK and Fu LW: Axitinib targeted cancer
stemlike cells to enhance efficacy of chemotherapeutic drugs via
inhibiting the drug transport function of ABCG2. Mol Med.
18:887–898. 2012.PubMed/NCBI
|
|
32
|
Lu L, Saha D, Martuza RL, Rabkin SD and
Wakimoto H: Single agent efficacy of the VEGFR kinase inhibitor
axitinib in preclinical models of glioblastoma. J Neurooncol.
121:91–100. 2015. View Article : Google Scholar
|
|
33
|
McKenzie BA, Zemp FJ, Pisklakova A,
Narendran A, McFadden G, Lun X, Kenchappa RS, Kurz EU and Forsyth
PA: In vitro screen of a small molecule inhibitor drug library
identifies multiple compounds that synergize with oncolytic myxoma
virus against human brain tumor-initiating cells. Neuro Oncol.
17:1086–1094. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gomez-Casal R, Bhattacharya C, Ganesh N,
Bailey L, Basse P, Gibson M, Epperly M and Levina V: Non-small cell
lung cancer cells survived ionizing radiation treatment display
cancer stem cell and epithelial-mesenchymal transition phenotypes.
Mol Cancer. 12:942013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Conley SJ, Gheordunescu E, Kakarala P,
Newman B, Korkaya H, Heath AN, Clouthier SG and Wicha MS:
Antiangiogenic agents increase breast cancer stem cells via the
generation of tumor hypoxia. Proc Natl Acad Sci USA. 109:2784–2789.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Piao Y, Liang J, Holmes L, Zurita AJ,
Henry V, Heymach JV and de Groot JF: Glioblastoma resistance to
anti-VEGF therapy is associated with myeloid cell infiltration,
stem cell accumulation, and a mesenchymal phenotype. Neuro Oncol.
14:1379–1392. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chinchar E, Makey KL, Gibson J, Chen F,
Cole SA, Megason GC, Vijayakumar S, Miele L and Gu JW: Sunitinib
significantly suppresses the proliferation, migration, apoptosis
resistance, tumor angiogenesis and growth of triple-negative breast
cancers but increases breast cancer stem cells. Vasc Cell.
6:122014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Clery JP, Gomez E, Sharma M, Khemani A,
Sharma C, Punzalan R, Navel M, Amezcua N, Jani J and Sharma J:
Optimal drug concentration screening and evaluation in cancer stem
cells & 3D tumor stem cell cultures drug response assays in
association with clinical efficacy for pancreatic cancer stem cell.
Cancer Res. 73:55452013. View Article : Google Scholar
|
|
39
|
Sharma J, DiMaggio CM, Taw N, Majdoub M,
Liles N, Velazquez G, Amezcua N, Sharma C and Sharma M: Human liver
cancer stem cells as a potential target for novel drug therapy and
drug discovery. The American Society for Cell Biology 2009 Annual
Meeting Regular; Abstracts, no. B48. 2009
|
|
40
|
Cleary JP, Raman D, Punzalan RR, et al:
Targeting cancer stem cells as potential new therapy for pancreatic
cancer. Cancer Res. 72:44082012. View Article : Google Scholar
|
|
41
|
Sharma CVR, Passarini JD, Majdoub MW,
DiMaggio CM, Sobhy OM, Punzalan RR, Sharma MRV, Harris-White ME,
Warden MW, Sharma SP and Sharma JP: Human triple-negative breast
cancer stem cells utilized for drug discovery therapeutics for
triple-negative breast cancer patients. Cancer Res. 70:33192010.
View Article : Google Scholar
|
|
42
|
Passarini J, Cleary JP, Kumar P, Newton T,
Sharma M, Sharma C, Panunzalan RR, Gill P, Sharma S, Srivastava R,
Shankar S and Sharma JP: Screening potential drug candidates for
treatment of glioblastoma patients utilizing an in-vivo mouse/rat
model system. Cancer Res. 71:33062011. View Article : Google Scholar
|
|
43
|
Ebert T, Bander NH, Finstad CL, Ramsawak
RD and Old LJ: Establishment and characterization of human renal
cancer and normal kidney cell lines. Cancer Res. 50:5531–5536.
1990.PubMed/NCBI
|
|
44
|
Breslin S and O'Driscoll L: Receptor
tyrosine kinase targeting in multicellular spheroids. Methods Mol
Biol. 1233:161–168. 2015. View Article : Google Scholar
|
|
45
|
Foty R: A simple hanging drop cell culture
protocol for generation of 3D spheroids. J Vis Exp.
51:27202011.PubMed/NCBI
|
|
46
|
Schneider CA, Rasband WS and Eliceiri KW:
NIH Image to ImageJ: 25 years of image analysis. Nat Methods.
9:671–675. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Abramoff MD, Magalhaes PJ and Ram SJ:
Image processing with ImageJ. Biophotonics Int. 11:36–42. 2004.
|
|
48
|
Nakayama GR, Caton MC, Nova MP and
Parandoosh Z: Assessment of the Alamar Blue assay for cellular
growth and viability in vitro. J Immunol Methods. 204:205–208.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Triglia D, Sherard Braa S, Yonan C and
Naughton GK: Cytotoxicity testing using neutral red and MTT assays
on a three-dimensional human skin substrate. Toxicol In Vitro.
5:573–578. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Vistica DT, Skehan P, Scudiero D, Monks A,
Pittman A and Boyd MR: Tetrazolium-based assays for cellular
viability: A critical examination of selected parameters affecting
formazan production. Cancer Res. 51:2515–2520. 1991.PubMed/NCBI
|
|
51
|
Machesky LM: Lamellipodia and filopodia in
metastasis and invasion. FEBS Lett. 582:2102–2111. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Négrier S and Raymond E: Antiangiogenic
treatments and mechanisms of action in renal cell carcinoma. Invest
New Drugs. 30:1791–1801. 2012. View Article : Google Scholar
|
|
53
|
O'Farrell AM, Abrams TJ, Yuen HA, Ngai TJ,
Louie SG, Yee KW, Wong LM, Hong W, Lee LB, Town A, et al: SU11248
is a novel FLT3 tyrosine kinase inhibitor with potent activity in
vitro and in vivo. Blood. 101:3597–3605. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Wiernik PH: FLT3 inhibitors for the
treatment of acute myeloid leukemia. Clin Adv Hematol Oncol.
8:429–436. 4442010.PubMed/NCBI
|
|
55
|
Karaman MW, Herrgard S, Treiber DK,
Gallant P, Atteridge CE, Campbell BT, Chan KW, Ciceri P, Davis MI,
Edeen PT, et al: A quantitative analysis of kinase inhibitor
selectivity. Nat Biotechnol. 26:127–132. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kitagawa D, Yokota K, Gouda M, Narumi Y,
Ohmoto H, Nishiwaki E, Akita K and Kirii Y: Activity-based kinase
profiling of approved tyrosine kinase inhibitors. Genes Cells.
18:110–122. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Mendel DB, Laird AD, Xin X, Louie SG,
Christensen JG, Li G, Schreck RE, Abrams TJ, Ngai TJ, Lee LB, et
al: In vivo antitumor activity of SU11248, a novel tyrosine kinase
inhibitor targeting vascular endothelial growth factor and
platelet-derived growth factor receptors: Determination of a
pharmacokinetic/pharmaco-dynamic relationship. Clin Cancer Res.
9:327–337. 2003.PubMed/NCBI
|
|
58
|
Potapova O, Laird AD, Nannini MA, Barone
A, Li G, Moss KG, Cherrington JM and Mendel DB: Contribution of
individual targets to the antitumor efficacy of the multitargeted
receptor tyrosine kinase inhibitor SU11248. Mol Cancer Ther.
5:1280–1289. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Gotink KJ, Broxterman HJ, Labots M, de
Haas RR, Dekker H, Honeywell RJ, Rudek MA, Beerepoot LV, Musters
RJ, Jansen G, et al: Lysosomal sequestration of sunitinib: A novel
mechanism of drug resistance. Clin Cancer Res. 17:7337–7346. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Gotink KJ, Broxterman HJ, Honeywell RJ,
Dekker H, de Haas RR, Miles KM, Adelaiye R, Griffioen AW, Peters
GJ, Pili R, et al: Acquired tumor cell resistance to sunitinib
causes resistance in a HT-29 human colon cancer xenograft mouse
model without affecting sunitinib biodistribution or the tumor
microvasculature. Oncoscience. 1:844–853. 2014. View Article : Google Scholar
|
|
61
|
Ma J, Chen CS, Blute T and Waxman DJ:
Antiangiogenesis enhances intratumoral drug retention. Cancer Res.
71:2675–2685. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Al Hazzouri A, Vaziri SA, Lynch M,
Schwartz B, Rini BI, Bukowski R and Ganapathy R: Anti-proliferative
effects of sorafenib in clear cell renal cell carcinoma (CCRCC)
cell lines: Relationship to von Hippel-Lindau protein (pVHL)
expression and hypoxia. J Clin Oncol. 24:46012006.
|
|
63
|
Juengel E, Kim D, Makarević J, Reiter M,
Tsaur I, Bartsch G, Haferkamp A and Blaheta RA: Molecular analysis
of sunitinib resistant renal cell carcinoma cells after sequential
treatment with RAD001 (everolimus) or sorafenib. J Cell Mol Med.
19:430–441. 2015. View Article : Google Scholar :
|