Introduction
Chemokine (C-X-C motif) ligand 16 (CXCL16) is a
member of CXC chemokine family, initially identified in dendritic
cells in T cell zone and cells in the splenic red pulp (1). Later, the expression of CXCL16 in
macrophages (2,3), dendritic (4), liver sinusoidal endothelial (5) and cancer cells (6–8) was
observed. There are two forms of CXCL16: membrane-bound CXCL16
inducing firm adhesion in chemokine (C-X-C motif) receptor 6
(CXCR6) expressing cells (9), and
soluble CXCL16 inducing migration in CXCR6 expressing cells
(10,11). CXCL16 is involved in progress of
many diseases (12–14), including cancer (6,7). For
instance, higher expression of CXCL16 reduced the overall survival
of cancer patients (6); CXCR6 is
involved in cancer the cell proliferation and invasion in
vitro (15). Our previous
research showed that CXCR6 contributed to breast cancer cell
migration via the regulation of hypoxia-inducible factor 1α
(HIF-1α) under hypoxia (16).
The effect of CXCL16 in cancer cell is well defined,
while the effect of CXCL16 on angiogenesis is rarely studied.
Previous research has reported that CXCL16 promotes human umbilical
vein endothelial cell (HUVEC) proliferation, chemotaxis, and tube
formation by activating ERK pathway in vitro (17). Whereas, the role of other pathways
and their involvement in CXCL16-induced angiogenesis is not clear.
It has been observed that CXCL16 improves the human aortic smooth
muscle cells proliferation and adhesion by activating the PI3K/Akt
pathway (18) whereas the binding
of dextran sulfate sodium to CXCL16 activates the p38 pathway in
murine peritoneal macrophages (19). Moreover, the activation of p38 and
Akt pathways induces angiogenesis (20–22).
In the present study, we investigated the molecular
mechanisms underlying CXCL16-induced angiogenesis. Our results
revealed that CXCL16 induced angiogenesis and HIF-1α expression by
activating the ERK, p38 and Akt pathways in HUVECs. Furthermore, we
found that CXCL16 increased VEGF and CXCL16 secretion by modulating
HIF-1α in HUVECs.
Materials and methods
Cell culture
HUVECs were cultured in complete medium consisted of
M199 medium, 20% FBS, 50 ng/ml endothelial cell growth supplement
(ECGS) (both from Sigma) and 100 ng/ml heparin. Cells were
incubated in humidified atmosphere with 5% CO2 at 37°C.
HUVECs at the 6–9th passage were used for experiments.
Cell proliferation assay
HUVECs were seeded into a 24-well plate in complete
medium and cultured overnight. After the replacement of medium with
M199 supplemented with 1% FBS and 100 ng/ml heparin, HUVECs were
pre-treated with 2.5 µM FR180204 (ERK-selective inhibitor),
5 µM SB202190 (p38 MAP kinase-selective inhibitor), 5
µM LY294002 (selective inhibitor of phosphatidylinositol
3-kinase) or 10 µM PX12 (HIF-1α-selective inhibitor) (all
from Tocris Bioscience) for 1 h. Then cells were exposed to 0, 1,
10 and 100 ng/ml CXCL16 polypeptide (PeproTech) for 48 h. Cells
were then counted in five randomly selected fields per group under
a microscope.
CXCL16 was dissolved in phosphate-buffered saline
(PBS); FR180204, SB202190, LY294002 and PX12 were dissolved in
dimethylsulfoxide (DMSO).
Cell migration assay
Transwell chamber migration system with
polycarbonate membranes (8.0-µM pore size; Corning) was used
for cell migration assay (23). The
medium used in the migration assay consisted of M199, 1% FBS and
100 ng/ml heparin. HUVECs (1×104 cells/ml) were seeded
into the upper chamber with 2.5 µM FR180204, 5 µM
SB202190, 5 µM LY294002 or 10 µM PX12. Medium with
CXCL16 (100 ng/ml) and FBS (10%) was added into the lower chamber.
HUVECs (1×104 cells/ml) were seeded into the upper
chamber along with various concentrations of CXCL16 (1, 10 or 100
ng/ml) added into the lower chamber. The cell migration system was
incubated at 37°C for 6 h. Cells were fixed with 4%
paraformaldehyde, stained with 0.2% cresyl violet solution and
counted in five randomly selected fields.
Tube formation assay
Tube formation assay was performed as previously
described (23), 96-well plates
were coated with 50 µl/well growth factor reducing Matrigel
(Becton, Dickinson and Company) and incubated at 37°C for 2 h.
HUVECs (1×104 cells/well) were incubated with CXCL16 (0,
1, 10 or 100 ng/ml) at 37°C for 12 h. Or, HUVECs (1×104
cells/well) were incubated with 2.5 µM FR180204, 5 µM
SB202190 or 5 µM LY294002 in the presence of CXCL16 (100
ng/ml) for 24 h. Or, HUVECs (1×104 cells/well) were
treated with or without 10−5 M topotecan in the presence
of CXCL16 (1 `or 100 ng/ml) for 12 h. Tubes were counted in five
randomly selected fields per group under a microscope.
Western blotting
HUVECs were treated with 100 ng/ml of CXCL16 for 0,
10, 20, 30, 40 and 50 min in a time-dependent study and with
different concentrations of CXCL16 (0, 1, 10 and 100 ng/ml) for 30
min in a dose-dependent study; or with 100 ng/ml of CXCL16 for 0,
3, 6, 9, and 12 h; or with 100 ng/ml of CXCL16 in the presence of
DMSO, 2.5 µM FR180204, 5 µM SB202190 or 5 µM
LY294002, respectively. Conditioned medium was collected and
concentrated with Amicon Ultra-15 (with Ultracel-10; Millipore).
Cells were washed with ice-cold PBS and then lysed with RIPA buffer
(Beyotime). Total protein concentration was evaluated with the BCA
protein assay reagent kit (Pierce, Rockford, IL, USA). Denatured
samples (40 µg/lane) were separated by 10–15% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Transferred
membranes were probed with murine anti-human antibodies:
anti-CXCL16 antibody (R&D Systems), anti-HIF-1α antibody
(Sigma), anti-p-ERK1/2 antibody, anti-p-Akt antibody, anti-p-p38
MAPK antibody, anti-ERK1/2 antibody, anti-Akt antibody, anti-p38
antibody (all from Cell Signaling Technology) and anti-actin
antibody (Sigma). Horseradish peroxidase (HRP)-linked anti-rabbit
IgG (Cell Signaling Technology) or HRP-linked anti-mouse IgG
(Sigma) was used as the secondary antibody. Immunoreactive proteins
on the membrane were visualized by enhanced chemiluminescence
western blotting detection reagents (Millipore, USA).
Real-time quantitative PCR
HUVECs were pre-treated with or without 10 µM
PX12 for 1 h, followed by the treatment of 100 ng/ml of CXCL16 for
3 or 6 h; or followed by incubation under normoxia or hypoxia for 6
h. Total cell RNA was isolated with TRIzol® reagent
(Invitrogen). Total RNA (2 µg) was used for first-strand
cDNA synthesis with RevertAid First Strand cDNA Synthesis kit
(Fermentas). Primers for RT-PCR sequences were as follows: HIF-1α
forward, 5′-CTCACCCAACGAAAAATTACAGAA-3′ and HIF-1α reverse,
5′-ATTGAGTGCAGGGTCAGCACTAC-3′ (24); CXCL16 forward,
5′-GGCAGCGTCACTGGAAGTTGTTAT-3′ and CXCL16 reverse,
5′-ACCGATGGTAAGCTCTCAGGTGTT-3′; β-actin forward,
5′-GAGCGGGAAATCGTGCGTGACATT-3′ and β-actin reverse,
5′-GAAGGTAGTTTCGTGGATGCC-3′ (25).
iQ™ SYBR-Green Supermix (Bio-Rad Laboratories) was used as a
fluorescent dye to detect the presence of double-stranded DNA. The
amount of mRNA was normalized to an internal control β-actin mRNA.
Relative mRNA levels were normalized to control.
VEGF enzyme-linked immunosorbent
assay
HUVECs were exposed to 100 ng/ml of CXCL16 for 0, 3,
6, 9, 12, 24 and 30 h. At each time point, conditioned medium were
collected and concentrated at 8,000 rpm at 4°C for 1 min. The
supernatant was collected and stored at −70°C. The enzyme-linked
immunosorbent assay (ELISA) was performed according to the
manufacturer's instruction for human VEGF.
Statistical analysis
All data represent at least three independent
experiments and are expressed as mean ± SEM. Student's t-test was
used for statistical analysis.
Results
Effects of CXCL16 on proliferation,
migration and tube formation in HUVECs
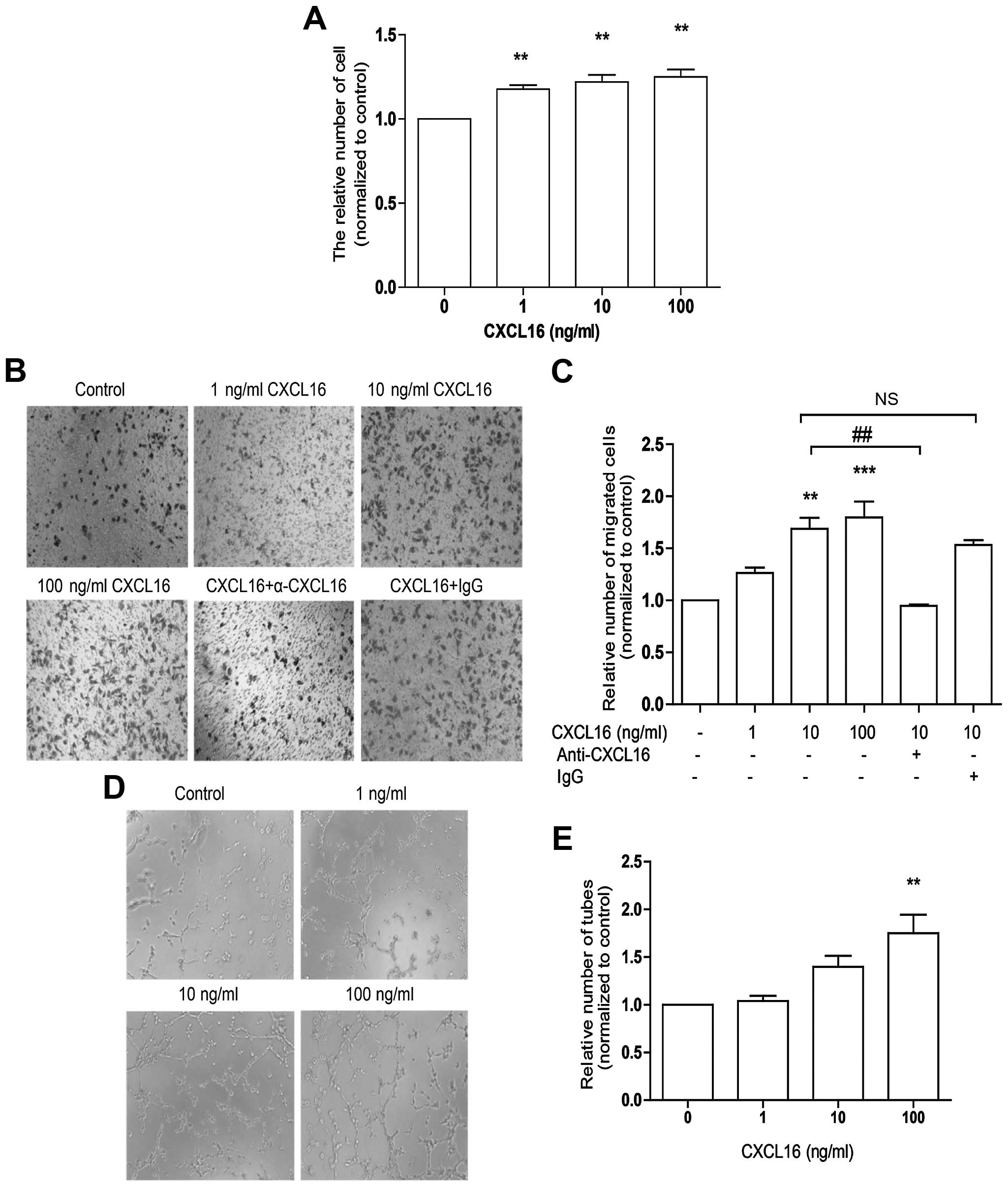
To study the effect of CXCL16 on angiogenesis in
vitro, HUVECs were treated with 1–100 ng/ml of CXCL16 for 48 h
for the cell proliferation assay, 6 h formigration assay and 12 h
for tube formation assay. In line with previous research (17), our data showed that CXCL16 promoted
HUVECs proliferation (Fig. 1A),
migration (Fig. 1B and C) and tube
formation (Fig. 1D and E) in a
dose-dependent manner. Our data confirm that CXCL16 promotes HUVECs
angiogenesis in vitro.
Effects of CXCL16 on the activation of
ERK, p-38 and Akt pathways in HUVECs
In murine peritoneal macrophages and human aortic
smooth muscle cells, CXCL16 activated ERK1/2, p38 and PI3K/Akt,
respectively (18,19). To determine whether CXCL16 could
activate ERK1/2, p38 and Akt pathways in HUVECs, we treated HUVECs
with CXCL16 (100 ng/ml) for 0, 10, 20, 30, 40 and 50 min. Western
blot results revealed that the phosphorylation levels of ERK1/2,
p38 and Akt were increased within 30 min and p-ERK1/2 and p-Akt
were increased time-dependently (Fig.
2A).
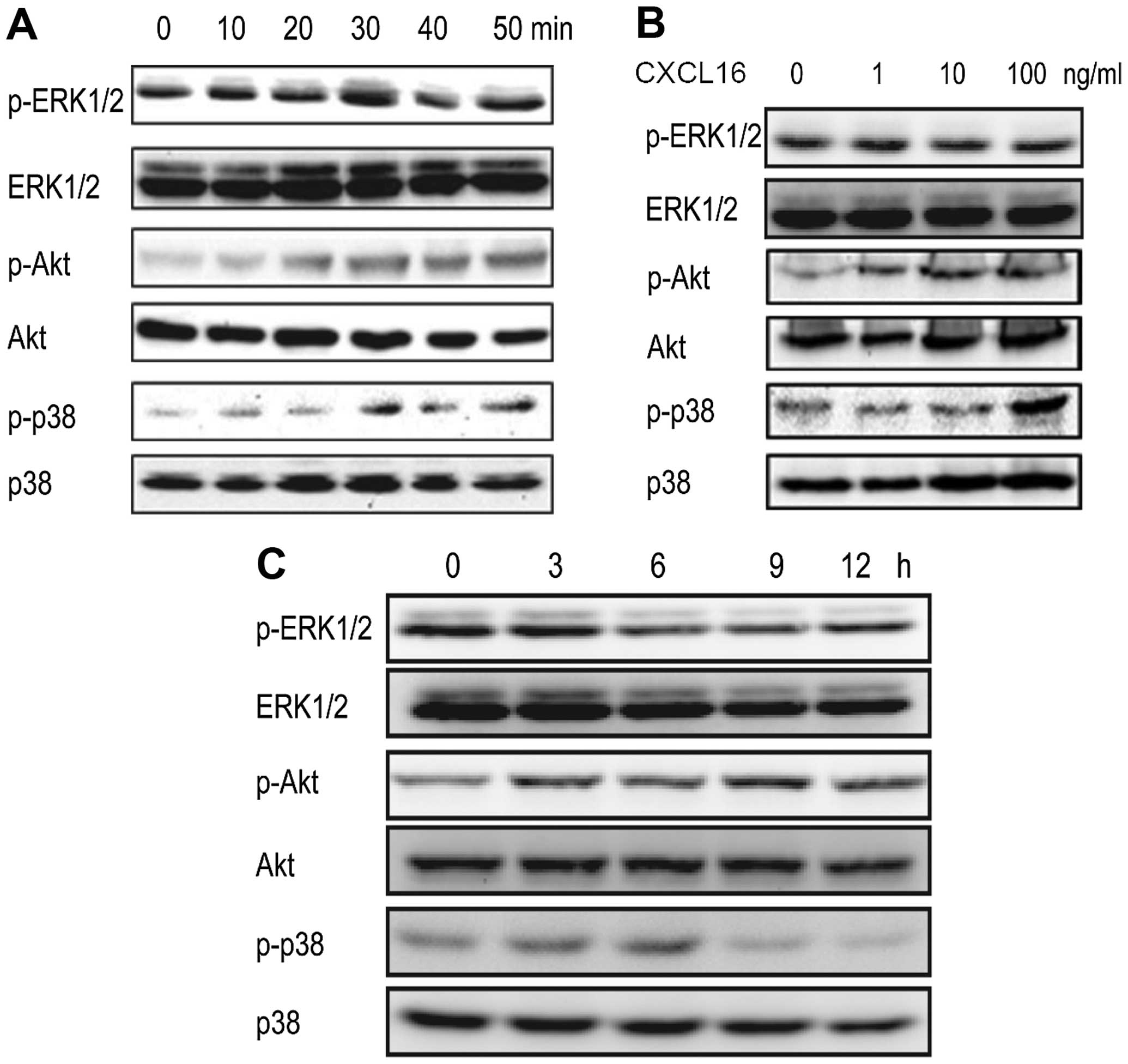 | Figure 2Activation of ERK1/2, Akt and p38
pathways induced by CXCL16. (A) CXCL16 activates ERK1/2, Akt and
p38 in a time-dependent manner in HUVECs. HUVECs were treated with
100 ng/ml of CXCL16 for 0, 10, 20, 30, 40 and 50 min. p-ERK1/2,
ERK1/2, p-Akt, Akt, p-p38 and p38 were checked by western blot
analyses. (B) CXCL16 activates ERK1/2 and Akt in a dose-dependent
manner. HUVECs were treated with 0, 1, 10 and 100 ng/ml of CXCL16
for 30 min. Phosphorylation of ERK1/2, Akt and p38 was analyzed by
western blot analyses. (C) Long-term activation of ERK1/2, Akt and
p38. HUVECs were treated with 100 ng/ml of CXCL16 for 0, 3, 6, 9
and 12 h and followed by western blot analysis. |
After these findings, we studied the activation of
ERK1/2, p38 and Akt pathways in HUVECs treated with different
concentrations of CXCL16 (0, 1, 10 and 100 ng/ml) for 30 min. It
was observed that p-ERK1/2 and p-Akt induced by CXCL16 were
increased dose-dependently, whereas, p-p38 was increased in the
presence of 100 ng/ml of CXCL16 (Fig.
2B).
To further elucidate the activation of ERK1/2, p38
and Akt pathways induced by CXCL16 in HUVECs against the time
frame, we treated HUVECs with CXCL16 (100 ng/ml) for 0, 3, 6, 9 and
12 h. Western blot results showed that all three pathways were
activated within 3 h (Fig. 2C).
However, the activation patterns of these pathways were different.
ERK pathway was activated within 30 min (Fig. 2A) and lasted until 3 h (Fig. 2C); Akt pathway activation reached
its maximum level within 1 h and decreased at 12 h (Fig. 2C); the highest levels of p-p38 were
observed at 6 h interval (Fig. 2C).
The data indicated that CXCL16 activates ERK, p38 and Akt pathways
in HUVECs.
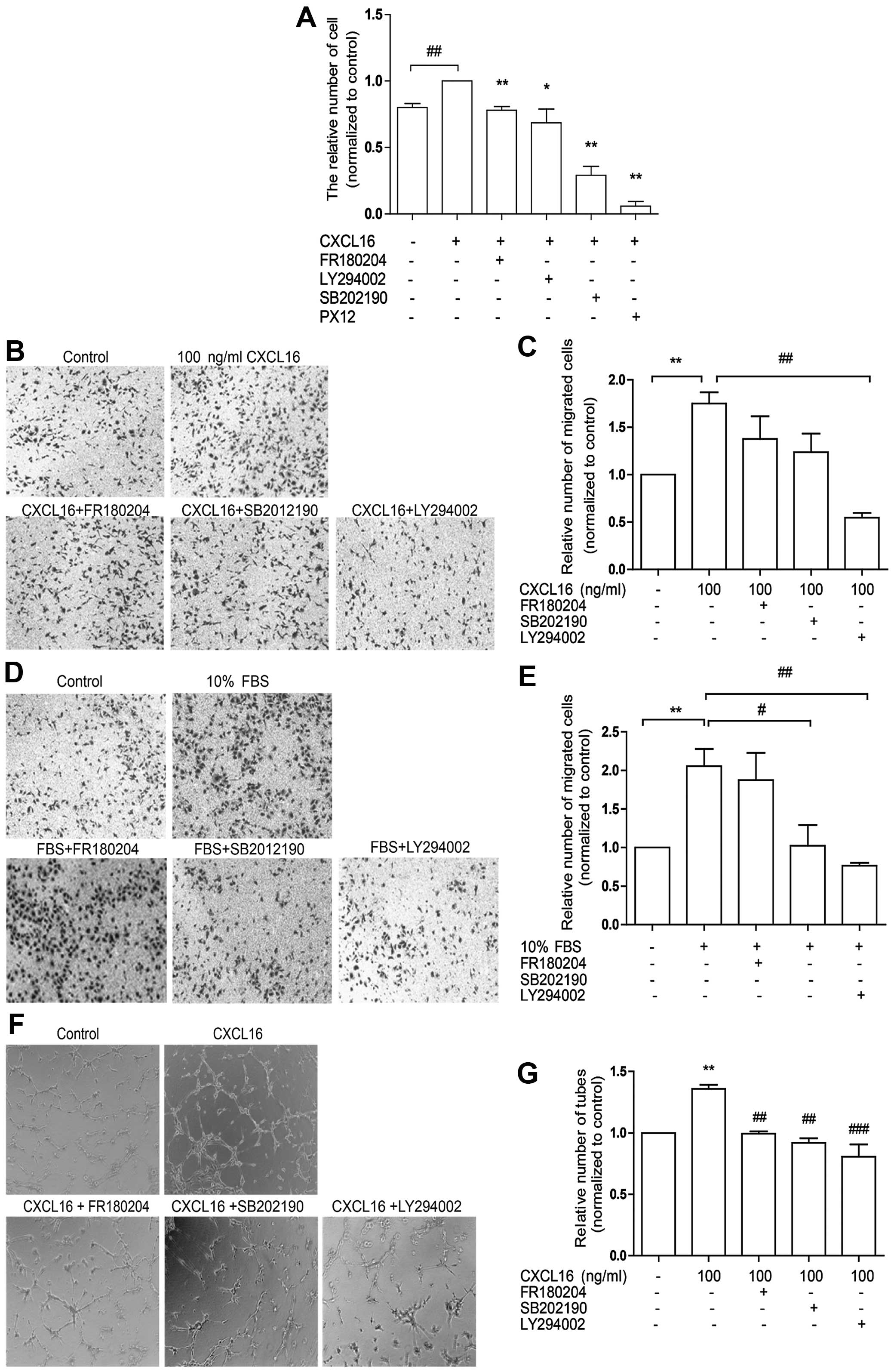
Inhibition of ERK, p38 and Akt pathways
reduces HUVEC angiogenesis induced by CXCL16
To confirm the involvement of ERK, p38 and Akt
pathways in HUVEC-associated angiogenesis induced by CXCL16, we
treated HUVECs with 100 ng/ml CXCL16 and FR180204 (inhibitor of ERK
pathway) or SB202190 (inhibitor of p38 pathway) or LY294002
(inhibitor of PI3K/Akt pathway) in proliferation, migration and
tube formation assays sequentially. We observed that FR180204,
SB202190 and LY294002 markedly attenuated CXCL16-induced HUVEC
proliferation (Fig. 3A) and tube
formation (Fig. 3F and G), whereas
LY294002 decreased HUVEC migration induced by CXCL16 (Fig. 3B and C) or FBS (Fig. 3D and E). It was also observed that
SB202190 was effective only in cell migration induced by FBS
(Fig. 3D and E). The data revealed
the involvement of ERK, p38 and Akt signaling in CXCL16-induced
HUVEC proliferation and tube formation and indicated that only Akt
pathway was associated with HUVEC migration induced by CXCL16.
Effect of CXCL16 on HIF-1α expression,
and CXCL16-stimulated HUVECs to secrete CXCL16 via ERK1/2, p38 and
Akt/HIF-1α pathway
Topotecan is a chemotherapeutic agent. We
investigated whether topotecan has an influence in CXCL16-induced
angiogenesis. Therefore, we treated HUVECs with different
concentrations of CXCL16 and 1 µM topotecan. Our findings in
tube formation assays revealed that topotecan antagonized the
positive effect of CXCL16 on tube formation in vitro
(Fig. 4A). Our previous study
showed that topotecan inhibits HIF-1α expression in vitro
(26). As shown in Fig. 3A, PX12 (HIF-1α inhibitor)
significantly attenuated HUVEC proliferation induced by CXCL16,
indicating the regulation of HIF-1α by CXCL16. Thus, we further
investigated whether CXCL16 influenced HIF-1α expression with
western blotting and RT-PCR assays. Our results showed that CXCL16
increased HIF-1α protein (Fig. 4B)
and mRNA level at 3 h (Fig. 4D),
however, the protein level of HIF-1α was reduced after 6 h.
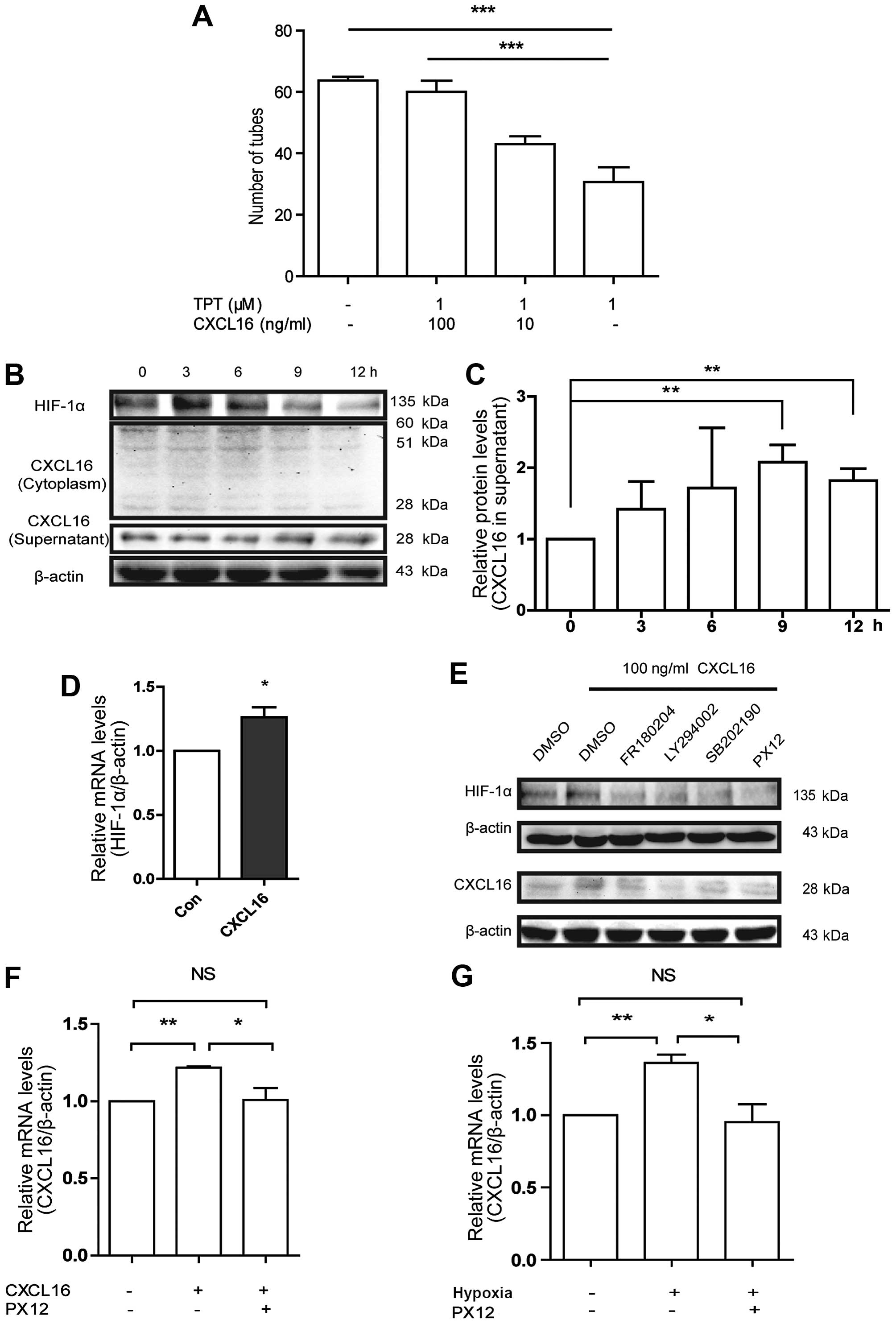 | Figure 4Effects of CXCL16 on HIF-1α in
HUVECs, and CXCL16-induced HUVECs to excrete CXCL16 by upregulating
HIF-1α through ERK, Akt and p38 pathways. (A) Involvement of HIF-1α
in CXCL16 induced tube formation in HUVECs. Cells were treated with
100 ng/ml CXCL16, or with TPT with different concentrations of
CXCL16 (100, 1 and 0 ng/ml) for 6 h. Relative numbers of tubes were
normalized to control (PBS). ***p<0.001, compared
with 100 ng/ml CXCL16 group. (B and C) CXCL16 increases HIF-1α
expression in HUVECs. Cells were treated with 100 ng/ml of CXCL16
for 0, 3, 6, 9 and 12 h. Total proteins from the cells were
examined for HIF-1α expression by western blot assays. Conditioned
medium and cell lysates were collected from HUVECs treated with 100
ng/ml of CXCL16 for 0, 3, 6, 9 and 12 h. Proteins were examined for
CXCL16 expression in cytoplasm and supernatant with murine
anti-CXCL16 antibodies by western blot assays. (D) CXCL16 increases
HIF-1α mRNA in HUVECs. Cells were treated with 100 ng/ml of CXCL16
for 3 h and followed by mRNA extraction and subsequent qRT-PCR
analysis. The mRNA level was normalized to control group.
*p<0.05. (E) Cell lysates were collected from HUVECs
treated with 100 ng/ml of CXCL16 in the presence of inhibitors
against ERK, p38, Akt and HIF-1α for 6 h. HIF-1α and CXCL16
expressions were analyzed by western blot analyses. (F and G)
CXCL16 induces CXCL16 secretion via the regulation of HIF-1α in
HUVECs. PX12 decreased CXCL16 mRNA level after CXCL16 peptide
treatment for 6 h (F) or hypoxia treatment for 12 h (G).
*P<0.05, **P<0.01,
***P<0.001. |
Regarding the data of CXCL16 activated ERK1/2, p38
and Akt pathways, we showed these pathways were correlated with
HIF-1α upregulation induced by CXCL16. Upon exposure of HUVECs to
FR180204, SB202190, LY294002 and PX12 in the presence of CXCL16 for
6 h, we found that all the inhibitors reduced the improvement in
HIF-1α expression induced by CXCL16 at 6 h (Fig. 4E). These results implicated that
CXCL16 increased HIF-1α expression through ERK1/2, p38 and Akt
pathways under normoxia.
All the results shown above were performed with the
exogenous CXCL16 peptide with molecular weight of 10.1 kDa. We also
investigated whether exogenous CXCL16 affected the expression of
endogenous CXCL16 in the same way. Therefore, we treated HUVECs
with 100 ng/ml of CXCL16 peptide for 0–12 h, and harvested the
supernatant and cells for western blot analysis. Our findings
indicated that CXCL16 peptide increased endogenous CXCL16 secretion
(Fig. 4B and C) and mRNA level of
CXCL16 at 6 h (Fig. 4F). These
results suggested that CXCL16 is probably self-regulated and
modulates angiogenesis in HUVECs.
To determine the pathways involved in CXCL16
secretion, we treated HUVECs with CXCL16 (100 ng/ml) and FR180204,
SB202190, LY294002 or PX12, respectively, for 12 h. These
inhibitors reversed the increase of CXCL16 secretion induced by
CXCL16 peptide (Fig. 4E). In
addition, we found that PX12 significantly reduced mRNA level of
CXCL16 induced by CXCL16 peptide (Fig.
4F). These results indicated that CXCL16 regulates its
expression by itself via ERK1/2, p38 and Akt pathways and HIF-1α
regulation in HUVECs under normoxia.
To confirm the expression of CXCL16 via HIF-1α, we
pre-treated HUVECs with PX12 or DMSO for 1 h, and then incubated
HUVECs under hypoxia or normoxia for 6 h; notably we found that
hypoxia increased the CXCL16 mRNA level that was previously
inhibited by PX12 (Fig. 4G). These
findings suggested that CXCL16 induces HUVEC secretion under
normoxia via activation of ERK, p38 and Akt signaling and
involvement of HIF-1α.
Effects of CXCL16 on tube formation are
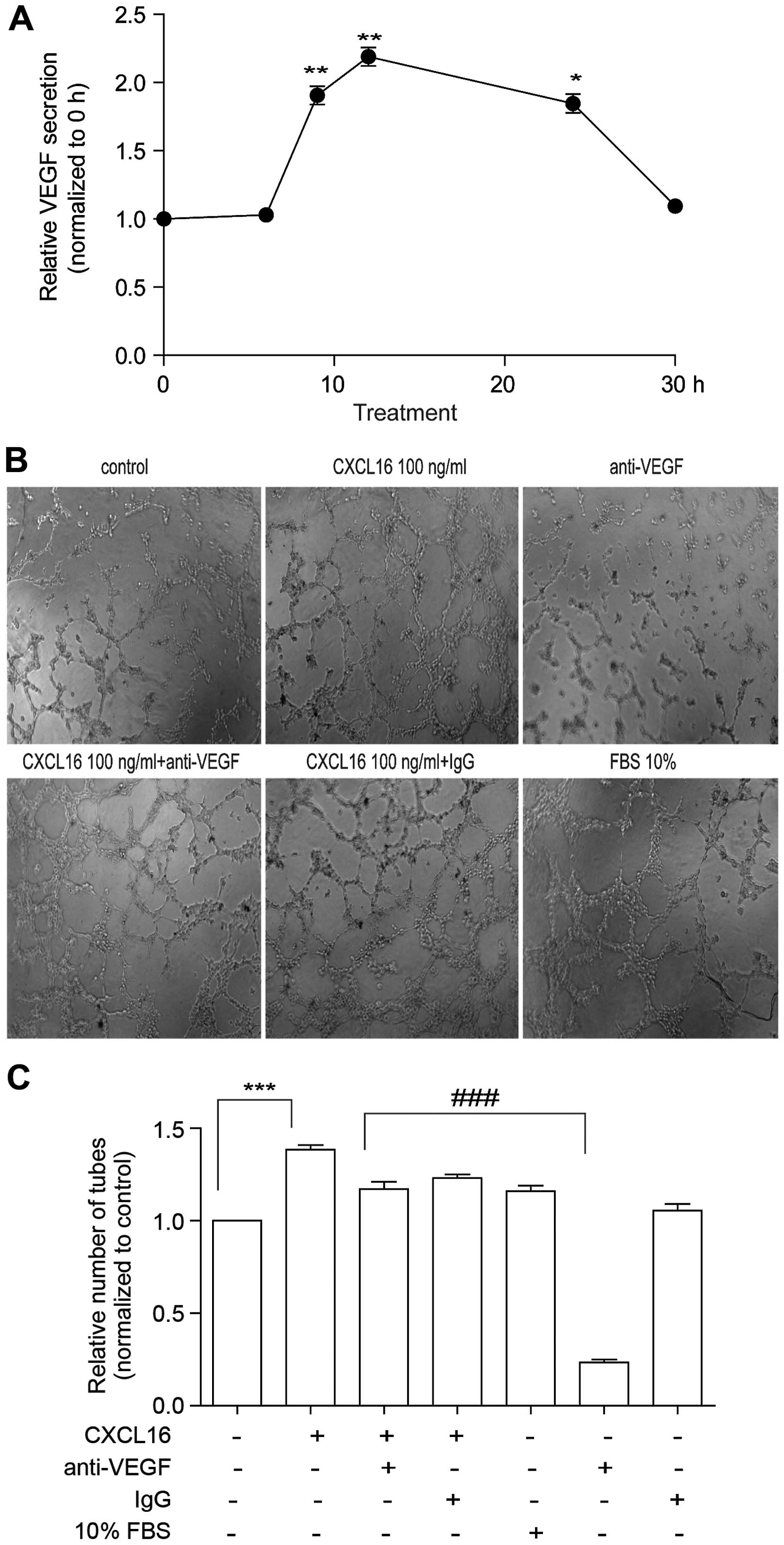
not due to VEGF secretion in HUVECs
As the secretion of VEGF is partially regulated by
HIF-1α in endothelial cells (27,28),
we examined the secretion of VEGF from HUVECs treated with CXCL16.
The HUVECs were exposed to CXCL16 (100 ng/ml) for different time
periods (0, 3, 6, 9, 12, 24 and 30 h), followed by the detection of
VEGF in the supernatant. ELISA results showed that the amount of
VEGF was significantly increased at 9 h, reaching its maximum
during 12 h and normalized at 30 h of CXCL16 treatment (Fig. 5A). The data indicate that CXCL16
promotes HUVECs to secrete VEGF possibly by regulating HIF-1α
expression.
We have found that CXCL16 can promote HUVECs to
secrete VEGF, which can also promote tube formation. So we added
CXCL16 100 ng/ml and, at the same time we added anti-VEGF antibody
(R&D Systems) to verify whether the effect of CXCL16 on tube
formation is due to VEGF secretion in HUVECs or due to some other
reason. Notably, we found that the tube formation was reduced to a
small extent (Fig. 5B and C). The
data indicated that the effect of CXCL16 on tube formation was not
due to VEGF secretion in HUVECs.
Discussion
Recent research has shown that CXCL16 and its
receptor play crucial roles in cancer growth and metastasis
(6,15,16).
Although many studies have confirmed the effect of CXCL16 in
tumorigenesis, the effect of CXCL16 on angiogenesis was not
clarified. The present study was aimed to investigate the signaling
pathways of CXCL16 during angiogenesis. We demonstrated an
autocrine signaling of CXCL16 that induced angiogenesis via
activating ERK1/2, p38 and Akt pathways and upregulating HIF-1α
expression in HUVECs.
Akt phosphorylation is crucial in angiogenesis both
in vitro and in vivo (20,29,30),
and ERK and p38 pathways were also considered to be associated with
angiogenesis (21,31,32).
It has been found that CXCL16 activates Elt in HUVECs within 30 min
(17). Consistent with these
findings, activations of Akt, ERK and p38 by CXCL16 was also
observed to be started within 30 min in HUVECs. However, the
activation of ERK, Akt and p38 was decreased after 6 h, which could
be due to the desensitization of CXCR6 after prolonged exposure to
CXCL16. By treating HUVECs with FR180204 (inhibitor of ERK
pathway), SB202190 (p-p38 inhibitor) and LY294002 (p-Akt
inhibitor), we confirmed the involvement of ERK, p38 and Akt
pathways in the angiogenesis induced by CXCL16 in HUVECs.
HIF-1α, a transcription factor is usually found at
low oxygen conditions and mediates cellular responses to hypoxia.
Previous studies have suggested that cytokines and growth factors,
such as epidermal growth factor (33), insulin and interleukin-1β (34,35)
induce HIF-1α expression under normoxia. In the present study, we
found that HIF-1α was upregulated by CXCL16 at both protein and
mRNA levels within 6 h under normoxia. However, at normoxia, HIF1-α
immediately degrades by cell type-specific regulation of von
Hippel-Lindau tumor-suppressor protein (36). The Akt pathway is involved in the
stabilization of HIF-1α under normoxia (37) and regulation of HIF-1α expression.
ERK and p38 pathways are likely regulating HIF-1α expression as
well (38,39). Consistent with these observations,
our data showed that SB202190 (p-p38 inhibitor) and LY294002 (p-Akt
inhibitor) suppressed HIF-1α expression induced by CXCL16.
HIF-1α induces the expression of VEGF and CXCL12 in
endothelial cells (38,39) under normoxia. In addition, CXCL16
improves secretion of VEGF in prostate cancer cells (15). Our data showed that CXCL16 induced
HUVECs to secrete VEGF which reached the maximum level within 9 h
under normoxia.
CXCL16 expression in HUVECs was measured. Since the
CXCL16, used for treatment, was an exogenous polypeptide with a
small molecular weight (10.1 kDa), we were able to distinguish
endogenous soluble CXCL16 (30 kDa) from the peptide in western
blotting and RT-PCR assays. We found that CXCL16 peptide improved
the secretion of endogenous soluble CXCL16 from HUVECs. However,
SB202190, FR180204 and LY294002 inhibited the secretion suggesting
the involvement of ERK, p38 and Akt pathways in CXCL16 secretion
induced by CXCL16 in HUVECs. Furthermore, we found that hypoxia
induced CXCL16 mRNA was increased which was resisted by the HIF-1α
pathway inhibitor PX12. It can be due to high level of HIF-1α
induced by CXCL16. The soluble form of CXCL16 was kept in
supernatant for 12 h, but HIF-1α, potential upstream of CXCL16, was
reduced after treatment with CXCL16 for 6 h, which may be due to
the lower level of CXCR6 or CXCR6 desensitization. Thus, our data
indicated that exogenous CXCL16 activated ERK, p38, and Akt/HIF-1α
pathway to induce CXCL16 secretion in HUVECs.
In solid tumor, cancer cells excreted CXCL16 to
promote angiogenesis, and endothelial cells excreted VEGF under
high level of CXCL16. However, CXCL16 exhibit a dual effect in
atherosclerotic lesions; CXCL16 not only promotes endothelial cell
survival but also guides the migration of macrophages into inflamed
tissues, thus resulting in more serious inflammation.
In conclusion, the present study exhibited a novel
signaling pathway of CXCL16 in HUVEC angiogenesis. CXCL16 promoted
angiogenesis in autocrine signaling in HUVECs under normoxia,
involving activation of ERK1/2, Akt and p38 pathways and subsequent
upregulation of HIF-1α. Moreover, CXCL16 increased VEGF secretion
in HUVECs which is most probably via the regulation of HIF-1α.
Acknowledgments
The present study was financially supported by the
'National Natural Science Funds' Project nos. 81102853 and
81071841, and the 2011 Program for Excellent Scientific and
Technological Innovation Team of Jiangsu Higher Education. For this
sponsorship we are highly thankful to both organizations.
References
|
1
|
Matloubian M, David A, Engel S, Ryan JE
and Cyster JG: A transmembrane CXC chemokine is a ligand for
HIV-coreceptor Bonzo. Nat Immunol. 1:298–304. 2000. View Article : Google Scholar
|
|
2
|
Shimaoka T, Kume N, Minami M, Hayashida K,
Kataoka H, Kita T and Yonehara S: Molecular cloning of a novel
scavenger receptor for oxidized low density lipoprotein, SR-PSOX,
on macrophages. J Biol Chem. 275:40663–40666. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
van der Voort R, van Lieshout AW, Toonen
LW, Slöetjes AW, Van Den Berg WB, Figdor CG, Radstake TR and Adema
GJ: Elevated CXCL16 expression by synovial macrophages recruits
memory T cells into rheumatoid joints. Arthritis Rheum.
52:1381–1391. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shimaoka T, Nakayama T, Kume N, Takahashi
S, Yamaguchi J, Minami M, Hayashida K, Kita T, Ohsumi J, Yoshie O,
et al: Cutting edge: SR-PSOX/CXC chemokine ligand 16 mediates
bacterial phagocytosis by APCs through its chemokine domain. J
Immunol. 171:1647–1651. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Heydtmann M, Lalor PF, Eksteen JA,
Hubscher SG, Briskin M and Adams DH: CXC chemokine ligand 16
promotes integrin-mediated adhesion of liver-infiltrating
lymphocytes to cholangiocytes and hepatocytes within the inflamed
human liver. J Immunol. 174:1055–1062. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gao Q, Zhao YJ, Wang XY, Qiu SJ, Shi YH,
Sun J, Yi Y, Shi JY, Shi GM, Ding ZB, et al: CXCR6 upregulation
contributes to a proinflammatory tumor microenvironment that drives
metastasis and poor patient outcomes in hepatocellular carcinoma.
Cancer Res. 72:3546–3556. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sung SY, Hsieh CL, Law A, Zhau HE, Pathak
S, Multani AS, Lim S, Coleman IM, Wu LC, Figg WD, et al:
Coevolution of prostate cancer and bone stroma in three-dimensional
coculture: Implications for cancer growth and metastasis. Cancer
Res. 68:9996–10003. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hojo S, Koizumi K, Tsuneyama K, Arita Y,
Cui Z, Shinohara K, Minami T, Hashimoto I, Nakayama T, Sakurai H,
et al: High-level expression of chemokine CXCL16 by tumor cells
correlates with a good prognosis and increased tumor-infiltrating
lymphocytes in colorectal cancer. Cancer Res. 67:4725–4731. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shimaoka T, Nakayama T, Fukumoto N, Kume
N, Takahashi S, Yamaguchi J, Minami M, Hayashida K, Kita T, Ohsumi
J, et al: Cell surface-anchored SR-PSOX/CXC chemokine ligand 16
mediates firm adhesion of CXC chemokine receptor 6-expressing
cells. J Leukoc Biol. 75:267–274. 2004. View Article : Google Scholar
|
|
10
|
Gough PJ, Garton KJ, Wille PT, Rychlewski
M, Dempsey PJ and Raines EW: A disintegrin and metalloproteinase
10-mediated cleavage and shedding regulates the cell surface
expression of CXC chemokine ligand 16. J Immunol. 172:3678–3685.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Abel S, Hundhausen C, Mentlein R, Schulte
A, Berkhout TA, Broadway N, Hartmann D, Sedlacek R, Dietrich S,
Muetze B, et al: The transmembrane CXC-chemokine ligand 16 is
induced by IFN-gamma and TNF-alpha and shed by the activity of the
disintegrin-like metalloproteinase ADAM10. J Immunol.
172:6362–6372. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lehrke M, Konrad A, Schachinger V, Tillack
C, Seibold F, Stark R, Parhofer IG and Broedl UC: CXCL16 is a
surrogate marker of inflammatory bowel disease. Scand J
Gastroenterol. 43:283–288. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ruth JH, Haas CS, Park CC, Amin MA,
Martinez RJ, Haines GK, Shahrara S, Campbell PL and Koch AE:
CXCL16-mediated cell recruitment to rheumatoid arthritis synovial
tissue and murine lymph nodes is dependent upon the MAPK pathway.
Arthritis Rheum. 54:765–778. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Postea O, Koenen RR, Hristov M, Weber C
and Ludwig A: Homocysteine up-regulates vascular transmembrane
chemokine CXCL16 and induces CXCR6+ lymphocyte
recruitment in vitro and in vivo. J Cell Mol Med. 12:1700–1709.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang J, Lu Y, Koch AE, Zhang J and
Taichman RS: CXCR6 induces prostate cancer progression by the
AKT/mammalian target of rapamycin signaling pathway. Cancer Res.
68:10367–10376. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lin S, Sun L, Hu J, Wan S, Zhao R, Yuan S
and Zhang L: Chemo-kine C-X-C motif receptor 6 contributes to cell
migration during hypoxia. Cancer Lett. 279:108–117. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhuge X, Murayama T, Arai H, Yamauchi R,
Tanaka M, Shimaoka T, Yonehara S, Kume N, Yokode M and Kita T:
CXCL16 is a novel angiogenic factor for human umbilical vein
endothelial cells. Biochem Biophys Res Commun. 331:1295–1300. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chandrasekar B, Bysani S and Mummidi S:
CXCL16 signals via Gi, phosphatidylinositol 3-kinase, Akt, I kappa
B kinase, and nuclear factor-kappa B and induces cell-cell adhesion
and aortic smooth muscle cell proliferation. J Biol Chem.
279:3188–3196. 2004. View Article : Google Scholar
|
|
19
|
Kwon KH, Ohigashi H and Murakami A:
Dextran sulfate sodium enhances interleukin-1 beta release via
activation of p38 MAPK and ERK1/2 pathways in murine peritoneal
macrophages. Life Sci. 81:362–371. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jiang BH, Zheng JZ, Aoki M and Vogt PK:
Phosphatidylinositol 3-kinase signaling mediates angiogenesis and
expression of vascular endothelial growth factor in endothelial
cells. Proc Natl Acad Sci USA. 97:1749–1753. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Binion DG, Otterson MF and Rafiee P:
Curcumin inhibits VEGF-mediated angiogenesis in human intestinal
microvascular endothelial cells through COX-2 and MAPK inhibition.
Gut. 57:1509–1517. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhu WH, Han J and Nicosia RF: Requisite
role of p38 MAPK in mural cell recruitment during angiogenesis in
the rat aorta model. J Vasc Res. 40:140–148. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhao R, Sun L, Lin S, Bai X, Yu B, Yuan S
and Zhang L: The saponin monomer of dwarf lilyturf tuber, DT-13,
inhibits angio-genesis under hypoxia and normoxia via
multi-targeting activity. Oncol Rep. 29:1379–1386. 2013.PubMed/NCBI
|
|
24
|
Chi JT, Wang Z, Nuyten DS, Rodriguez EH,
Schaner ME, Salim A, Wang Y, Kristensen GB, Helland Å,
Børresen-Dale AL, et al: Gene expression programs in response to
hypoxia: Cell type specificity and prognostic significance in human
cancers. PLoS Med. 3:e472006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Petering H, Kluthe C, Dulkys Y, Kiehl P,
Ponath PD, Kapp A and Elsner J: Characterization of the CC
chemokine receptor 3 on human keratinocytes. J Invest Dermatol.
116:549–555. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhou L, Sun L, Lin S, Fang D, Zhao R, Zhu
J, Liu J, Chen L, Shi W, Yuan S, et al: Inhibition of angiogenic
activity of hypoxic fibroblast cell line MRC-5 in vitro by
topotecan. Med Oncol. 28:653–659. 2011. View Article : Google Scholar
|
|
27
|
Kim HG, Hwang YP and Jeong HG:
Metallothionein-III induces HIF-1alpha-mediated VEGF expression in
brain endothelial cells. Biochem Biophys Res Commun. 369:666–671.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ahluwalia A and Tarnawski AS: Critical
role of hypoxia sensor - HIF-1α in VEGF gene activation.
Implications for angiogenesis and tissue injury healing. Curr Med
Chem. 19:90–97. 2012. View Article : Google Scholar
|
|
29
|
Zubilewicz A, Hecquet C, Jeanny JC,
Soubrane G, Courtois Y and Mascarelli F: Two distinct signalling
pathways are involved in FGF2-stimulated proliferation of
choriocapillary endothelial cells: A comparative study with VEGF.
Oncogene. 20:1403–1413. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang HQ, Bai L, Shen BR, Yan ZQ and Jiang
ZL: Coculture with endothelial cells enhances vascular smooth
muscle cell adhesion and spreading via activation of beta1-integrin
and phosphatidylinositol 3-kinase/Akt. Eur J Cell Biol. 86:51–62.
2007. View Article : Google Scholar
|
|
31
|
Gee E, Milkiewicz M and Haas TL: p38 MAPK
activity is stimulated by vascular endothelial growth factor
receptor 2 activation and is essential for shear stress-induced
angiogenesis. J Cell Physiol. 222:120–126. 2010. View Article : Google Scholar
|
|
32
|
Mavria G, Vercoulen Y, Yeo M, Paterson H,
Karasarides M, Marais R, Bird D and Marshall CJ: ERK-MAPK signaling
opposes Rho-kinase to promote endothelial cell survival and
sprouting during angiogenesis. Cancer Cell. 9:33–44. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhong H, Chiles K, Feldser D, Laughner E,
Hanrahan C, Georgescu MM, Simons JW and Semenza GL: Modulation of
hypoxia-inducible factor 1alpha expression by the epidermal growth
factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human
prostate cancer cells: Implications for tumor angiogenesis and
therapeutics. Cancer Res. 60:1541–1545. 2000.PubMed/NCBI
|
|
34
|
Treins C, Giorgetti-Peraldi S, Murdaca J,
Semenza GL and Van Obberghen E: Insulin stimulates
hypoxia-inducible factor 1 through a phosphatidylinositol
3-kinase/target of rapamycin-dependent signaling pathway. J Biol
Chem. 277:27975–27981. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Stiehl DP, Jelkmann W, Wenger RH and
Hellwig-Burgel T: Normoxic induction of the hypoxia-inducible
factor 1alpha by insulin and interleukin-1beta involves the
phosphatidylinositol 3-kinase pathway. FEBS Lett. 512:157–162.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zheng X, Ruas JL, Cao R, Salomons FA, Cao
Y, Poellinger L and Pereira T: Cell-type-specific regulation of
degradation of hypoxia-inducible factor 1 alpha: Role of
subcellular compartmentalization. Mol Cell Biol. 26:4628–4641.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lai VK, Afzal MR, Ashraf M, Jiang S and
Haider HK: Non-hypoxic stabilization of HIF-Ialpha during
coordinated interaction between Akt and angiopoietin-1 enhances
endothelial commitment of bone marrow stem cells. J Mol Med.
90:719–730. 2012. View Article : Google Scholar
|
|
38
|
Hur E, Chang KY, Lee E, Lee SK and Park H:
Mitogen-activated protein kinase kinase inhibitor PD98059 blocks
the trans-activation but not the stabilization or DNA binding
ability of hypoxia-inducible factor-1alpha. Mol Pharmacol.
59:1216–1224. 2001.PubMed/NCBI
|
|
39
|
Roos TU, Heiss EH, Schwaiberger AV,
Schachner D, Sroka IM, Oberan T, Vollmar AM and Dirsch VM: Caffeic
acid phenethyl ester inhibits PDGF-induced proliferation of
vascular smooth muscle cells via activation of p38 MAPK,
HIF-1alpha, and heme oxygenase-1. J Nat Prod. 74:352–356. 2011.
View Article : Google Scholar : PubMed/NCBI
|