Introduction
Bladder cancer is the 11th most common cancer
worldwide, accounting for an estimated 429,793 new diagnosed cases
and 165,084 deaths each year (1,2).
Bladder cancer is becoming the leading cause of cancer-related
death among all urinary malignancies (3–5).
Cigarette smoke (CS) is a well-established risk
factor for bladder cancer (3,6,7).
Zeegers et al concluded that cigarette smokers have an
approximately 3-fold higher risk of bladder cancer (8), and it has been estimated that CS
accounts for 50% of bladder cancer cases among both men and women
(9). CS contains at least 60
carcinogens such as cigarette-specific nitrosamines, specific
polycyclic aromatic hydrocarbons (PAHs), and multiple alkylated
agents which are capable of initiating tumorigenesis (10–12).
There have been many epidemiological investigations of CS, but
there is limited research on the mechanism of CS leading to bladder
cancer.
Accumulating evidence has confirmed that the
deregulation of the cell cycle in normal cells is central to cancer
initiation (13). The constituents
of CS can impact cell cycle progression and proliferation, and
accelerate tumor progression in multiple cancer types, including
bladder cancer (14,15). Ho et al demonstrated that
exposure to nicotine increased the expression of cyclin D1 and
proliferating cell nuclear antigen (PCNA) via the nuclear factor-κB
(NF-κB) signaling pathway (16).
Other researchers found that upon exposure to cigarette smoke
extract (CSE) the expression levels of p21 and p27 were
significantly reduced (17,18).
There are five members of the NF-κB family: p50/p105
(NF-κB1), p52/p100 (NF-κB2), c-Rel, RelB, and p65 (RelA). In most
cell types, NF-κB complexes exist as homodimeric and heterodimeric,
which are predominantly cytoplasmic and transcriptionally inactive
by the inhibitory protein IκB mainly IκBα. In response to
environmental stimuli, the IκB proteins then undergo rapid
ubiquitination and proteasome-mediated degradation by IκB kinases
(IKK) and releasing complex to translocate to the nucleus and alter
gene transcription (19,20). NF-κB plays an essential role in cell
proliferation and cancer development (21–24),
and regulates the expression of cell cycle genes, such as cyclin D1
and c-myc (19,25). However, little information is
available in regards to the role of NF-κB activity in CSE-induced
proliferation of normal human urothelial cells.
The present study aimed to investigate the
proliferative effect of CSE on normal human urothelial SV-HUC-1
cells. Moreover, the role of NF-κB activity in CSE-induced
proliferation in these cells was also explored.
Materials and methods
Materials
An SV-40 immortalized human urothelial cell line
(SV-HUC-1) was purchased from the Chinese Academy of Typical
Culture Collection Cell Bank. F12K medium was purchased from Gibco
(New York, NY, USA). Fetal bovine serum (FBS) was obtained from PAA
Laboratories (Pasching, Austria).
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
was purchased from Sigma-Aldrich. p-IKKα/β, IκB, p65, p21, cyclin
D1, PCNA antibodies were all purchased from Cell Signaling
Technology (Beverly, MA, USA). P50 was purchased from Santa Cruz
Biotechnology (Santa Cruz, CA, USA). Primers were synthesized
according to published sequences from Invitrogen (Carlsbad, CA,
USA). Ammonium pyrrolidinedithiocarbamate (PDTC) was purchased from
Beyotime (Shanghai, China). A nuclear and cytoplasmic protein
extraction kit was purchased from KeyGen (Nanjing, China). Sources
of other materials are noted accordingly in the text.
Cell culture and treatment
The SV-HUC-1 cells were cultured in F12K medium with
10% FBS and antibiotics (100 U/ml penicillin and 100
µg/ml streptomycin) in a humidified atmosphere of 5%
CO2 at 37°C. The medium was changed every other day.
When the cells reached 80–90% confluency, they were treated with
various concentrations of cigarette smoke extract (CSE) or with
PDTC (2 µM) for 7 days.
Preparation of the cigarette smoke
extract
CSE was prepared daily immediately before use
according to a previously reported method (26). Briefly, one filterless 3R4F
reference cigarette (12 mg tar and 1.0 mg nicotine/cigarette) was
combusted, and the mainstream smoke was continuously drawn through
a glass syringe containing 10 ml of FBS-free F12K that was
pre-warmed to 37°C at a rate of 5 min/cigarette. The resulting
suspension was adjusted to pH 7.4 and then filtered through a
0.22-µm-pore size filter. The obtained solution was referred
to as a 100% CSE solution and was further diluted to the desired
concentration with culture medium. Control solution was prepared
using the same protocol, except that the cigarette was unlit.
SV-HUC-1 cells were exposed to various concentrations of CSE within
30 min of preparation.
MTT assay
Briefly, SV-HUC-1 cells (1×103/well) were
seeded in 96-well plates with F12K medium containing 10% FBA for up
to 24 h. The cells were treated with different concentrations of
CSE for 7 days. Cells viability was measured by MTT assay. Twenty
microliters of MTT solution (5 mg/ml) was added to each well, and
the plates were further incubated for 4 h at 37°C. Then the MTT was
removed and the precipitants were solubilized in DMSO. Absorbance
was measured at 490 nm using a microplate reader. All measurements
were performed in triplicate.
Cell cycle analysis
The distribution of SV-HUC-1 cells at different
stages in the cell cycle was calculated by flow cytometric
analysis. Briefly, we plated 1×106 cells on 6-cm dishes
for 24 h. Then the cells were incubated at 37°C overnight. SV-HUC-1
cells were treated with 0, 0.1, 0.25, and 0.5% CSE for 7 days and
then trypsinized and washed twice with cold PBS and mixed with 75%
ethanol at 4°C overnight. Then the cells were centrifuged and
resuspended in 500 µl PBS. After addition of 10 µl
RNAse (10 mg/ml), the cells were left for 30 min at 37°C in the
dark and stained with 10 µl propidium iodide (1 mg/ml). The
cells were immediately evaluated by flow cytometry. The percentages
of cells in each cell cycle phase (G0/G1, S, G2/M) were measured
using Multicycle V.3.0 software, with a minimum of 1×105
cells/sample being evaluated.
Quantitative reverse
transcription-polymerase chain reaction (qRT-PCR)
For RNA analysis, 1×106 cells per dish
were plated in 6-cm plastic tissue culture dishes for 24 h. Then
SV-HUC-1 cells were either exposed to different concentrations of
CSE, or were pretreated for 30 min with PDTC (2 µM) before
exposure to CSE for 7 days. Finally, the cells were harvested, and
total RNA was extracted using TRIzol reagent (Invitrogen) according
to the manufacturer's protocol. Total RNA was reverse-transcribed
using the Easy RT-PCR kit according to the manufacturer's
instructions. GAPDH was analyzed in parallel as an internal
control. Real-time PCR assays were performed using Power SYBR Green
Master Mix and an ABI 7300 real-time PCR detection system (both
from Applied Biosystems, USA). The primer sequences used for the
RT-PCR were: cyclin D1 forward, 5′-CGTGGCCTCTAAGATGAAGG-3′ and
reverse, 5′-TGCGGATGATCTGTTTGTTC-3′; p21 forward,
5′-GACACCACTGGAGGGTGACT-3′ and reverse, 5′-CAGGTCCACATGGTCTTCCT-3′;
and GAPDH forward, 5′-GCTGCCCAACGCACCGAATA-3′ and reverse
5′-GAGTCAACGGATTTGGTCGT-3′. All of the primers were synthesized by
RiboBio (Guangzhou, China). Fold changes in expression of each gene
were calculated by a comparative threshold cycle (Ct) method using
the formula 2−(ΔΔCt).
Preparation of nuclear and cytoplasmic
protein extraction
The NF-κB activity in the nuclear and cytoplasmic
fractions was determined using the KeyGen kit (KeyGen, Nanjing,
China) according to the instructions of the manufacturer. Briefly,
cultured cells were collected by centrifugation, washed in cold PBS
three times and suspended in 100 µl of buffer A (containing
DTT 1 mM and PMSF 0.5 mM) and incubated on ice for 15 min. Then 5
µl of buffer was added to the suspension and briely
vortexed. Following this, the nuclear proteins were pelleted by
centrifugation at low speed. The (cytoplasmic extract) supernatant
was collected and stored at −20°C. The nuclear pellet was
resuspended in 50 µl of buffer C (containing DTT 1 mM and
PMSF 0.5 mM). The suspension was incubated for 30 min at 4°C and
volatility every 10 min to 15 sec followed by centrifugation at
16,000 × g for 10 min. The supernatant containing the nuclear
protein extract was transferred to a fresh microcentrifuge tube and
stored at −20°C.
Western blot analysis
Western blot analyses were used for assessment of
the protein levels of NF-κB p65, p50, IκBα, p-IKKα/β, cyclin D1,
p21 and PCNA. Cytosolic or nuclear proteins (40–60 µg) were
loaded and separated on 10% SDS-PAGE and were transferred to PVDF
membranes (Millipore, Billerica, MA, USA). After blocking with 5%
milk, the membranes were subsequently probed with primary
antibodies at a dilution of 1:500 for 12 h at 4°C, and then
incubated with the secondary antibodies at a dilution of 1:1,000.
Finally, the membranes were developed using an enhanced
chemiluminescence detection kit (Amersham Biosciences, USA) and
exposed to film. GAPDH was used as a loading control.
Statistical analysis
All data are presented as mean ± standard deviation.
Statistical analyses were performed by one-way analysis of
variance, using SPSS 17.0. A value of P<0.05 was considered
statistically significant, All of the experiments were performed in
triplicate and performed at least thrice to confirm their
reproducibility.
Results
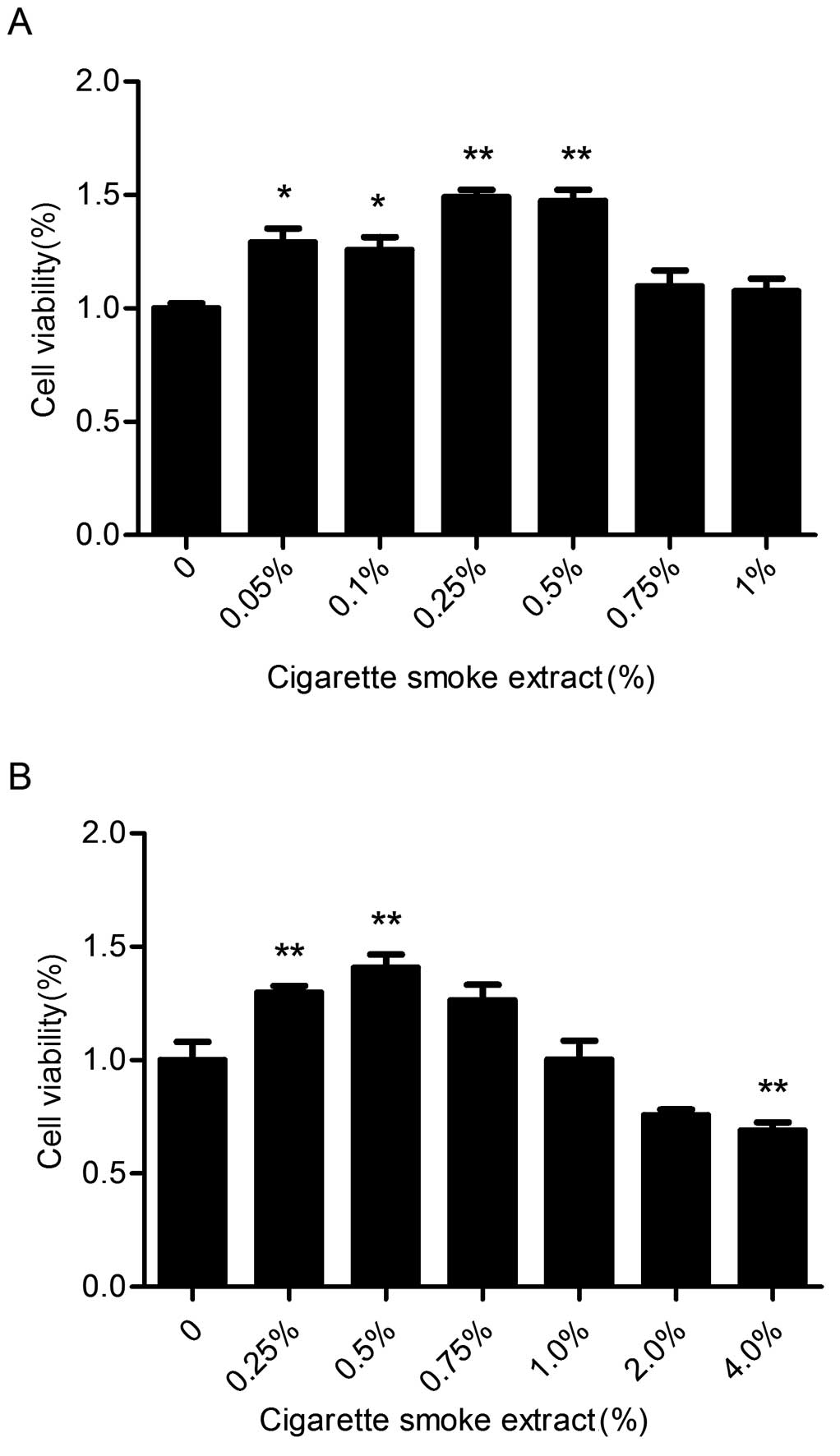
CSE induces the proliferation of SV-HUC-1
cells
The growth of normal bladder epithelial cells
(SV-HUC-1) was detected by MTT assay at various concentrations of
CSE (0, 0.05, 0.1, 0.25, 0.5, 0.75, 1.0, 2.0 and 4.0%) for 7 days.
As shown in Fig. 1, CSE caused a
significant increase in cell viability at concentrations from 0.05
to 0.5%. Therefore, CSE concentrations of 0.1, 0.25, and 0.5% were
selected in our following experiments.
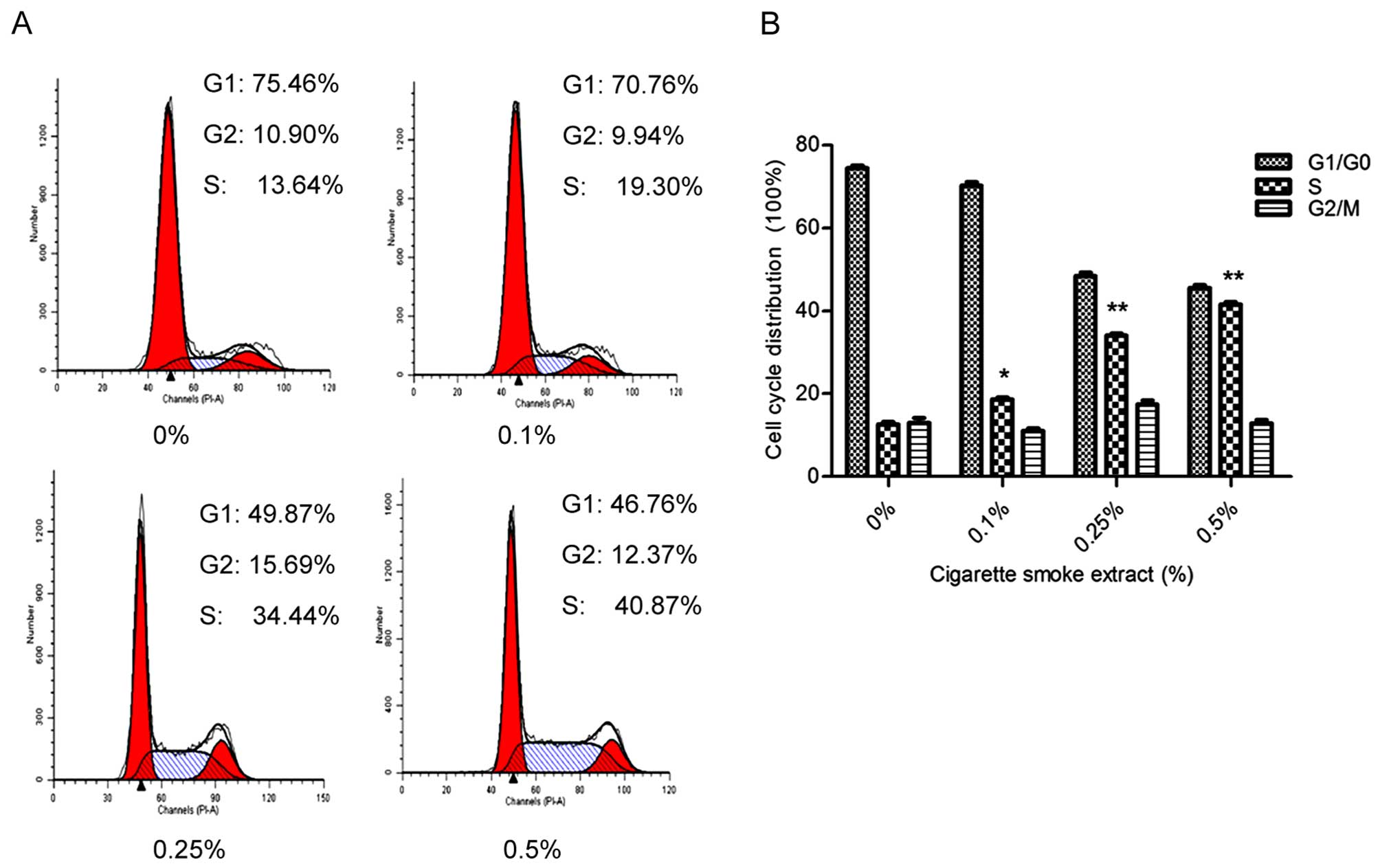
CSE triggers cell cycle progression in
SV-HUC-1 cells
In order to investigate the effect of CSE on the
cell cycle, we used flow cytometry to analyze the proportion of
cells in each phase. As shown in Fig.
2A and B, flow cytometry showed that treatment with 0.5% CSE
accelerated G1 to S phase progression, with 40.87% of cells
entering into the S phase, which was significantly higher than that
of the control value (13.86%). The effect was in a dose-related
manner. The results demonstrated that exposure of SV-HUC-1 cells to
CSE induced proliferation by modulating cell cycle distribution. As
shown in Fig. 2C, western blot
analysis showed that CSE exposure increased the cyclin D1 and PCNA
protein expression level and decreased the p21 protein expression
level. As shown in Fig. 2D, results
of qRT-PCR revealed that CSE exposure enhanced the level of mRNA
expression of cyclin D1, while the expression level of p21 was
significantly decreased.
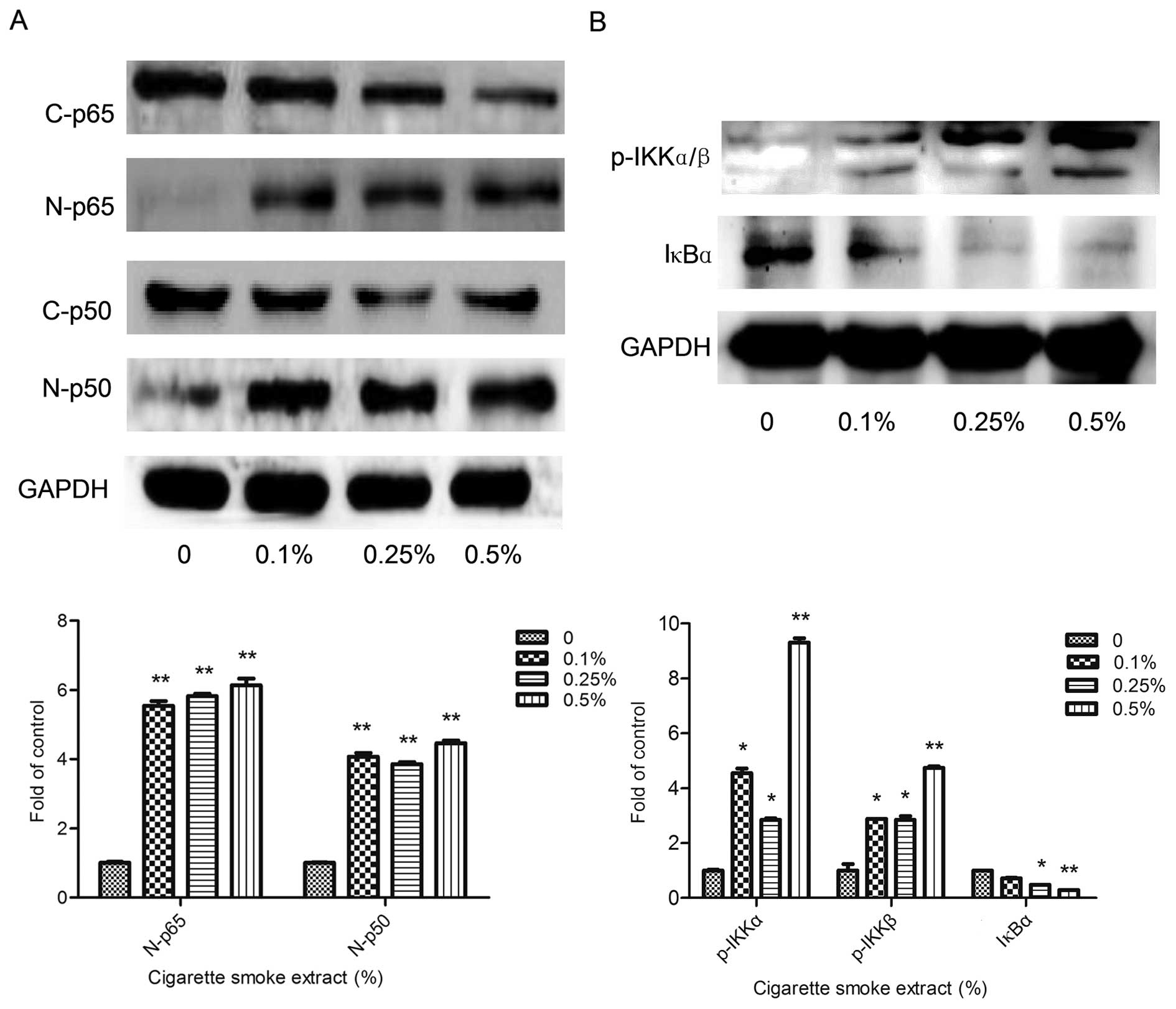
CSE activates the NF-κB pathway in
SV-HUC-1 cells
To determine whether the CSE-induced proliferation
is correlated with the NF-κB pathway, we examined the protein
levels of p65 and p50 in the nuclear extract of the SV-HUC-1 cells
(Fig. 3A). The nuclear protein
levels of both p65 and p50 were significantly increased.
Furthermore, we also examined the upstream of the NF-κB pathway. As
shown in Fig. 3B, the expression of
IκB kinase (p-IKKα/β) was clearly increased in the cytoplasm, and
IκB was subsequently degradation and the expression was obviously
reduced.
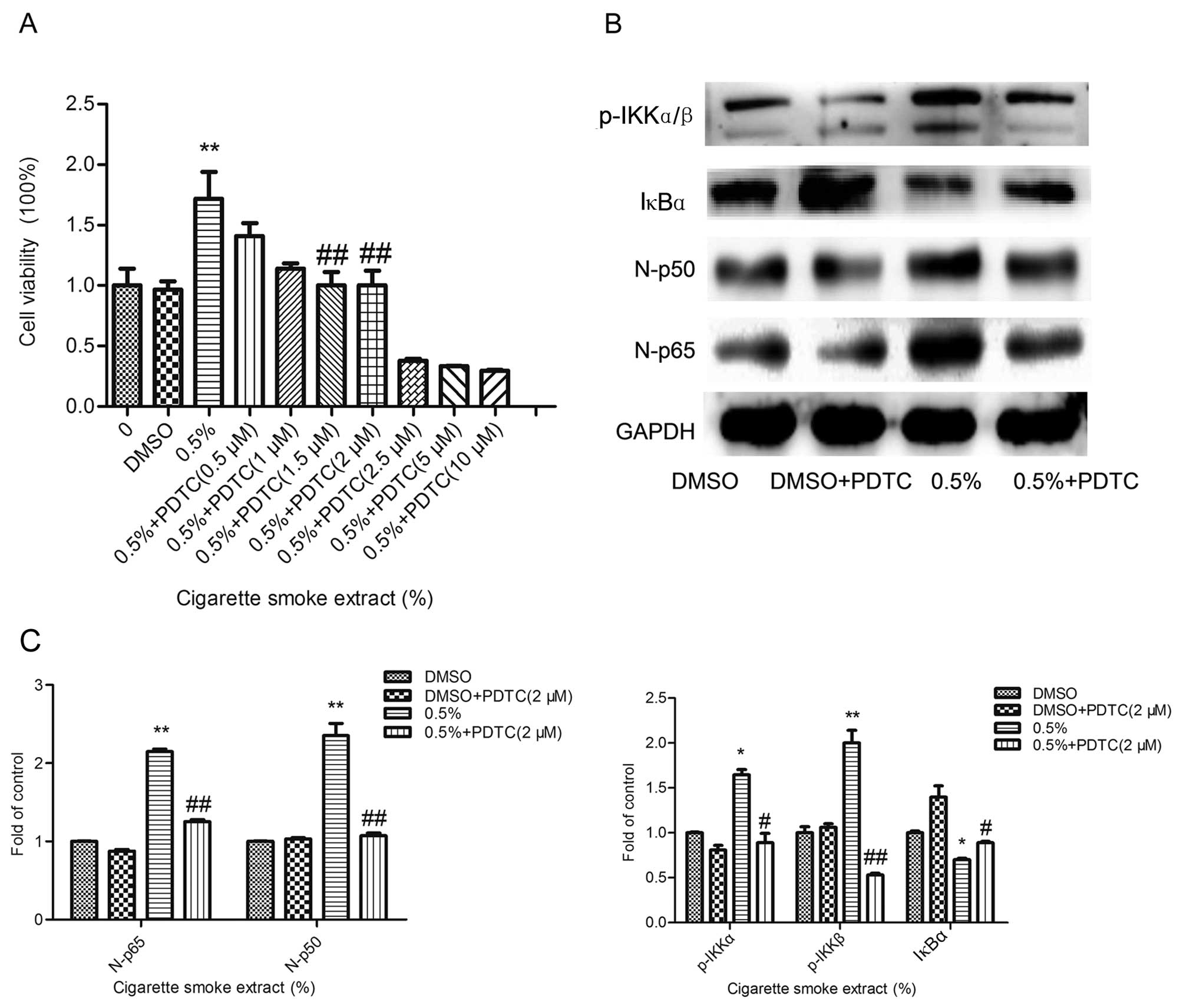
Inhibition of NF-κB reverses CSE-induced
SV-HUC-1 cell proliferation
To further determine the effect of the altered NF-κB
signaling pathway activity on CSE-induced SV-HUC-1 cell
proliferation, the inhibition of the NF-κB pathway with ammonium
pyrrolidinedithiocarbamate (PDTC, 2 µM), a NF-κB-specific
inhibitor was used to pretreat the cells. As shown in Fig. 4A, an MTT assay was executed to test
the cell viability and the inhibitory effect of the inhibitor. As
shown in Fig. 4B and C, the western
blot results showed that the activities of p65 and p50 proteins
were significantly reduced. This confirmed that the NF-κB pathway
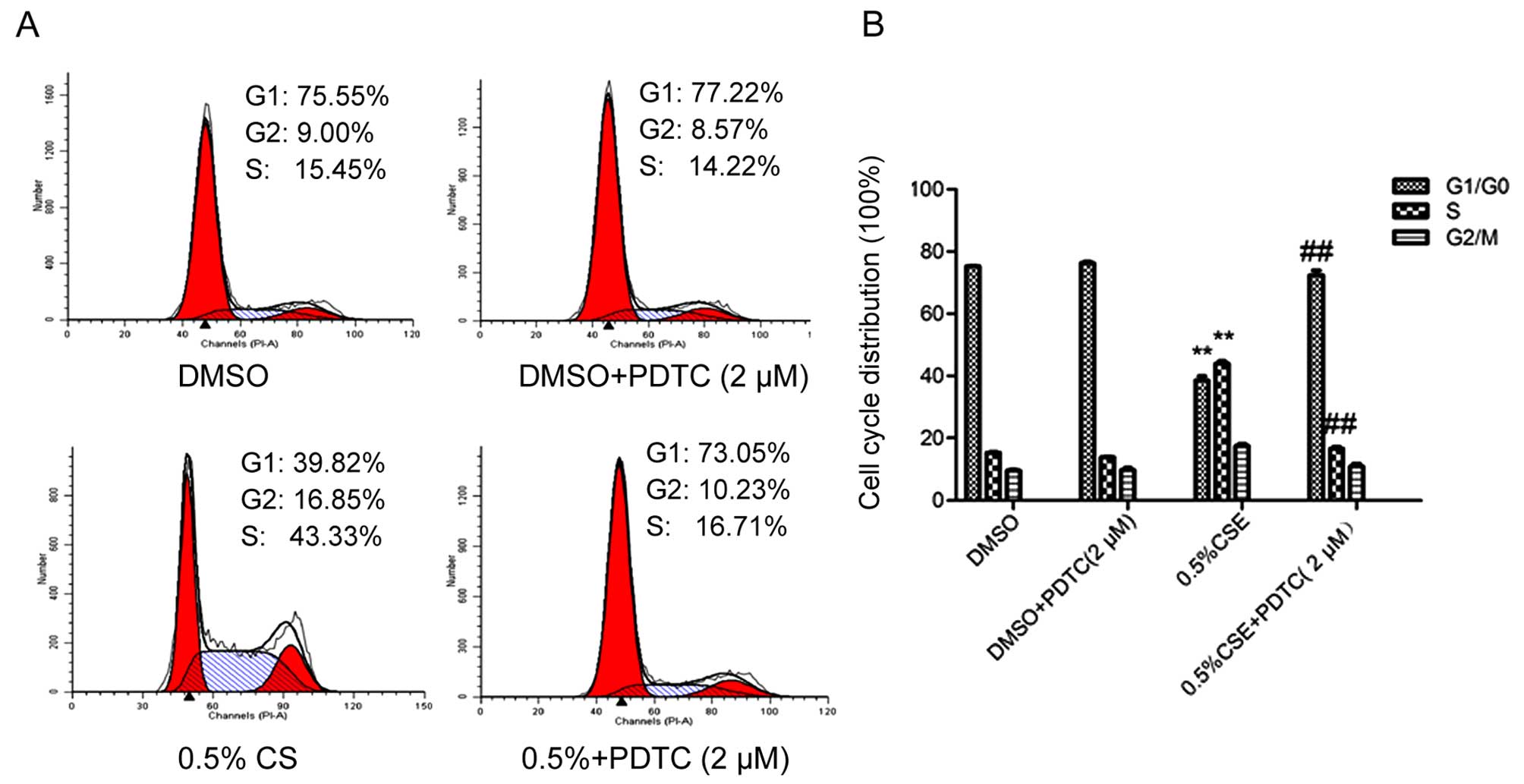
was effectively inhibited. As shown in Fig. 5A and B, flow cytometry revealed that
inhibition of the NF-κB pathway decreased the percentage (16.71%)
of cells in the S phase as compared with the control (43.33%). As
shown in Fig. 5C, cyclin D1 and
PCNA protein levels were downregulated and accordingly the effect
on p21 was reversed, when activation of the NF-κB pathway was
inhibited. In addition, as shown in Fig. 5D, qRT-PCR showed that the cyclin D1
mRNA level was decreased following inhibition of the NF-κB pathway,
consistent with the change in the protein level. Inhibition of the
NF-κB pathway reversed the effect of CSE on the SV-HUC-1 cells.
These data suggest that the NF-κB pathway plays an important role
in CSE-induced proliferation of normal human urothelial SV-HUC-1
cells.
Discussion
The hypothesis that CS is closely related to the
development of bladder cancer is supported by studies with rodents,
non-human primates and human cancer cell lines (8–12). The
underlying molecular mechanisms by which CS causes bladder cancer
development remain to be established. In the present study, we
revealed that exposure to CSE induced cell proliferation in normal
human urothelial SV-HUC-1 cells. Most importantly, we showed that
CSE-induced cell proliferation was reversed by NF-κB inhibition.
These data suggest the important role of NF-κB activity in
CSE-associated cell proliferation and provide critical information
in regards to the molecular mechanisms of CSE-related bladder
tumorigenesis as well as the identification of a potential target
of bladder cancer intervention.
Dysregulation of the cell cycle and unlimited
proliferation play an important role in initiation of tumorigenesis
(28,29). Evidence has revealed that exposure
of cells to carcinogens induce dysregulation of the cell cycle
(14,30,31),
suggesting the important role of dysregulation of the cell cycle in
the initiation of tumorigenesis. Schaal and Chellappan demonstrated
that exposure of non-neuronal cells to tobacco carcinogen nicotine
induced dysregulation of the cell cycle, and early precancerous
lesions during the tumorigenic process, participating in cancer
initiation and promoting the clonal expansion of premalignant cells
(14). It has been documented that
CS promoted the progression of the cell cycle, resulting in loss of
growth inhibition which increased the potential of limitless
replication (32–34). In agreement with previous studies,
we showed in the present study that exposure of SV-HUC-1 cells to
CSE induced proliferation by modulating cell cycle distribution.
Compared with the control group (13.64%), CSE induced cell
proliferation as determined by MTT assay and the percentage of
cells in the S phase (40.87%) was increased significantly in the
CSE-treated group. Moreover, we also showed that CSE exposure
resulted in increased expression of PCNA and cyclin D1, as well as
decreased expression of p21 in normal human urothelial SV-HUC-1
cells, suggestive of proliferation induction. Take together; our
data revealed that CSE triggered proliferation in normal human
urothelial cells.
To explore the molecular mechanisms of CSE-induced
proliferation, the role of the NF-κB pathway in regulating
CSE-mediated cell proliferation was investigated in our study. A
large number of reports have demonstrated that NF-κB regulates cell
proliferation (35–38). Many studies revealed that NF-κB can
directly regulate cell cycle genes and trigger the expression of
proliferation-related proteins, such as cyclin D1, PCNA and p21
(39,40). Cyclin D1 forms active holoenzymes
with CDK4 or CDK6 by phosphorylating the retinoblastoma protein
(pRB) to play an important role in regulating transition from the
G1 phase to the S phase in the cell cycle (41,42).
Cyclin D1 expression is regulated by NF-κB (42). Proliferating cell nuclear antigen
(PCNA) plays a crucial role in modulating DNA replication and cell
proliferation in AML cells (44).
Moreover, PCNA can also serve as a potential chemotherapy target
(45). The cyclin dependent kinase
inhibitor p21 is a negative regulator of the cell cycle through
inactiving Cdks and directly interacts with PCNA to inhibit
replication. A close relationship between NF-κB and p21 has been
demonstrated (46). In line with
these previous studies, we found that CSE-induced NF-κB activation
was associated with downregulation of p21, and upregulation of
cyclin D1 and PCNA. In addition, we also showed in the present
study that CSE increased the levels of phosphorylated IKK and
reduced the IκB level, suggesting activation of the NF-κB pathway
by CSE. Meanwhile, we found that inhibition of NF-κB activity
reversed CSE-induced proliferation in SV-HUC-1 cells. Collectively,
our data indicated that the NF-κB pathway positively regulates
CSE-induced proliferation in SV-HUC-1 cells.
In summary, we demonstrated that exposure of normal
human urothelial SV-HUC-1 cells to cigarette smoke extract induced
cell proliferation. Moreover, NF-κB activity played an important
role in CSE-triggered cell proliferation. The findings of this
study provide new insight into the molecular mechanisms of
cigarette smoke-related bladder cancer carcinogenesis.
Abbreviations:
|
SV-HUC-1
|
SV-40 immortalized human uroepithelial
cell line
|
|
CS
|
cigarette smoke
|
|
CSE
|
cigarette smoke extract
|
|
MTT
|
thiazolyl blue tetrazolium bromide
|
|
RT-PCR
|
reverse transcription-polymerase chain
reaction
|
Acknowledgments
This study was supported by grants from the National
Natural Science Foundation of China (nos. 81373005, 81072330, and
81202194) and by the Priority Academic Program Development of
Jiangsu Higher Education Institutions (PAPD).
References
|
1
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: GLOBOCAN 2008, Cancer incidence and
mortality worldwide. (IARC CancerBase No. 10. [Internet]).
International Agency for Research on Cancer; Lyon, France: 2010,
Available from http://globocan.iarc.fr.
Accessed May 6, 2012.
|
|
2
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar
|
|
3
|
Burger M, Catto JW, Dalbagni G, Grossman
HB, Herr H, Karakiewicz P, Kassouf W, Kiemeney LA, La Vecchia C,
Shariat S, et al: Epidemiology and risk factors of urothelial
bladder cancer. Eur Urol. 63:234–241. 2013. View Article : Google Scholar
|
|
4
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Letašiová S, Medve'ová A, Šovčíková A,
Dušinská M, Volkovová K, Mosoiu C and Bartonová A: Bladder cancer,
a review of the environmental risk factors. Environ Health.
11(Suppl 1): S112012. View Article : Google Scholar
|
|
7
|
Boffetta P: Tobacco smoking and risk of
bladder cancer. Scand J Urol Nephrol Suppl. 218:45–54. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zeegers MP, Tan FE, Dorant E and van Den
Brandt PA: The impact of characteristics of cigarette smoking on
urinary tract cancer risk: A meta-analysis of epidemiologic
studies. Cancer. 89:630–639. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Freedman ND, Silverman DT, Hollenbeck AR,
Schatzkin A and Abnet CC: Association between smoking and risk of
bladder cancer among men and women. JAMA. 306:737–745. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hecht SS: Tobacco smoke carcinogens and
lung cancer. J Natl Cancer Inst. 91:1194–1210. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Phillips DH: Smoking-related DNA and
protein adducts in human tissues. Carcinogenesis. 23:1979–2004.
2002. View Article : Google Scholar
|
|
12
|
Hecht SS: Tobacco carcinogens, their
biomarkers and tobacco-induced cancer. Nat Rev Cancer. 3:733–744.
2003. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sobus SL and Warren GW: The biologic
effects of cigarette smoke on cancer cells. Cancer. 120:3617–3626.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Schaal C and Chellappan SP:
Nicotine-mediated cell proliferation and tumor progression in
smoking-related cancers. Mol Cancer Res. 12:14–23. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cardinale A, Nastrucci C, Cesario A and
Russo P: Nicotine: Specific role in angiogenesis, proliferation and
apoptosis. Crit Rev Toxicol. 42:68–89. 2012. View Article : Google Scholar
|
|
16
|
Ho YS, Chen CH, Wang YJ, Pestell RG,
Albanese C, Chen RJ, Chang MC, Jeng JH, Lin SY and Liang YC:
Tobacco-specific carcinogen
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) induces cell
proliferation in normal human bronchial epithelial cells through
NFkappaB activation and cyclin D1 up-regulation. Toxicol Appl
Pharmacol. 205:133–148. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shin VY, Jin HC, Ng EK, Yu J, Leung WK,
Cho CH and Sung JJ: Nicotine and
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone induce
cyclooxygenase-2 activity in human gastric cancer cells:
Involvement of nicotinic acetylcholine receptor (nAChR) and
beta-adrenergic receptor signaling pathways. Toxicol Appl
Pharmacol. 233:254–261. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Norton JD: ID helix-loop-helix proteins in
cell growth, differentiation and tumorigenesis. J Cell Sci.
113:3897–3905. 2000.PubMed/NCBI
|
|
19
|
Aggarwal BB: Nuclear factor-kappaB: The
enemy within. Cancer Cell. 6:203–208. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Orlowski RZ and Baldwin AS Jr: NF-kappaB
as a therapeutic target in cancer. Trends Mol Med. 8:385–389. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bassères DS and Baldwin AS: Nuclear
factor-kappaB and inhibitor of kappaB kinase pathways in oncogenic
initiation and progression. Oncogene. 25:6817–6830. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hoesel B and Schmid JA: The complexity of
NF-κB signaling in inflammation and cancer. Mol Cancer. 12:862013.
View Article : Google Scholar
|
|
23
|
Lin Y, Bai L, Chen W and Xu S: The
NF-kappaB activation pathways, emerging molecular targets for
cancer prevention and therapy. Expert Opin Ther Targets. 14:45–55.
2010. View Article : Google Scholar
|
|
24
|
Dobrovolskaia MA and Kozlov SV:
Inflammation and cancer: When NF-kappaB amalgamates the perilous
partnership. Curr Cancer Drug Targets. 5:325–344. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Guttridge DC, Albanese C, Reuther JY,
Pestell RG and Baldwin AS Jr: NF-kappaB controls cell growth and
differentiation through transcriptional regulation of cyclin D1.
Mol Cell Biol. 19:5785–5799. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gál K1, Cseh A, Szalay B, Rusai K, Vannay
A, Lukácsovits J, Heemann U, Szabó AJ, Losonczy G, Tamási L and
Müller V: Effect of cigarette smoke and dexamethasone on Hsp72
system of alveolar epithelial cells. Cell Stress Chaperones.
16:369–378. 2011. View Article : Google Scholar :
|
|
27
|
Alguacil J, Kogevinas M, Silverman DT,
Malats N, Real FX, García-Closas M, Tardón A, Rivas M, Torà M,
García-Closas R, et al: Urinary pH, cigarette smoking and bladder
cancer risk. Carcinogenesis. 32:843–847. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Baldi A, De Luca A, Esposito V, Campioni
M, Spugnini EP and Citro G: Tumor suppressors and cell-cycle
proteins in lung cancer. Patholog Res Int.
2011:6050422011.PubMed/NCBI
|
|
29
|
Crawford JM: The origins of bladder
cancer. Lab Invest. 88:686–693. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li X, Xie W, Xie C, Huang C, Zhu J, Liang
Z, Deng F, Zhu M, Zhu W, Wu R, et al: Curcumin modulates
miR-19/PTEN/AKT/p53 axis to suppress bisphenol A-induced MCF-7
breast cancer cell proliferation. Phytother Res. 28:1553–1560.
2014. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bishop RK, Valle OC and Spencer JV: Human
cytomegalovirus interleukin-10 promotes proliferation and migration
of MCF-7 breast cancer cells. Cancer Cell Microenviron.
2:e6782015.PubMed/NCBI
|
|
32
|
Li T, Song T, Ni L, Yang G, Song X, Wu L,
Liu B and Liu C: The p-ERK-p-c-Jun-cyclinD1 pathway is involved in
proliferation of smooth muscle cells after exposure to cigarette
smoke extract. Biochem Biophys Res Commun. 453:316–320. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Massagué J: G1 cell-cycle control and
cancer. Nature. 432:298–306. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yao J, Zhao L, Zhao Q, Zhao Y, Sun Y,
Zhang Y, Miao H, You QD, Hu R and Guo QL: NF-κB and Nrf2 signaling
pathways contribute to wogonin-mediated inhibition of
inflammation-associated colorectal carcinogenesis. Cell Death Dis.
5:e12832014. View Article : Google Scholar
|
|
36
|
Alvira CM: Nuclear factor-kappa-B
signaling in lung development and disease: One pathway, numerous
functions. Birth Defects Res A Clin Mol Teratol. 100:202–216. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li J, Yan M, Wang Z, Jing S, Li Y, Liu G,
Yu J and Fan Z: Effects of canonical NF-κB signaling pathway on the
proliferation and odonto/osteogenic differentiation of human stem
cells from apical papilla. Biomed Res Int. 2014:3196512014.
|
|
38
|
Perkins ND: The diverse and complex roles
of NF-κB subunits in cancer. Nat Rev Cancer. 12:121–132.
2012.PubMed/NCBI
|
|
39
|
Helisch A, Förster GJ, Reber H, Buchholz
HG, Arnold R, Göke B, Weber MM, Wiedenmann B, Pauwels S, Haus U, et
al: Pre-therapeutic dosimetry and biodistribution of
86Y-DOTA-Phe1-Tyr3-octreotide versus 111In-pentetreotide in
patients with advanced neuroendocrine tumours. Eur J Nucl Med Mol
Imaging. 31:1386–1392. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kim HJ, Hawke N and Baldwin AS: NF-kappaB
and IKK as therapeutic targets in cancer. Cell Death Differ.
13:738–747. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhang XY, Xu YJ, Liu XS and Zhang ZX:
Cigarette smoke extract promotes proliferation of airway smooth
muscle cells in asthmatic rats via regulating cyclin D1 expression.
Chin Med J (Engl). 123:1709–1714. 2010.
|
|
42
|
Musgrove EA, Caldon CE, Barraclough J,
Stone A and Sutherland RL: Cyclin D as a therapeutic target in
cancer. Nat Rev Cancer. 11:558–572. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hinz M, Krappmann D, Eichten A, Heder A,
Scheidereit C and Strauss M: NF-kappaB function in growth control:
Regulation of cyclin D1 expression and G0/G1-to-S-phase transition.
Mol Cell Biol. 19:2690–2698. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Villar HO, Uyeno ET, Toll L, Polgar W,
Davies MF and Loew GH: Molecular determinants of benzodiazepine
receptor affinities and anticonvulsant activities. Mol Pharmacol.
36:589–600. 1989.PubMed/NCBI
|
|
45
|
Morgan RG, Ives SJ, Lesniewski LA, Cawthon
RM, Andtbacka RH, Noyes RD, Richardson RS and Donato AJ:
Age-related telomere uncapping is associated with cellular
senescence and inflammation independent of telomere shortening in
human arteries. Am J Physiol Heart Circ Physiol. 305:H251–H258.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chung S, Sundar IK, Hwang JW, Yull FE,
Blackwell TS, Kinnula VL, Bulger M, Yao H and Rahman I: NF-κB
inducing kinase, NIK mediates cigarette smoke/TNFα-induced histone
acetylation and inflammation through differential activation of
IKKs. PLoS One. 6:e234882011. View Article : Google Scholar
|