Introduction
Liver cancer has high incidence worldwide, and
especially in China (1).
Hepatocellular carcinoma represents 70–90% of primary liver cancers
occurring worldwide, it is highly aggressive with poor prognosis
resulting in high mortality. Although the risk of liver cancer has
been reduced with the use of antiviral treatment, they are costly
and not in wide use (2,3), there are no well-established effective
adjuvant therapies for hepatocellular carcinoma (4). Recent advances in understanding the
molecular pathogenesis of hepatocellular carcinoma have shown great
promise to develop more effective treatment for hepatocellular
carcinoma (5,6). For this purpose, researchers have
focused on the mechanism of hepatocellular carcinoma progression
and metastasis, to identify better molecular targets and
agents.
It has demonstrated that multiple steps are involved
in the process of cancer progression and metastasis (7–9).
Metastasis, one of the most complicated and major pathologic
processes, is responsible for poor prognosis of cancer patients.
Transforming growth factor β1 (TGF-β1) is one of the important
cytokines involved. By stimulation of TGF-β, hepatocellular
carcinoma HepG2 cells showed morphological, molecular and
functional changes, including the formation of spindle shape, the
loss of cell contact, and the downregulation of E-cadherin
expression, but upregulation of Vimentin expression, showing a
typical epithelial-mesenchymal transition (EMT) (10,11).
EMT plays an important role in various cancer metastasis, and is
thought to be a prerequisite for tumor cell invasion and metastasis
(12–15). Although increased evidence shows
that TGF-β could induce the EMT of various cancer cells and that
EMT is pivotal for controlling tumor invasiveness and metastasis,
the regulation mechanism of EMT is still unclear (16). Studies of the regulation mechanism
of EMT are important for developing more effective anti-metastatic
strategies.
Sulforaphane (SFN), a naturally occurring
isothiocyanate derived from cruciferous vegetables, especially
broccoli, which has been widely used for treatment of inflammatory
diseases, shows antitumor effects in vitro and in
vivo studies (17–20). Increased evidence shows that SFN is
an important candidate cancer preventive agent with high activity
in diverse cancers, including thyroid cancer (17), breast cancer (19), colon cancer (21,22),
prostate cancer (18), bladder
cancer (23,24) and leukemia (25). Mechanically, it was demonstrated
that SFN inhibited thyroid cancer cell growth and invasiveness,
promoting mitochondrial-mediated apoptosis via reactive oxygen
species (ROS)-dependent pathway (1). SFN induces apoptosis in bladder cancer
cells through a ROS-mediated mitochondrial pathway (23,24).
Although the antitumor effect of SFN on hepatocellular carcinoma
remains unknown, ROS has been also reported to mediate many
cellular effects of hepatoma cells, including the EMT of HepG2
cells induced by the tumor promoter 12-O-tetradecanoyl
phorbol-13-acetate (26).
The purpose of this study was to evaluate the effect
of SFN on cell apoptosis, migration and invasion of hepatocellular
carcinoma, as well as EMT induced by TGF-β. We also tested whether
ROS involved in the antitumor mechanisms of SFN by the ROS
scavenger N-acetyl-L-cysteine (NAC) and to test its in vivo
therapeutic potential in hepatocellular carcinoma.
Materials and methods
Cell culture
Human hepatocellular carcinoma cell line HepG2 was
obtained from ATCC (Rockville, MD, USA). Cells were cultured in
RPMI-1640 medium with 10% fetal bovine serum (FBS) at 37°C in a
humidified atmosphere containing 5% CO2 and experiments
were done using 70–80% confluent cultures.
Cell proliferation assay
HepG2 cell survival was evaluated using the MTT
(3-(4,5-dimethyl-thiazol-2-y1) 2,5-diphenyl-tetrazolium bromide)
(Sigma, USA) colorimetric assay. After different doses (vehicle,
10, 20, 40, 60 and 80 µM) of SFN treatment for 48 h, MTT
assay was performed on hepatocellular carcinoma cell line HepG2 to
evaluate cell growth ability and to calculate the 50% inhibitory
concentration of SFN. Then, the cell proliferation in the presence
of indicated SFN (IC50) or vehicle control for indicated
time-points. In brief, prior to the treatment, HepG2 cells were
seeded in 96-well tissue culture plates at 2×104 cells
per well. After treatment, cells were washed with PBS and incubated
in 100 µl of 5 mg/ml MTT solution (Invitrogen Inc., USA) for
3 h. MTT is converted into purple colored formazan in living cells
which was then solubilized with dimethylsulfoxide (DMSO)
(Invitrogen Inc.) and absorbance of solution was evaluated at 450
nm using the microplate reader Thermo Plate (Rayto Life and
Analytical Science C. Ltd., Germany).
Cell cycle analysis
After treatment, cells were harvested, trypsinized
and rinsed 3 times with buffer solution with adjusted concentration
of 1×106 cells/ml and prepared using Cycletest™ Plus DNA
Reagent kit (Becton-Dickinson, USA) according to the manufacturer's
instructions. Cell cycle status was analyzed by flow cytometer
using propidium iodide (PI) as a specific fluorescent dye probe.
The PI fluorescence intensity of 10,000 cells was measured for each
sample using a Becton-Dickinson FACSCalibur flow cytometer.
Cell apoptosis assay
Cell apoptosis was determined by Annexin V assay.
After transfection for 48 h, cells were harvested by trypsinization
and washed with PBS, and suspended in Annexin V binding buffer.
FITC-conjugated Annexin V and propidium iodide (PI; Beyotime,
China) were added to the cells successively. After incubation,
Annexin V binding buffer was added, and cells were analyzed by a
FACScan (Becton-Dickinson) flow cytometry. Annexin V(+)/P(−) and
Annexin V(+)/P(+) represent the cells in early apoptosis and late
apoptosis/necrosis, respectively.
Cell migration and invasion assay
Cell migration and invasion were assayed using a
Transwell chamber (Millipore, USA). For the invasion assay,
Transwell chamber was coated with 30 µl Matrigel and was
placed into 24-well plate and incubated for 30 min at 37°C. After
48 h of transfection, HepG2 cells were trypsinized and seeded in
chambers at the density of 8×104 cells per well and
cultured in medium with RPMI-1640 medium with 2% serum, while 600
µl of 10% FBS-RPMI-1640 with or without TGF-β (10 ng/ml) was
added to the lower chamber. Twenty-four hours later, migrated cells
were fixed with 100% methanol for 30 min and stained by crystal
violet for 20 min. Non-migrated cells were removed by cotton swabs.
Cell images were obtained under a phase-contrast microscope
(Olympus, Tokyo, Japan).
Electron microscopy
Morphological changes in cells were emulated by
scanning electron microscopy. Cells were grown on plastic cover
slips. After indicated treatments, fixed in 2.5% glutaraldehyde in
phosphate-buffered saline (PBS, pH 7.4) for 2 h, rinsed with PBS,
and dehydrated through graded ethanol (30, 50, 70, 80, 90 and 100%
for 20 min each). Cells were transferred to amylacetate for 10 min,
critical point dried, and then coated with gold. Cells were viewed
in a scanning electron microscope (Olympus).
Western blotting
Western blotting was applied to detect markers of
EMT at protein level. After indicated treatments, HepG2 cells
cultured on the glass slide were washed twice in ice-cold PBS, and
then lysed by RIPA buffer with 1% phenylmethanesulfonyl fluoride
(PMSF) and complete™ protease inhibitor cocktail (Roche Molecular
Biochemical, Indianapolis, IN, USA) for 30 min. After
centrifugation at 12,000 rpm for 5 min, supernatant was collected
and protein concentrations were determined using the Bio-Rad kit
(Bio-Rad Laboratories, USA). Cell lysates were separated by
SDS-PAGE gel, transferred to polyvinylidene difluoride (PVDF;
Millipore). Membranes were blotted with 10% non-fat milk, washed in
TBS-Tween and incubated with primary polyclonal antibodies
anti-E-cadherin or anti-Vimentin (1:400; Santa Cruz, USA) overnight
at 4°C. After washing with TBS-Tween, membranes were incubated with
secondary antibody (horseradish peroxidase conjugated IgG) for 60
min at room temperature. Polyclonal anti-GAPDH (1:800; Bioss,
China) was used as an internal control. Then, they were washed
again with TBS-Tween before using the enhanced chemiluminescence
detection system (Amersham Pharmacia Biotech, USA). Blots were
imaged by Molecular Image® ChemiDoc™ XRS+ with Image
Lab™ Software (Bio-Rad Laboratories, Inc.).
Xenograft tumor model in nude mice
Five-week-old female Balb/c athymic nude mice
(Vitalriver Laboratory Animals, Beijing, China) were subcutaneously
injected in the right flank with 3.0×106 HepG2 cells in
0.1 ml PBS. Once tumors grew to~5 mm in diameter, tumor volume (V)
was measured by caliper daily and calculated using the formula
V=(L×W2)/2, where L was the length and W was the width
of the tumor. The mice were randomly divided into two groups (n=6)
for treatment with SFN (50 mg/kg) or vehicle control through i.p.
injection every 2 days, respectively. Growth curves were plotted
using average tumor volume within each experimental group every 2
days. Thirteen days later, the mice were euthanized, and the
dissected tumors were collected and prepared for subsequent
analyses. All animal experiments were approved by the animal
center.
Statistical analysis
The data are presented as the mean ± SD from the
three independent experiments. Immunoblot signals were quantitated
using densitometry and ImageJ software version 1.4 (NIH, USA).
Statistical analysis was performed by the SPSS version 18.0 (SPSS
Inc., Chicago, IL, USA) using analysis of variance (ANOVA) followed
by the Tukey's t-test. P<0.05 was considered statistically
significant.
Results
SFN suppresses proliferation of
hepatocellular carcinoma HepG2 cells
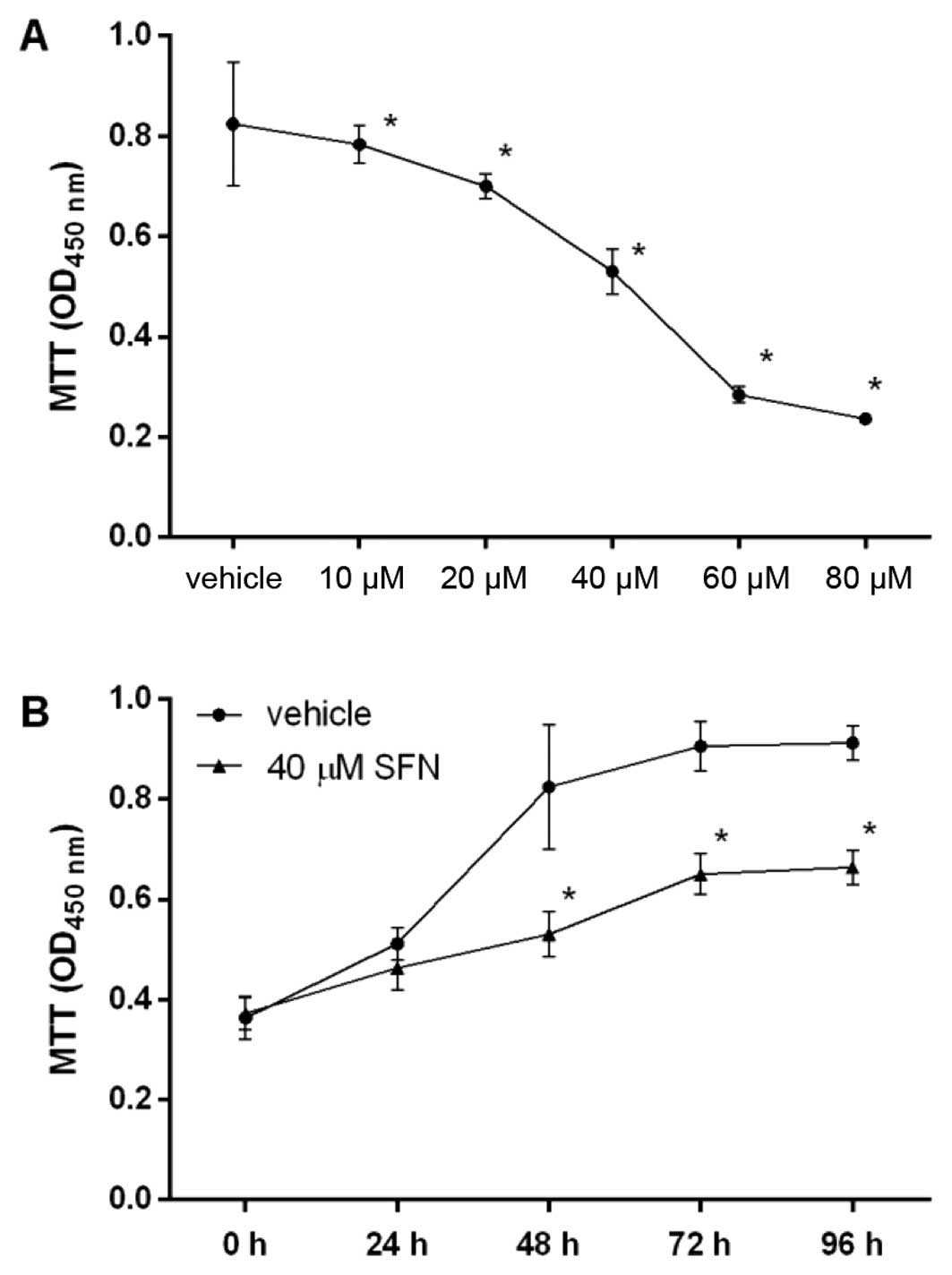
The dose and time course of the effect of SFN on
cell proliferation was examined by MTT assay (Fig. 1). We found that SFN significantly
inhibited cell proliferation of HepG2 in a dose-dependent manner,
with an IC50 value of 40.05 µM. HepG2 also
responded to SFN in a time-dependent manner. SFN significantly
inhibited proliferation of HepG2 cells at 40.05 µM after 48
h. Thus, SFN inhibited the proliferation of hepatocellular
carcinoma HepG2 cells both dose- and time-dependently.
SFN promotes cell cycle arrest and
apoptosis in hepatocellular carcinoma HepG2 cells
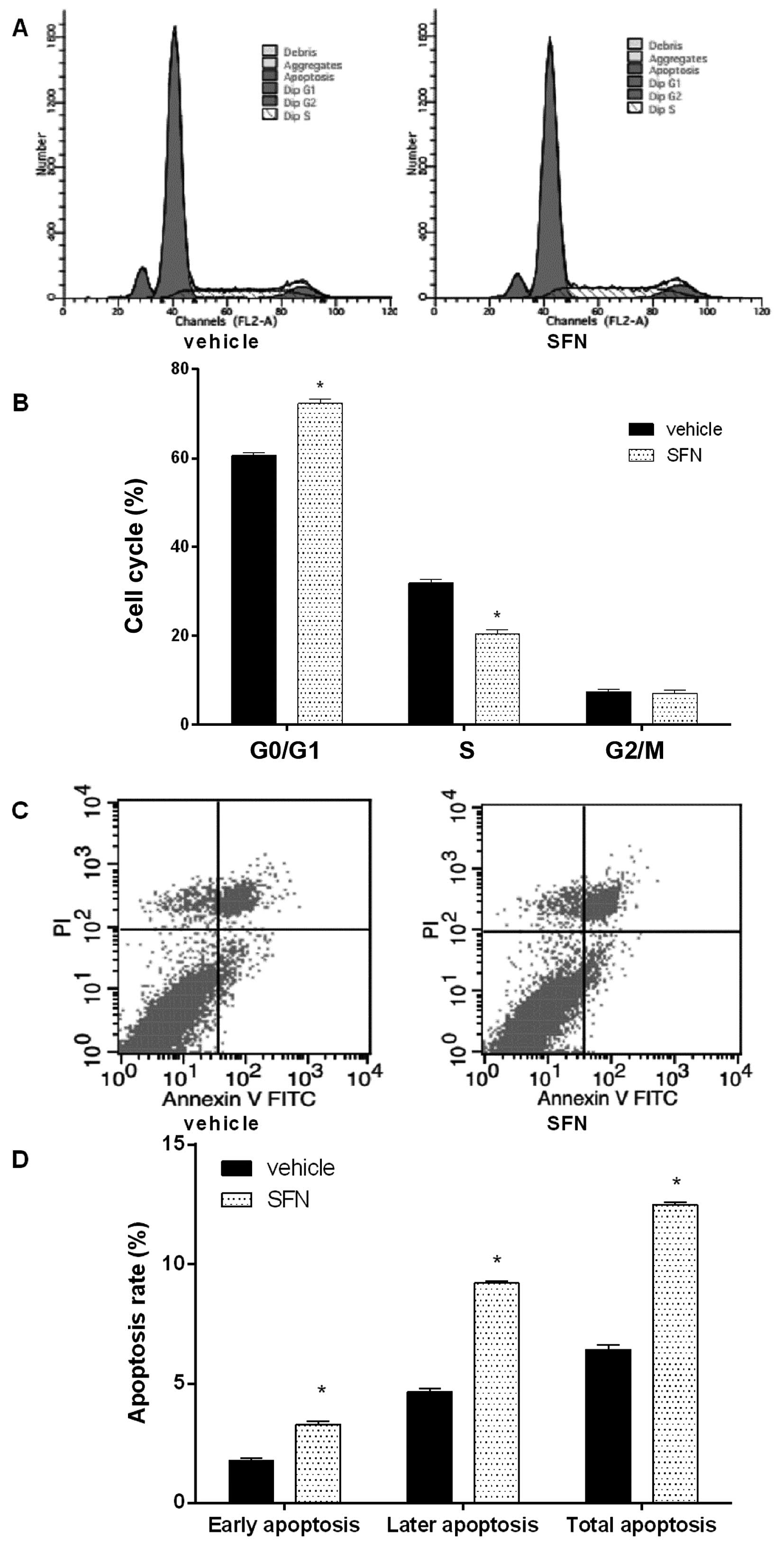
As the growth inhibition of cancer cells is usually
associated with cell cycle arrest, the effect of SFN on cell cycle
distribution of HepG2 was examined (Fig. 2A and B). We found that cell cycle
was arrested at the G0/G1 phase when HepG2 cells were treated with
40 µM SFN for 48 h.
Next, we tested the effect of SFN on HepG2 cell
apoptosis (Fig. 2C and D). SFN
markedly increased both the early and late apoptosis, compared with
control.
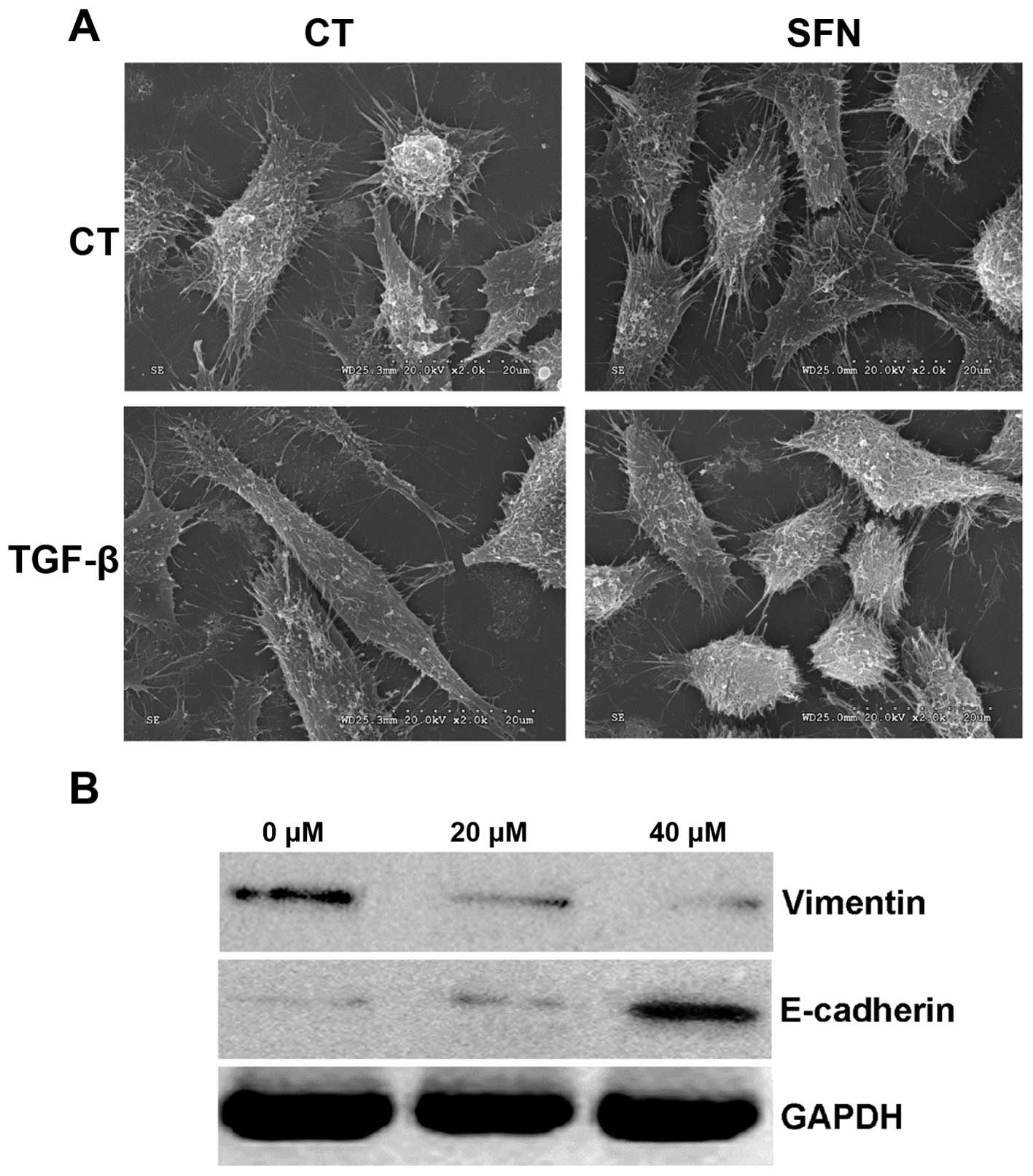
SFN changes the morphology and
TGF-β-induced EMT of HepG2 cells
Morphology of HepG2 cells was changed in the
presence of SFN (Fig. 3A). SFN
suppressed the typical morphology changes of EMT induced by TGF-β,
and inhibited the formation of fibroblast-like mesenchymal cells in
HepG2 cells.
Phenotypic transition of SFN-inhibited EMT was
further evaluated by changes of the mesenchymal marker Vimentin,
and the epithelial marker E-cadherin. As expected, SFN
significantly suppressed the expression of Vimentin, and
significantly elevated the expression of E-cadherin (Fig. 3B). The effect of SFN on the
expression of these EMT markers was in a dose-dependent. Overall,
SFN inhibited TGF-β-induced EMT of HepG2.
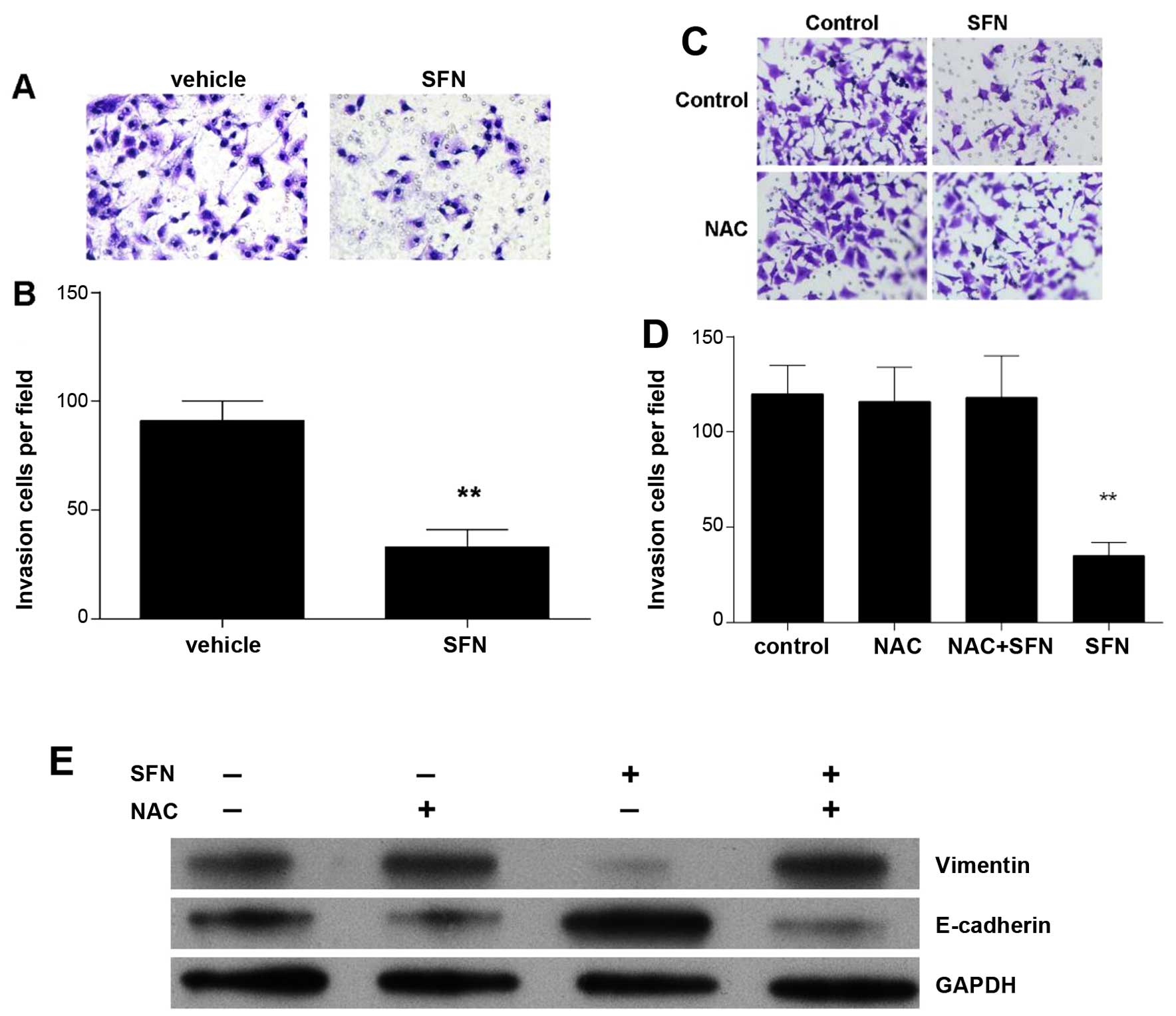
SFN inhibits HepG2 cell migration and
invasion
Using Transwell assay, the effects of SFN on cell
migration and invasive ability was determined (Fig. 4A). We found a significantly
decreased number of migrated HepG2 cells treated with 40 µM
of SFN (Fig. 4B).
ROS-dependent mechanism mediated the
effect of SFN on EMT in HepG2 cells
To evaluate whether ROS was involved in
TGF-β-induced HepG2 cell migration and invasion, and EMT, ROS
scavenger N-acetyl-L-cysteine (NAC, 20 mM) was added to treat the
HepG2 cells for 24 h in the present of TGF-β (Fig. 4C–E). The inhibitory effect of SFN on
migration and invasion were almost completely abolished by NAC in
HepG2 cells (Fig. 4C and D). Also,
NAC abolished the decrease of Vimentin, and the increase of
E-cadherin (Fig. 4E). It was
suggested that ROS-dependent mechanism mediated the effect of SFN
on EMT of HepG2 cells.
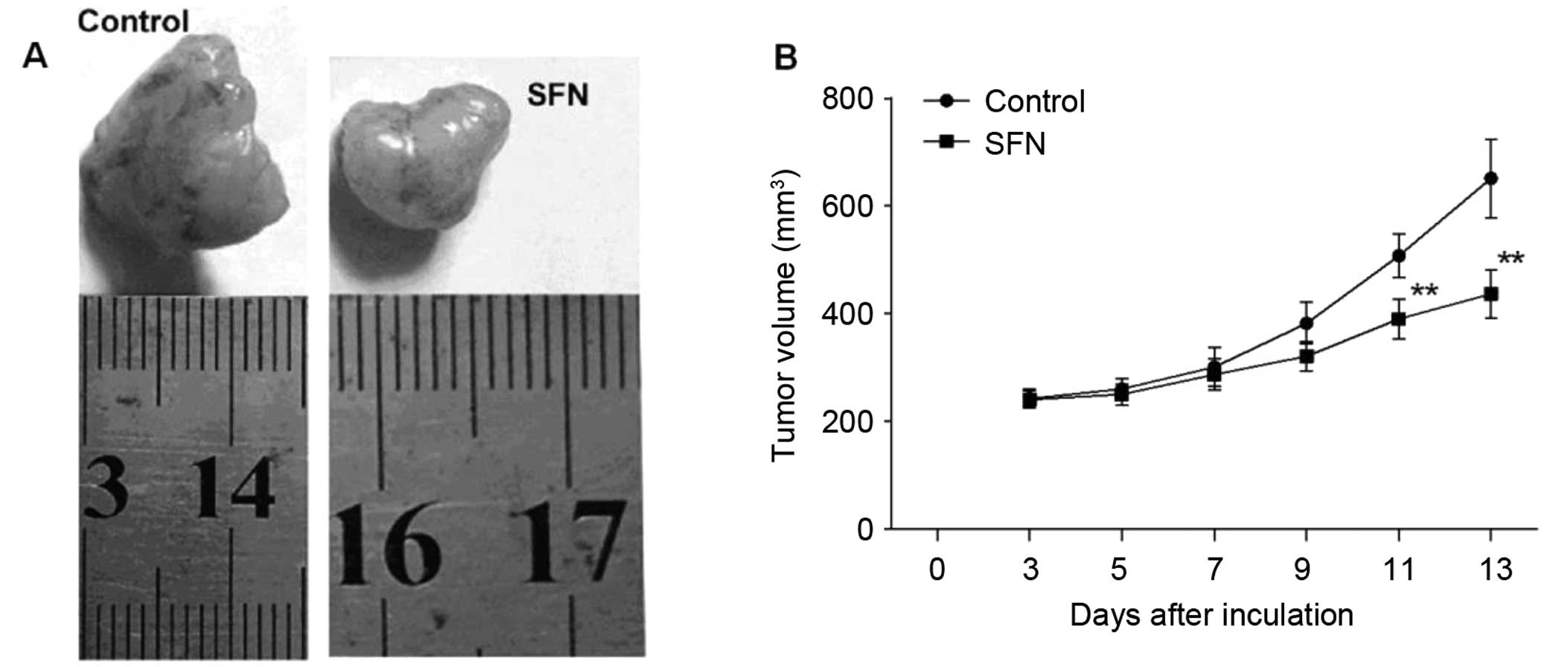
SFN suppresses xenograft tumor
growth
We showed a significant inhibitory effect of SFN on
cell growth of HepG2 cells. Here, we further determined the effect
of SFN on the growth of HepG2 cell-derived xenograft tumors in nude
mice (Fig. 5). HepG2 cell-derived
xenograft tumor grew slowly in the mice treated with 50 mg/kg SFN,
whereas grew progressively in the control mice with a larger tumor
volume than the SFN group. Taken together, our results indicate
that SFN is an effective agent for hepatocellular carcinoma
treatment.
Discussion
SFN has been widely used for treatment of
inflammatory diseases in vitro and in vivo (17–20).
In a previous clinical study conducted on healthy volunteers
(27), it was reported that no
obvious abnormal events (toxicities) were observed, suggesting the
safety of SFN for humans. Recent studies on the role of SFN in
cancer progression indicated an antitumor effect of SFN. SFN has
been reported to suppress proliferation and diverse cancer cells by
causing apoptosis, cell cycle arrest, or both (28). Furthermore, SFN influenced the cell
growth and metastasis in diverse cancers, including thyroid cancer
(17), breast cancer (19), colon cancer (21,22),
prostate cancer (18), bladder
cancer (23,24) and leukemia (25). SFN inhibited cell growth and
invasiveness of thyroid cancer, and promoted mitochondrial-mediated
apoptosis via reactive oxygen species (ROS)-dependent pathway
(17). SFN induces apoptosis in
bladder cancer cells through the ROS-mediated mitochondrial pathway
(23,24). However, the antitumor effect of SFN
on hepatocellular carcinoma remains incompletely understood. It was
demonstrated that SFN induces the generation of ROS in hepatoma
cells, and sensitize many tumor necrosis factor-related
apoptosis-inducing ligand (TRAIL)-resistant hepatoma cells to
TRAIL-induced apoptosis by upregulating DR5 (29). ROS has been reported to mediate many
cellular effects of hepatoma cells, including the EMT of HepG2
cells induced by the tumor promoter 12-O-tetradecanoyl
phorbol-13-acetate (26). Our
present study demonstrates that SFN significantly suppressed
hepatocellular carcinoma cell growth in vitro and in
vivo, and the migration, invasion and EMT of hepatocellular
carcinoma cells.
As reported (17),
the exact mechanism of the potent antitumor effect of SFN on cancer
cells is still unclear, and the mechanism of SFN in cell cycle and
apoptosis seems very complex. In T-cell leukemia and melanoma
cells, SFN could induce a positive effect on p53 expression with a
consequent decrease in Bcl-2 and increase in Bax (30,31),
whereas it is not required for the SFN activity in leukemia U937
cells and colon cancer cells (25,32).
In SFN-treated thyroid cancer cells, the cells were arrested in
G2/M phase, which was mediated by a ROS-dependent pathway, but not
a p53-dependent mechanism (17). In
this study, our results showed that the cell proliferation of HepG2
were significantly decreased by treatment of SFN for 48 h, the 50%
inhibitory concentrations (IC50) for HepG2 cells was
40.05 mM. By using 40 mM SFN, the observations suggested that SFN
suppressed cell proliferation of HepG2 with treatment concentration
and suppressed cell growth by decreasing the cells in S phase.
SFN-treated HepG2 cells were arrested in G0/G1 phase. SFN also
promoted cell apoptosis. However, the underlying mechanism is still
unclear, further studies will be necessary to determine the changes
in p53 proteins and ROS signaling pathway by which sulforaphane
induces the cell arrest and cell apoptosis in hepatocellular
carcinoma.
In recent years, EMT has been highlighted to be
involved in cancer progression (33). The process of EMT is characterized
as the loss of epithelial phenotype and the gain of mesenchymal
phenotype (34). A recent study has
showed that SFN increased expression of E-cadherin, and decreased
expression of Vimentin, contributing to inhibition of EMT process
of thyroid cancer cells (17). It
was consistent with our finding that SFN significantly promoted the
expression of E-cadherin, and reduced expression of Vimentin in
hepatocellular carcinoma cells. The present study also showed the
EMT of hepatocellular carcinoma cells was mediated by a
ROS-dependent mechanism. The use of NAC efficiently abolished the
role of SFN in migration and invasion, and in regulation of the
expression of E-cadherin and Vimentin in hepatocellular carcinoma
cells.
In conclusion, the present study shows that SFN
inhibits hepatocellular carcinoma cell proliferation, migration and
invasion, as well as EMT via a ROS-dependent pathway. The effect of
SFN on the growth of hepatocellular carcinoma cells was confirmed
by the xenograft tumor growth model. All our finding implicated
that SFN is a promising and safe strategy for treating
hepatocellular carcinoma.
Acknowledgments
This work was supported in part by the Key Project
of Hainan Province, China (no. 1321320 and 67A1006) and by the
Scientific Research Project of Hainan Provincial Health Department
(no. Qiongwei-2012pt-92).
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mittal S and El-Serag HB: Epidemiology of
hepatocellular carcinoma: Consider the population. J Clin
Gastroenterol. 47(Suppl): S2–S6. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lu T, Seto WK, Zhu RX, Lai CL and Yuen MF:
Prevention of hepatocellular carcinoma in chronic viral hepatitis B
and C infection. World J Gastroenterol. 19:8887–8894. 2013.
View Article : Google Scholar :
|
|
4
|
Zhu Z, Zhang X, Wang G and Zheng H: Role
of microRNAs in hepatocellular carcinoma. Hepat Mon. 14:e186722014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jang E, Kim BJ, Lee KT, Inn KS and Lee JH:
A survey of therapeutic effects of Artemisia capillaris in liver
diseases. Evid Based Complement Alternat Med. 2015:7281372015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mancuso A and Perricone G: Hepatocellular
carcinoma and liver transplantation: State of the art. J Clin
Transl Hepatol. 2:176–181. 2014. View Article : Google Scholar
|
|
7
|
Song IH: Cancer metastasis and metastasis
suppressors. Korean J. 43:1–7. 2004.
|
|
8
|
Stracke ML and Liotta LA: Multi-step
cascade of tumor cell metastasis. In Vivo. 6:309–316.
1992.PubMed/NCBI
|
|
9
|
Hu CT, Wu JR, Chang TY, Cheng CC and Wu
WS: The transcriptional factor Snail simultaneously triggers cell
cycle arrest and migration of human hepatoma HepG2. J Biomed Sci.
15:343–355. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Huber MA, Kraut N and Beug H: Molecular
requirements for epithelial-mesenchymal transition during tumor
progression. Curr Opin Cell Biol. 17:548–558. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xu XM, Yuan GJ, Li QW, Shan SL and Jiang
S: Hyperthermia inhibits transforming growth factor beta-induced
epithelial-mesenchymal transition (EMT) in HepG2 hepatocellular
carcinoma cells. Hepatogastroenterology. 59:2059–2063.
2012.PubMed/NCBI
|
|
12
|
Scanlon CS, Van Tubergen EA, Inglehart RC
and D'Silva NJ: Biomarkers of epithelial-mesenchymal transition in
squamous cell carcinoma. J Dent Res. 92:114–121. 2013. View Article : Google Scholar :
|
|
13
|
Shih JY and Yang PC: The EMT regulator
slug and lung carcinogenesis. Carcinogenesis. 32:1299–1304. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Szarvas T, vom Dorp F, Ergün S and Rübben
H: Matrix metalloproteinases and their clinical relevance in
urinary bladder cancer. Nat Rev Urol. 8:241–254. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sullivan NJ, Sasser AK, Axel AE, Vesuna F,
Raman V, Ramirez N, Oberyszyn TM and Hall BM: Interleukin-6 induces
an epithelial-mesenchymal transition phenotype in human breast
cancer cells. Oncogene. 28:2940–2947. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang L, Tian Z, Yang Q, Li H, Guan H, Shi
B, Hou P and Ji M: Sulforaphane inhibits thyroid cancer cell growth
and invasiveness through the reactive oxygen species-dependent
pathway. Oncotarget. 6:25917–25931. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xu C, Shen G, Yuan X, Kim JH,
Gopalkrishnan A, Keum YS, Nair S and Kong AN: ERK and JNK signaling
pathways are involved in the regulation of activator protein 1 and
cell death elicited by three isothiocyanates in human prostate
cancer PC-3 cells. Carcinogenesis. 27:437–445. 2006. View Article : Google Scholar
|
|
19
|
Jackson SJ and Singletary KW:
Sulforaphane: A naturally occurring mammary carcinoma mitotic
inhibitor, which disrupts tubulin polymerization. Carcinogenesis.
25:219–227. 2004. View Article : Google Scholar
|
|
20
|
Bergantin E, Quarta C, Nanni C, Fanti S,
Pession A, Cantelli-Forti G, Tonelli R and Hrelia P: Sulforaphane
induces apoptosis in rhabdomyosarcoma and restores
TRAIL-sensitivity in the aggressive alveolar subtype leading to
tumor elimination in mice. Cancer Biol Ther. 15:1219–1225. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jeong WS, Kim IW, Hu R and Kong AN:
Modulatory properties of various natural chemopreventive agents on
the activation of NF-kappaB signaling pathway. Pharm Res.
21:661–670. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jeong WS, Kim IW, Hu R and Kong AN:
Modulation of AP-1 by natural chemopreventive compounds in human
colon HT-29 cancer cell line. Pharm Res. 21:649–660. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jo GH, Kim GY, Kim WJ, Park KY and Choi
YH: Sulforaphane induces apoptosis in T24 human urinary bladder
cancer cells through a reactive oxygen species-mediated
mitochondrial pathway: The involvement of endoplasmic reticulum
stress and the Nrf2 signaling pathway. Int J Oncol. 45:1497–1506.
2014.PubMed/NCBI
|
|
24
|
Park HS, Han MH, Kim GY, Moon SK, Kim WJ,
Hwang HJ, Park KY and Choi YH: Sulforaphane induces reactive oxygen
species-mediated mitotic arrest and subsequent apoptosis in human
bladder cancer 5637 cells. Food Chemical Toxicol. 64:157–165. 2014.
View Article : Google Scholar
|
|
25
|
Choi WY, Choi BT, Lee WH and Choi YH:
Sulforaphane generates reactive oxygen species leading to
mitochondrial perturbation for apoptosis in human leukemia U937
cells. Biomed Pharmacother. 62:637–644. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wu WS, Tsai RK, Chang CH, Wang S, Wu JR
and Chang YX: Reactive oxygen species mediated sustained activation
of protein kinase C alpha and extracellular signal-regulated kinase
for migration of human hepatoma cell HepG2. Mol Cancer Res.
4:747–758. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shapiro TA, Fahey JW, Dinkova-Kostova AT,
Holtzclaw WD, Stephenson KK, Wade KL, Ye L and Talalay P: Safety,
tolerance, and metabolism of broccoli sprout glucosinolates and
isothiocyanates: A clinical phase I study. Nutr Cancer. 55:53–62.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Singh SV, Srivastava SK, Choi S, Lew KL,
Antosiewicz J, Xiao D, Zeng Y, Watkins SC, Johnson CS, Trump DL, et
al: Sulforaphane-induced cell death in human prostate cancer cells
is initiated by reactive oxygen species. J Biol Chem.
280:19911–19924. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kim H, Kim EH, Eom YW, Kim WH, Kwon TK,
Lee SJ and Choi KS: Sulforaphane sensitizes tumor necrosis
factor-related apoptosis-inducing ligand (TRAIL)-resistant hepatoma
cells to TRAIL-induced apoptosis through reactive oxygen
species-mediated up-regulation of DR5. Cancer Res. 66:1740–1750.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fimognari C, Nüsse M, Cesari R, Iori R,
Cantelli-Forti G and Hrelia P: Growth inhibition, cell-cycle arrest
and apoptosis in human T-cell leukemia by the isothiocyanate
sulforaphane. Carcinogenesis. 23:581–586. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hamsa TP, Thejass P and Kuttan G:
Induction of apoptosis by sulforaphane in highly metastatic B16F-10
melanoma cells. Drug Chem Toxicol. 34:332–340. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rudolf E and Cervinka M: Sulforaphane
induces cytotoxicity and lysosome- and mitochondria-dependent cell
death in colon cancer cells with deleted p53. Toxicol In Vitro.
25:1302–1309. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Thiery JP: Epithelial-mesenchymal
transitions in tumour progression. Nat Rev Cancer. 2:442–454. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Guarino M, Rubino B and Ballabio G: The
role of epithelial-mesenchymal transition in cancer pathology.
Pathology. 39:305–318. 2007. View Article : Google Scholar : PubMed/NCBI
|