Introduction
Recent research into the inflammatory
microenvironment of malignant cancer tissues demonstrates that a
tight association exists between inflammation and tumor metastasis
progression (1,2). Despite attempts in the last few
decades to elucidate the inflammatory cytokines underlying invasion
and metastasis in the tumor micro-environment (TME) (3), knowledge of the role of
pro-inflammatory cytokines in cancer is limited. Mounting evidence
demonstrates that inflammation induces activity by several factors
in the TME, including growth factors, survival factors, and
extracellular matrix-modifying enzymes, which control proliferative
signaling, leading to activation of the epithelial-to-mesenchymal
transition (EMT), a crucial process in cancer cell migration and
metastasis. Transforming growth factor-β (TGF-β) was the first
factor described as an inducer of EMT (4). TGF-β is a pleiotropic cytokine that
has a wide variety of biological functions and is activated by
Smad2/3/4, which induces translocation of TGF-β from the receptor
to the nucleus. Dysregulation of the TGF-β pathway contributes to a
variety of diseases, including fibrotic kidney diseases, colorectal
cancer, Alzheimer's disease, and diabetes (5,6). Based
on the importance of the TGF-β pathway in the progression of
cancer, a better understanding of its mechanisms and targets could
facilitate the development of improved chemopreventive agents.
EMT is a fundamental process in embryogenesis and a
pivotal event in the initial steps of the metastatic cascade that
allows cells to acquire migratory, invasive, and stem cell-like
properties. During EMT, epithelial cells lose polarity and
cell-to-cell contacts, followed by dramatic remodeling of the
cytoskeleton. Epithelial markers such as E-cadherin promote
cell-to-cell contact, whereas mesenchymal markers such as vimentin,
fibronectin, and N-cadherin induce spindle-shaped, fibroblast-like
morphology and cell invasion (7,8). In
addition, several key transcriptional repressors, including Snail,
Slug, and Twist, are activated via multiple cellular signaling
pathways, including NF-κB, Wnt, and Hedgehog, during EMT (9–11).
Therefore, reversing and/or preventing the EMT process is thought
to be an effective strategy for limiting the dissemination of
cancer.
Recently, dietary chemopreventive agents have gained
much attention in the area of cancer research. Grape seed extract
and red wine contain strong antioxidants and polyphenols that
reduce free radical damage. Adequate intake of grape seed extract
and red wine has a preventive effect against some cancers. Among
dietary chemopreventive agents, rhapontigenin, a stilbene
derivative isolated from the rhizome of traditional herbal medicine
Rheum undulatum (Polygonaceae), has been recognized as an
analog of resveratrol with multiple biological functions, including
anti-allergic, anti-hyperglycemic, and anti-angiogenic activities.
Although several studies have described the diverse effects and
mechanisms of dietary chemoprevention, the mechanism underlying
prevention of cancer invasion and metastasis by Rha has not been
reported. We hypothesize that Rha has diverse biological functions
that contribute to its anticancer effects.
Based on a previous study (4) showing that TGF-β induces EMT in cancer
cells, TGF-β was used in the present study to induce a
pro-inflammatory microenvironment, which allowed dissection of the
anticancer mechanism of Rha via assessment of changes produced in
the TME. The aim of this study was to assess inhibition of
TGF-β-triggered EMT in cancer cells as a mechanism for the
anti-metastatic effect of Rha. We discovered that Rha inhibits
TGF-β-induced EMT and derived invasion in renal cancer cells while
suppressing activation of the PI3K/AKT/mTOR/Snail signaling
pathway.
Materials and methods
Cell culture
Human 769-P renal carcinoma cells, A549 human lung
epithelial cells, HeLa cervix adenocarcinoma cells, and PC3
prostate adenocarcinoma cells were obtained from the American Type
Culture Collection (ATCC, USA) and cultivated in Dulbecco's
modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS), 2
mM glutamine, 100 U/ml penicillin, and 100 μg/ml
streptomycin in a 37°C CO2 incubator.
Antibodies and chemical reagents
LY294002 (LY), SB203580 (SB), PD98059 (PD), and
MG132 were obtained from Sigma (USA). SP600125 was obtained from
Cayman. β-catenin inhibitor BH535 was obtained from Calbiochem.
TGF-β was obtained from Peprotech (USA). Antibodies against HIF-1α,
E-cadherin, N-cadherin, and β-catenin were obtained from BD
Biosciences. Antibodies against fibronectin, vimentin, tubulin, and
actin were obtained from Sigma. Antibodies against HA were obtained
from Covance. Antibodies against CA9, S6K, S6, cyclin D1, and GAPDH
were obtained from GeneTex. Antibodies against mTOR, mTOR-Ser2448,
Akt-Ser473, Akt, S6K-Thr389, S6-Ser235/236, GSK-3β, GSK3β-Ser9,
Smad2-Ser465/467, active β-catenin, and Snail were obtained from
Cell Signaling Technology.
RNA interference
The sequences targeted by the anti-Snail siRNA were
5′-GGUGUGACUAAUGCAATT-3′ and 5′-UUGCAUAGUUAGUCACACCTT-3′. siRNAs
were synthesized by and purchased from Biotools (USA). Transfection
of siRNA was conducted using Lipofectamine RNAiMAX (Invitrogen,
USA) at a final concentration of 0.1–0.3 μM. ON-TARGETplus
SMARTpool siRNAs (Dharmacon, Chicago, IL, USA) targeting human Akt
or mTOR were transfected as siRNA duplexes for 48 h. Knockdown of
HIF-1α achieved by transfection with the pSUPER-based plasmid
expressing sh-HIF-1α, which was a generous gift from Dr K.J. Wu
(Taiwan). Transfection of sh- HIF-1α siRNA was performed according
to the manufacturer's protocol. Briefly, cells in the exponential
phase of growth were plated in 6-well tissue culture plates at a
density of 1.5×105 cells/well, grown for 24 h, and
transfected with siRNA using Lipofectamine 2000 and OPTI-MEM serum
free medium. After 48 h, cells were harvested and lysed for western
blotting.
Immunoblotting and
immunoprecipitation
Cells were lysed in TEGN buffer (10 mM Tris, pH 7.5,
1 mM EDTA, 420 mM NaCl, 10% glycerol, and 0.5% Nonidet P-40)
containing 1 mM Na3VO4 and 1 mM protease
inhibitor cocktail (Roche). For immunoprecipitation, lysates were
diluted 1:1 with TEG buffer (10 mM Tris, pH 7.5, 1 mM EDTA, and 20%
glycerol) and rocked with antibodies and 20 μl of 50%
protein G beads (Upstate) for 3 h at 4°C. The immunoprecipitates
were washed four times in TEG:TEGN buffer (1:1), boiled in protein
sample dye (2 M β-mercaptoethanol, 12% SDS, 0.5 M Tris, pH 6.8, 0.5
mg/ml bromophenol blue, and 30% glycerol), analyzed by
SDS-polyacrylamide gel electrophoresis (PAGE), and subjected to
western blotting.
Transient transfections
Transient transfections of cancer cell lines were
performed using Lipofectamine 2000 (Invitrogen). Whole cell
extracts were prepared at specific time-points after transfection
and subjected to SDS-PAGE/western blotting or immunoprecipitation
assays as described previously (12).
Immunofluorescence
Cells were plated on cover slips 1 day before drug
exposure. After treating the cells with TGF-β with or without Rha
or LY for the specified times, the cells were fixed with 3%
paraformaldehyde and 2% sucrose in PBS for 10 min. Coverslips were
permeabilized with Triton X-100 buffer (0.05% Triton X-100, 20 mM
HEPES-KOH, pH 7.9, 50 mM NaCl, 3 mM MgCl2, 300 mM
sucrose) for 10 min and blocked in PBS containing 1% BSA for 1 h at
room temperature, after which the cells were washed three times
with PBS and incubated with Alexa Fluor 488- or
rhodamine-conjugated secondary antibodies (Molecular Probes) in
Hoechst 33342 stain containing PBS for 1 h. Finally, the treated
cells were washed with PBS three times and mounted in Fluoromount™
Aqueous Mounting Medium (Sigma).
Invasion assay
Cells (5×105) in 6-well plates were
pre-treated with or without LY294002 for 30 min, followed by
exposure to TGF-β for 24 h. Later, the cells were trypsinized and
counted, after which 1.25×105 cells were seeded in
Matrigel-coated Transwell inserts (24-well, 8-μm pore size;
BD Biosciences, Canada) with serum-free medium with inhibitors or
inhibitors/TGF-β. DMEM with 10% FBS was added to the lower chamber
to act as a chemo-attractant. After incubation for 48 h, the inner
cells that did not invade the lower chamber via the pores were
removed with a cotton swab. The cells that adhered to the underside
of the membrane were fixed in methanol and stained with 0.5%
crystal violet. For each sample, six random fields were counted and
examined under a microscope with a x100 objective to determine the
number of cells that had invaded across the membrane. Three
independent experiments were conducted.
Quantitative measurements and statistical
analyses
Quantitative measurements of the obtained results
were performed using ImageQuant software following the
manufacturer's protocol (Molecular Dynamics) and presented as mean
+ standard error of the mean (SEM) for three independent
experiments (n=3). Statistical analyses were performed using
Student's t-test. Results were considered significant at p-value
<0.05 (P<0.05).
Results
Cancer cells treated with TGF-β undergo
EMT
We investigated the mechanism underlying
TGF-β-induced EMT in A549 (lung), HeLa (cervical), and 769-P
(renal) cancer cells, all of which have been established as
suitable models for studying EMT in vitro (12–15).
As expected, TGF-β treatment resulted in the acquisition of
spindle-fibroblastic morphology, downregulation of epithelial
marker E-cadherin, and upregulation of mesenchymal markers
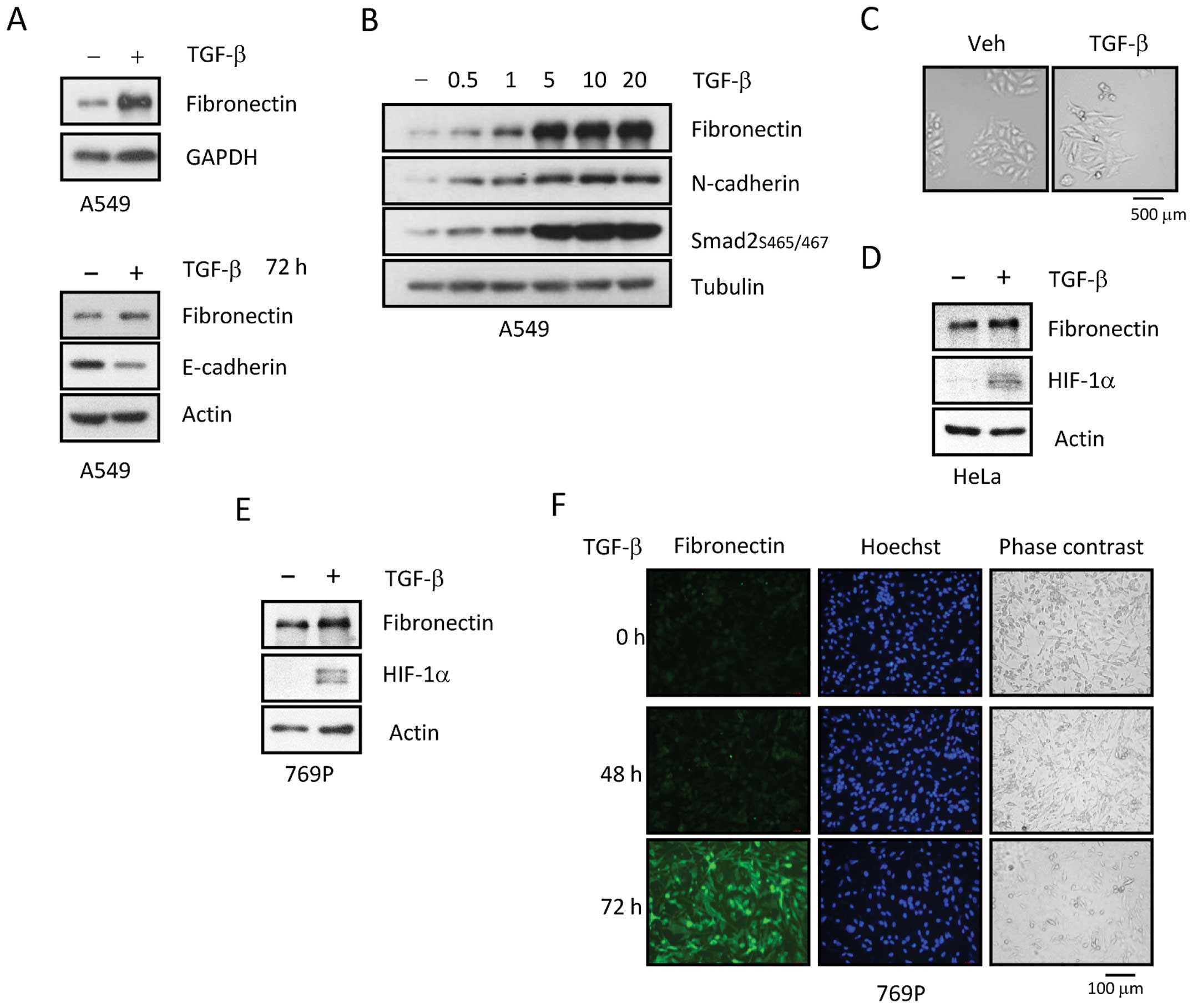
fibronectin (FN), vimentin and N-cadherin (Fig. 1A and C). FN induction was observed
at day 1 (Fig. 1A, upper panel).
TGF-β dose-dependently induced expression of FN and N-cadherin, as
well as phosphorylation of Smad2 (Fig.
1B). E-cadherin reduction by TGF-β occurred on day 3 in in A549
cells (Fig. 1A, bottom panel).
Additionally, TGF-β treatment for 2 days led to upregulation of
HIF-1α expression in 769-P cells (Fig.
1E). Changes in protein expression were confirmed by
immunofluorescence (IFA) experiments. When 769-P cells were
stimulated with TGF-β for 2 days, FN expression was slightly
increased. IFA experiments also confirmed upregulation of FN in
769-P cells (Fig. 1F). Similar
protein alterations were observed in HeLa cells (Fig. 1D).
TGF-β mediated EMT alteration and HIF1a
expression via the PI3K/AKT pathway
Several studies suggest a role for PI3K in TGF-β
signaling (16–18). The involvement of various signaling
pathways in regulating TGF-β-induced EMT was assessed. A series of
pharmacological inhibitors, including LY (PI3K inhibitor), PD (MAPK
inhibitor), SB (p38 inhibitor), and SP (JNK inhibitor), were used
to determine the involvement of various signaling pathways in
regulating TGF-β-mediated responses. Serine/threonine kinase Akt, a
well-known downstream effector of PI3K, is involved in cell
survival signaling. Akt phosphorylation at serine 473 was strongly
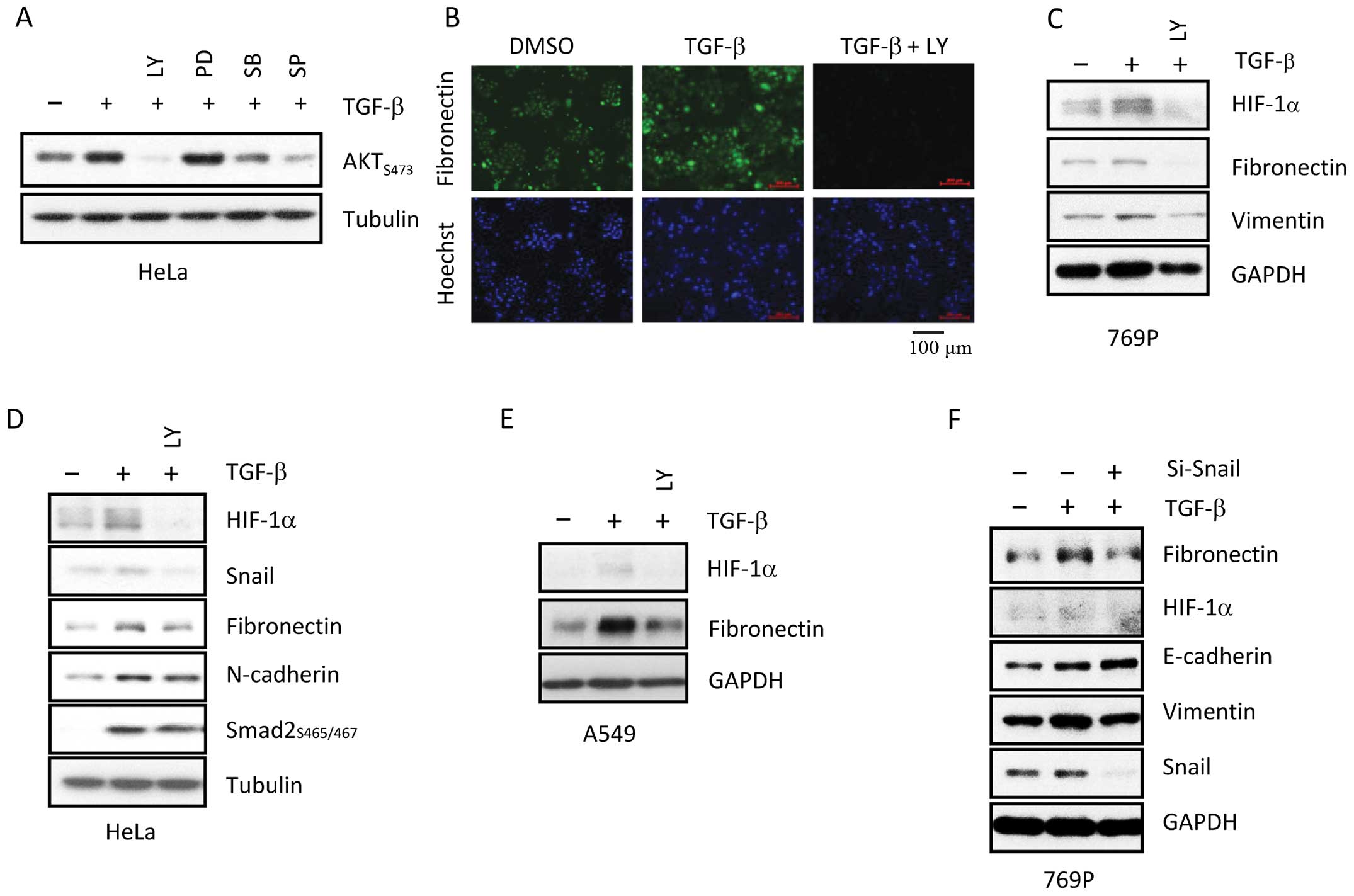
activated by TGF-β. When cells were treated with LY, PD, SB, or SP,
AKT phosphorylation at serine 473 was downregulated in HeLa cells
(Fig. 2A). Relative to SB and SP,
LY more effectively abolished phosphorylation of Akt, indicating
that TGF-β activation may occur via the PI3K/Akt signaling.
Next, in order to verify the importance of the PI3K
pathway in TGF-β regulation, 769-P cells were pre-treated with or
without PI3K inhibitor LY for 30 min, followed by co-treatment with
TGF-β for 20 h. As shown in Fig. 2B and
C, LY dramatically blocked TGF-β-mediated expression of EMT
markers in 769-P cells. Additionally, TGF-β-induced expression of
HIF-1α and EMT-related genes Snail, FN, and N-cadherin were
abrogated by LY in HeLa and A549 cells, implicating the PI3K
signaling pathway in the regulation of TGF-β-mediated responses
(Fig. 2D and E). To assess the role
of Snail in TGF-β-induced EMT (16,18),
Snail protein was knocked-down via small interfering RNA (siRNA) in
769-P cells. In 769-P cells lacking Snail protein, TGF-β-induced
EMT was abolished, demonstrating the crucial role of Snail in the
EMT (Fig. 2F). These results
suggest that Snail may play a crucial role in TGF-β-induced
EMT.
The 26S proteasome is responsible for
Rhapontigenin-induced degradation of Snail
To verify our hypothesis regarding Rha, we assessed
the impact of Rha on TGF-β-mediated Snail expression, which is a
transcriptional regulator active during EMT. Therefore, various
cancer cell types were exposed to Rha in the presence or absence of
TGF-β. Together with previous studies, the results described above
show that TGF-β can alter expression of EMT-related genes,
including Snail (10,11,19,20).
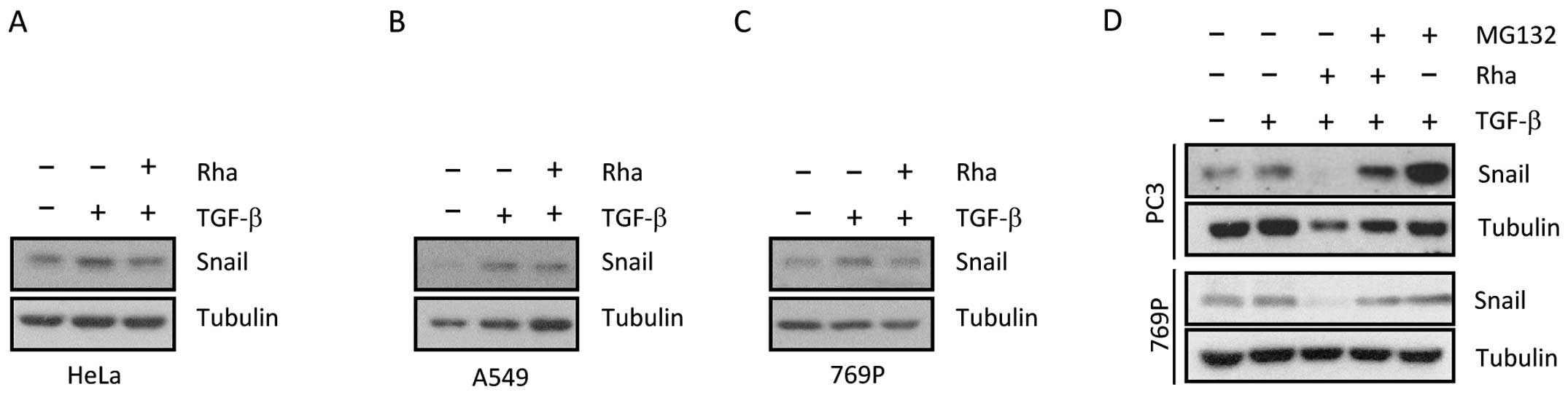
As shown in Fig. 3A–C, TGF-β
triggered increased Snail expression in diverse cancer cells.
However, when cancer cells were pre-treated with Rha, TGF-β-induced
Snail expression was blocked by Rha in all tested types of cancer
cells. To examine whether the proteasome degradation pathway is
involved in Rha-induced HIF-1α protein degradation, 769-P cells
were stimulated with TGF-β or co-treated with Rha for 20 h,
followed by exposure to 26S proteasome inhibitor MG132 for 6 h. As
shown in Fig. 3D, MG132 completely
blocked the reduction in TGF-β-induced Snail expression induced by
Rha, suggesting that Snail was recruited to the 26S proteasome for
degradation (Fig. 3D, upper).
Similar results were observed in PC3 cells (Fig. 3D, bottom), indicating that the
ability of MG132 to reverse Snail expression by Rha was
ubiquitous.
Rha induces ubiquitination and
degradation of HIF-1a
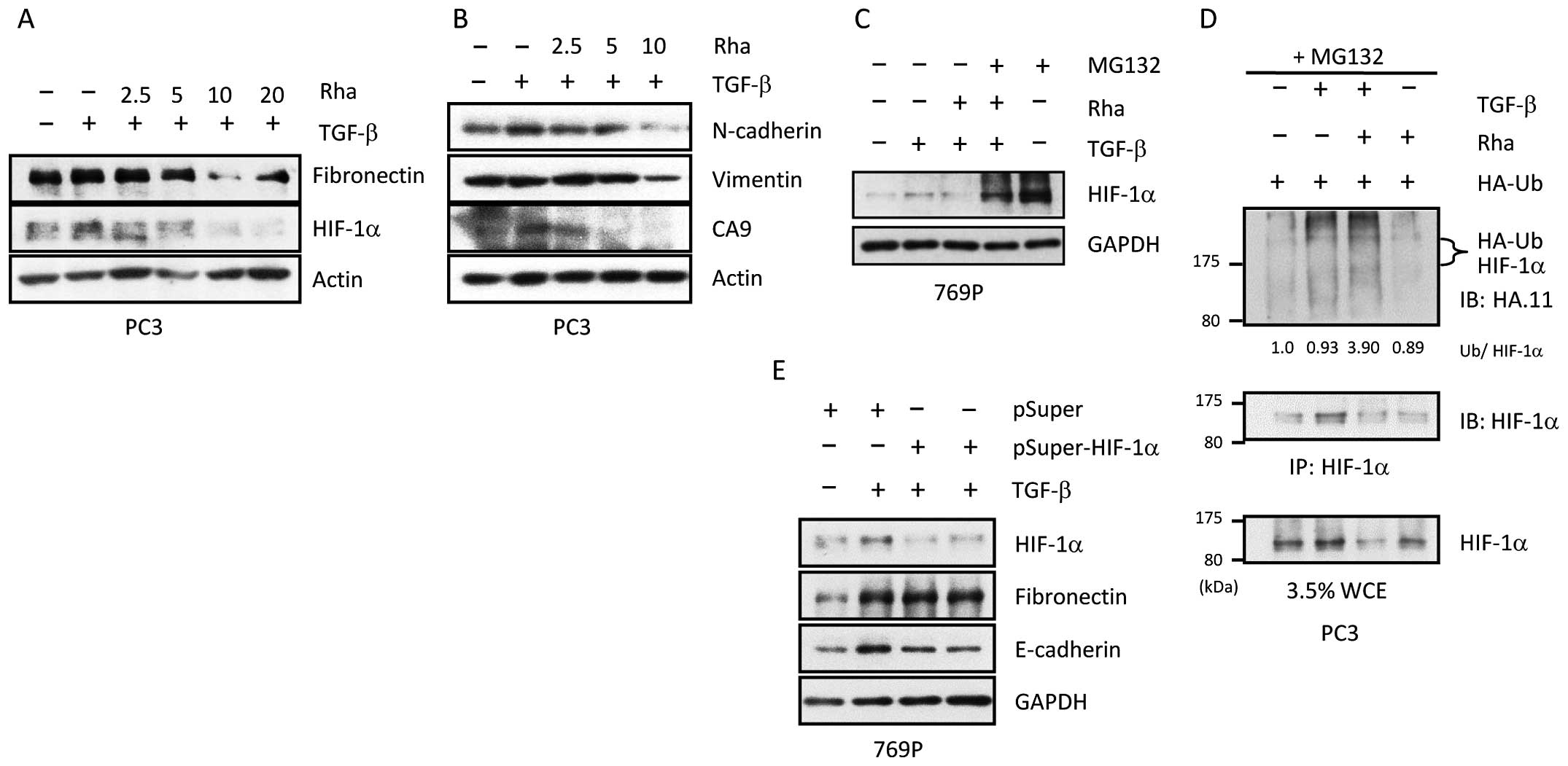
In previous studies, Rha was recognized as an
inhibitor of HIF-1α accumulation in PC3 cells. Therefore, TGF-β
mediated downregulation of HIF-1α expression by Rha was
investigated. Rha reduced protein levels of HIF-1α in a
concentration-dependent manner (Fig.
4A). In addition, protein levels of EMT-related markers
N-cadherin, vimentin, and the noted HIF-1α target gene carbonic
anhydrous IX (CA9) were reduced by Rha in a concentration-dependent
manner (Fig. 4B). The mechanism
underlying the effect of Rha on HIF-1α was assessed. As shown in
Fig. 4C, MG132 completely abolished
Rha-induced degradation of HIF-1α, indicating that the reduction in
HIF-1α expression induced by Rha was partially due to degradation
by the 26S proteasome. Next, to determine whether HIF-1α was
recruited to the 26S proteasome as a result of ubiquitination, an
immunoprecipitation (IP) experiment was performed, in which PC3
cells were transfected with a plasmid containing HA-tagged
ubiquitin for 24 h, followed by exposure to TGF-β in the presence
or absence of Rha for 24 h. Finally, the cells were treated with
MG132 for 6 h. As shown in Fig. 4D,
PC3 cells co-treated with TGF-β and Rha had approximately 3-fold
the level of HA-tagged ubiquitin as those treated only with TGF-β.
Because of the changes in EMT markers that accompanied HIF-1α
expression, we assessed the involvement of HIF-1α in TGF-β-mediated
changes in EMT markers. Therefore, 769-P cells were transfected
with pSuper-HIF-1α or the pSuper control plasmid for 24 h, followed
by exposure to TGF-β with or without Rha for 24 h. As shown in
Fig. 4E, HIF-1α silencing had
little impact on alterations in EMT markers, indicating that HIF-1α
was not a critical factor in TGF-β-mediated EMT. Collectively,
these results indicate that Rha treatment promotes TGF-β-induced
HIF-1α ubiquitination, subsequently leading to HIF-1α degradation
by the 26S proteasome. However, HIF-1α itself is not required for
TGF-β-induced EMT.
TGF-β-induced HIF-1α expression requires
AKT and mTOR
Previous studies demonstrated the importance of
glycogen synthase kinase-3β (GSK-3β) and mTOR as downstream
effectors of Akt following activation by TGF-β in diverse cell
types (21,22). However, the functional relevance of
Akt and its downstream effector pathways in TGF-β signaling remains
largely unknown. We tested the involvement of pathways downstream
of Akt in the regulation of TGF-β-mediated HIF-1α expression. The
role of GSK-3β in the regulation of TGF-β-mediated HIF-1α
expression was investigated. When cells are exposed to Wnt, GSK-3β
is phosphorylated and becomes inactive. When Wnt signaling is
blocked, active GSK-3β leads to phosphorylation, ubiquitination,
and degradation of β-catenin. In brief, β-catenin retains the
GSK-3β consensus motif for ubiquitination (23,24).
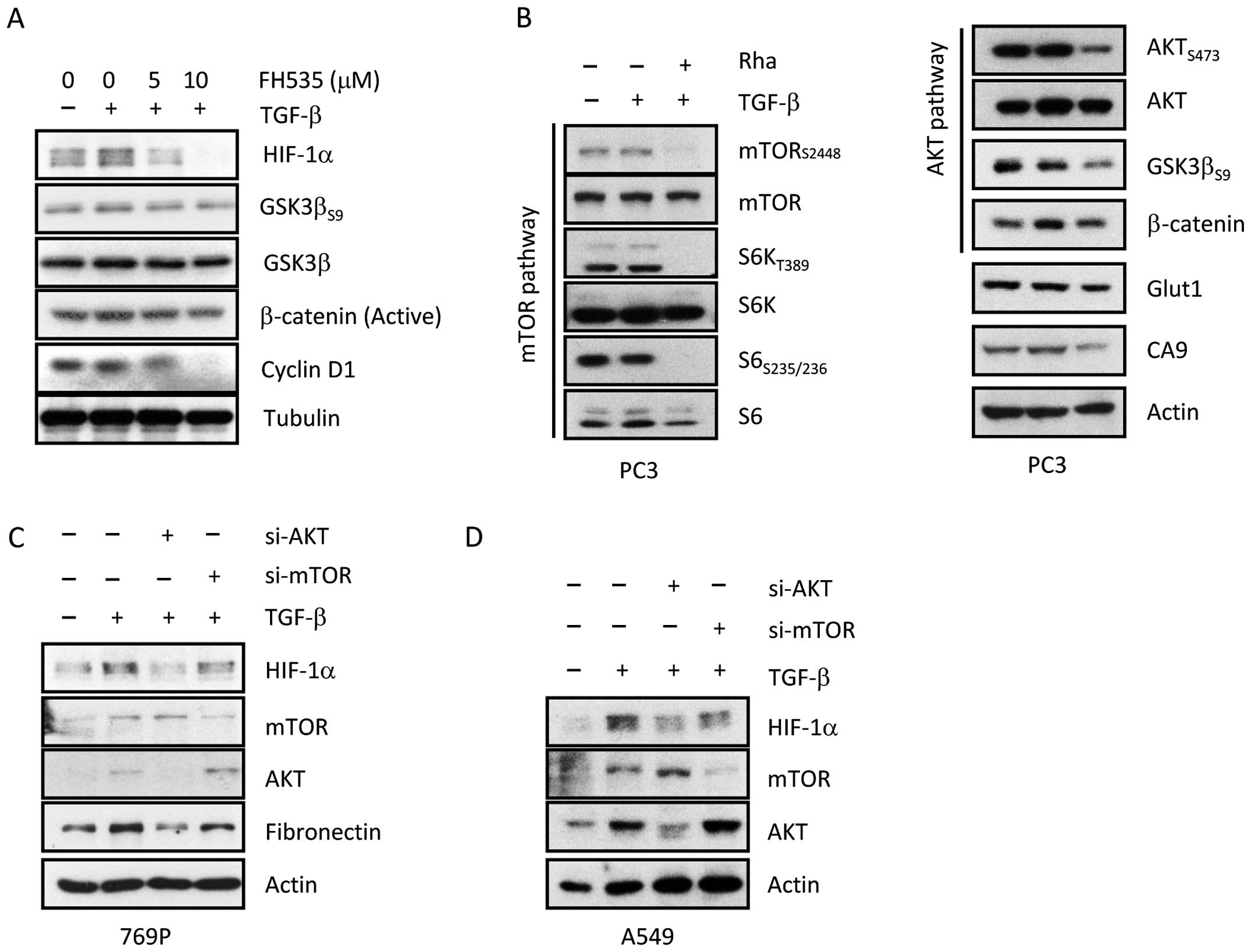
As shown in Fig. 5A, Wnt/β-catenin
inhibitor FH535 inhibited TGF-β-induced expression of HIF-1α and
phosphorylation of GSK-3β at serine 9 in a dose-dependent manner.
As the concentration of FH535 was increased, the decreased protein
level of HIF-1α was concomitant with the decrease in
phosphorylation of GSK-3β at serine 9 and active
(non-phosphorylated) β-catenin in PC3 cells. In addition, FH535
treatment also significantly decreased the protein level of cyclin
D1 in a dose-dependent manner in PC3 cells, demonstrating the
association between β-catenin and cyclin D1 (25,26).
We next examined the inhibitory effect of Rha on the mTOR and Akt
signaling pathways. As shown in Fig.
5B, Rha abolished mTOR signaling by reducing phosphorylation of
mTOR at Ser2448, S6K at Thr389, and S6 at Ser235/236. In addition,
phosphorylation of molecules involved in Akt signaling, such as Akt
(Ser473), GSK-3β (Ser9), and β-catenin, was also attenuated by Rha
treatment. Furthermore, expression of HIF-1α downstream gene CA9
was also reduced by Rha treatment. These results suggest that Akt
and mTOR may be important for TGF-β-induced expression of HIF-1α;
therefore, to confirm this hypothesis, 769-P cells were transfected
with siRNA against Akt and mTOR for 24 h, followed by exposure to
TGF-β in the presence or absence of Rha for 24 h, after which the
cells were collected for western blotting. As shown in Fig. 5C, knockdown of Akt and mTOR by siRNA
markedly attenuated TGF-β mediated HIF-1α expression in 769-P
cells, indicating that Akt and mTOR are required for TGF-β mediated
expression of HIF-1α. However, the effect of Akt inhibition was
more significant than that of mTOR inhibition. Similar effects were
observed in A549 cells (Fig.
5D).
Rha inhibits TGF-β-mediated EMT via the
PI3K/AKT pathway
Taken together with previous studies, the results
described above prompted us to investigate whether the alterations
in EMT induced by TGF-β enhanced invasiveness and metastasis
(5,15,16,27–30).
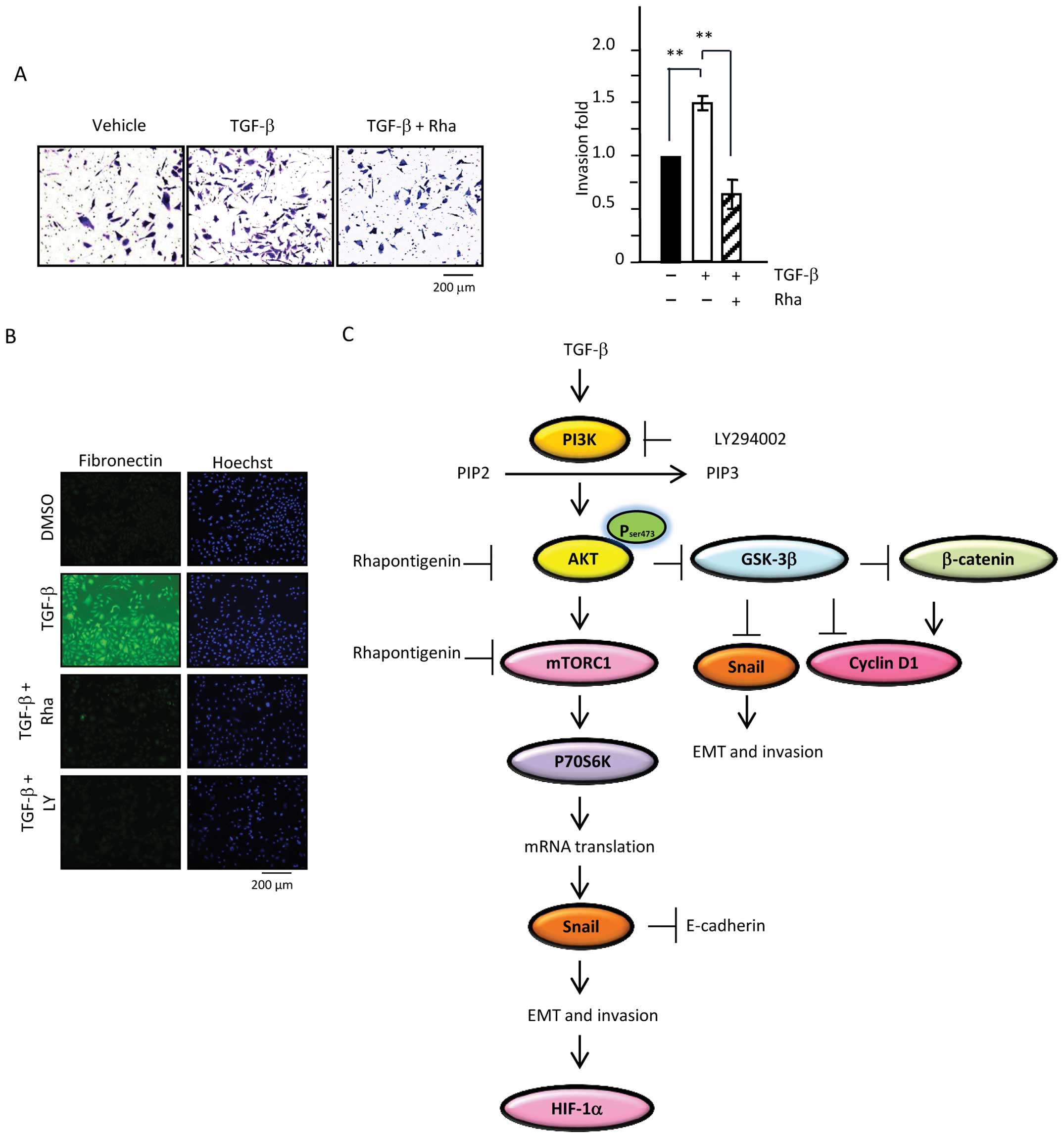
Therefore, IFA was performed. As shown in Fig. 6B, cells exposed to TGF-β for 48 h
had higher expression of mesenchymal marker FN. In contrast, cells
treated with LY or Rha in the presence of TGF-β lacked FN protein.
Because of the connection between increased FN expression and cell
invasiveness, cell invasion assays were employed to confirm the
inhibitory effect of Rha. 769-P cells were exposed to TGF-β in the
presence or absence of Rha for 20 h, followed by re-trypsinization
and re-seeding of the treated cells into an invasion chamber
harboring Matrigel (1.25×105 cells). After 48 h of
incubation, the invasive ability of the cells that had penetrated
through the inserts was assessed by crystal violent staining
microscopic examination. Cells stimulated with TGF-β had an
enhanced invasive ability in comparison with that of the control
cells. Moreover, cells exposed to TGF-β and Rha had less invasive
ability than cells treated with TGF-β only (Fig. 6A). Taken together, our results
demonstrate that TGF-β is a contributing factor in cancer
progression and metastasis. Moreover, our results suggest that Rha
may suppress TGF-β-mediated invasion via inhibition of the
PI3K/Akt/mTOR/GSK-3β/β-catenin signaling pathway (Fig. 6C).
Discussion
In this study, we investigated the potential link
between inflammation and the TME. In 1863, Virchow reported that
cancers tended to occur at sites of chronic inflammation (2). Later, a wide array of evidence
confirmed the connection between inflammation and cancer, as best
demonstrated by inflammatory bowel disease (IBD) and renal fibrosis
(27,31,32).
As a result of such studies, it has been accepted that the state of
the TME plays an important role in cancer development.
The results described above demonstrate that TGF-β
plays a pivotal role in the EMT and subsequent cancer cell
invasion. In order to survive and adapt to stressful environments,
stromal cells secrete cytokines that allow them to avoid detection
by the immune system, thus promoting the development of
inflammation-related cancer. Furthermore, invasion and metastasis
are crucial biological processes of malignant tumors. During the
progression to metastasis, the EMT allows cancer cells to acquire
mesenchymal-like characteristics that enhance invasiveness.
For decades, the transcription factor HIF-1α has
been implicated in cell survival, the EMT, and anticancer drug
resistance (33,34). Therefore, HIF-1α is an attractive
target for chemopreventive agents. Under normoxic conditions,
HIF-1α is activated by hormones, cytokines, and other signaling
molecules (33,35). The mechanism underlying
cytokine-induced HIF-1α expression is different from that by which
it is upregulated during hypoxia. Growth factor-mediated HIF-1α
activation is associated with increased protein synthesis and
reduced protein degradation via regulation of PI3K and MAPK
signaling (36–39), in accordance with our observation
that TGF-β mediated HIF-1α expression in the cancer cells tested in
our study. In contrast, hypoxia-mediated HIF-1α expression is
usually achieved by decreasing HIF-1α degradation and blocking
recognition of HIF-1α by E3 ligase VHL (33).
Numerous studies have demonstrated that the
stability of Snail is regulated primarily by Akt/GSK3β and NF-κB
(19,40,41).
We found that Rha decreased the stability of Snail by suppressing
PI3K/Akt/mTOR signaling. Snail-mediated FN upregulation and
E-cadherin downregulation are hallmarks of the EMT. In addition,
activation of PI3K/Akt signaling has been reported as a
characteristic of EMT (42,43). Our results indicate that the
ubiquitin-protea-some pathway was involved in regulating turnover
of HIF-1α and Snail in diverse cell lines. Furthermore, our
findings demonstrate that Snail and HIF-1α were recruited to the
26S proteasome for degradation as a result of Rha exposure. Taken
together, our results revealed that Snail is required for TGF-β
induced EMT, whereas HIF-1α is not.
The present study demonstrates that Snail plays a
pivotal role in TGF-β mediated EMT and is associated with
TGF-β-driven cell invasion. Based on these results, we have
identified mechanisms that underlie the effects of Rha on TGF-β and
HIF-1α signaling in cancer cells. First, Rha inhibited
TGF-β-triggered invasion. Second, Rha, like LY, strongly inhibited
TGF-β-induced EMT via PI3K/Akt/mTOR signaling (Fig. 6C). Therefore, targeting
PI3K/Akt/mTOR and HIF-1α could be useful therapeutic strategies for
preventing tumor metastasis (Fig.
6D). Our experiments demonstrated the link between
PI3K/Akt/mTOR signaling and TGF-β, which can be regulated by Rha.
We showed that Rha increased proteolytic degradation of HIF-1α and
suppressed TGF-β-mediated EMT. Our results demonstrate for the
first time that Rha inhibits cancer progression and metastasis by
suppressing the EMT via regulation of the PI3K/Akt/mTOR pathway.
Additionally, Rha was found to reduce HIF-1α protein expression in
a dose-dependent manner. Given the multiple functions of Rha,
future studies should be aimed at confirming its potential as a
chemopreventive agent.
Acknowledgments
This study was supported by the grants from the
Ministry of Science and Technology (MOST 104-2325-B-002-040) as
well as (Most-103-2320-B-002-063-MY3).
References
|
1
|
Grivennikov SI and Karin M: Inflammation
and oncogenesis: A vicious connection. Curr Opin Genet Dev.
20:65–71. 2010. View Article : Google Scholar :
|
|
2
|
Quail DF and Joyce JA: Microenvironmental
regulation of tumor progression and metastasis. Nat Med.
19:1423–1437. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Landskron G, De la Fuente M, Thuwajit P,
Thuwajit C and Hermoso MA: Chronic inflammation and cytokines in
the tumor microenvironment. J Immunol Res. 2014:1491852014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Akhurst RJ and Hata A: Targeting the TGFβ
signalling pathway in disease. Nat Rev Drug Discov. 11:790–811.
2012. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Das P and Golde T: Dysfunction of TGF-beta
signaling in Alzheimer's disease. J Clin Invest. 116:2855–2857.
2006. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jou J and Diehl AM: Epithelial-mesenchymal
transitions and hepatocarcinogenesis. J Clin Invest. 120:1031–1034.
2010. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kang Y and Massagué J:
Epithelial-mesenchymal transitions: Twist in development and
metastasis. Cell. 118:277–279. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Savagner P: The epithelial-mesenchymal
transition (EMT) phenomenon. Ann Oncol. 21(Suppl 7): vii89–vii92.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sun XD, Liu XE and Huang DS: Curcumin
reverses the epithelial-mesenchymal transition of pancreatic cancer
cells by inhibiting the Hedgehog signaling pathway. Oncol Rep.
29:2401–2407. 2013.PubMed/NCBI
|
|
11
|
Kim YH, Kim G, Kwon CI, Kim JW, Park PW
and Hahm KB: TWIST1 and SNAI1 as markers of poor prognosis in human
colorectal cancer are associated with the expression of ALDH1 and
TGF-β1. Oncol Rep. 31:1380–1388. 2014.PubMed/NCBI
|
|
12
|
Yeh YH, Yang YC, Hsieh MY, Yeh YC and Li
TK: A negative feedback of the HIF-1α pathway via
interferon-stimulated gene 15 and ISGylation. Clin Cancer Res.
19:5927–5939. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chaudhry P, Fabi F, Singh M, Parent S,
Leblanc V and Asselin E: Prostate apoptosis response-4 mediates
TGF-β-induced epithelial-to-mesenchymal transition. Cell Death Dis.
5:e10442014. View Article : Google Scholar
|
|
14
|
Huang J, Yao X, Zhang J, Dong B, Chen Q,
Xue W, Liu D and Huang Y: Hypoxia-induced downregulation of miR-30c
promotes epithelial-mesenchymal transition in human renal cell
carcinoma. Cancer Sci. 104:1609–1617. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xu J, Lamouille S and Derynck R:
TGF-beta-induced epithelial to mesenchymal transition. Cell Res.
19:156–172. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bakin AV, Tomlinson AK, Bhowmick NA, Moses
HL and Arteaga CL: Phosphatidylinositol 3-kinase function is
required for transforming growth factor beta-mediated epithelial to
mesenchymal transition and cell migration. J Biol Chem.
275:36803–36810. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang HY, Wang ZQ, Li YY, Wang F, Zeng QR,
Gao Y, Xuan XY and Li SS: Transforming growth factor-β1-induced
epithelial-mesenchymal transition in human esophageal squamous cell
carcinoma via the PTEN/PI3K signaling pathway. Oncol Rep.
32:2134–2142. 2014.PubMed/NCBI
|
|
18
|
Zhang YE: Non-Smad pathways in TGF-beta
signaling. Cell Res. 19:128–139. 2009. View Article : Google Scholar :
|
|
19
|
Brandl M, Seidler B, Haller F, Adamski J,
Schmid RM, Saur D and Schneider G: IKK(α) controls canonical
TGF(β)-SMAD signaling to regulate genes expressing SNAIL and SLUG
during EMT in panc1 cells. J Cell Sci. 123:4231–4239. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lin Y, Dong C and Zhou BP: Epigenetic
regulation of EMT: The Snail story. Curr Pharm Des. 20:1698–1705.
2014. View Article : Google Scholar :
|
|
21
|
Kitagishi Y, Kobayashi M, Kikuta K and
Matsuda S: Roles of PI3K/AKT/GSK3/mTOR pathway in cell signaling of
mental illnesses. Depress Res Treat. 2012:7525632012.
|
|
22
|
Shiojima I and Walsh K: Regulation of
cardiac growth and coronary angiogenesis by the Akt/PKB signaling
pathway. Genes Dev. 20:3347–3365. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Aberle H, Bauer A, Stappert J, Kispert A
and Kemler R: Beta-catenin is a target for the ubiquitin-proteasome
pathway. EMBO J. 16:3797–3804. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
McCubrey JA, Steelman LS, Bertrand FE,
Davis NM, Abrams SL, Montalto G, D'Assoro AB, Libra M, Nicoletti F,
Maestro R, et al: Multifaceted roles of GSK-3 and Wnt/β-catenin in
hematopoiesis and leukemogenesis: Opportunities for therapeutic
intervention. Leukemia. 28:15–33. 2014. View Article : Google Scholar :
|
|
25
|
Shtutman M, Zhurinsky J, Simcha I,
Albanese C, D'Amico M, Pestell R and Ben-Ze'ev A: The cyclin D1
gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad
Sci USA. 96:5522–5527. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tetsu O and McCormick F: Beta-catenin
regulates expression of cyclin D1 in colon carcinoma cells. Nature.
398:422–426. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Blobe GC, Schiemann WP and Lodish HF: Role
of transforming growth factor beta in human disease. N Engl J Med.
342:1350–1358. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Padua D and Massagué J: Roles of TGFbeta
in metastasis. Cell Res. 19:89–102. 2009. View Article : Google Scholar
|
|
29
|
Son H and Moon A: Epithelial-mesenchymal
transition and cell invasion. Toxicol Res. 26:245–252. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tsai JH and Yang J: Epithelial-mesenchymal
plasticity in carcinoma metastasis. Genes Dev. 27:2192–2206. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Colotta F, Allavena P, Sica A, Garlanda C
and Mantovani A: Cancer-related inflammation, the seventh hallmark
of cancer: Links to genetic instability. Carcinogenesis.
30:1073–1081. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Meng XM, Tang PM, Li J and Lan HY:
TGF-β/Smad signaling in renal fibrosis. Front Physiol. 6:822015.
View Article : Google Scholar
|
|
33
|
Semenza GL: Targeting HIF-1 for cancer
therapy. Nat Rev Cancer. 3:721–732. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang Q, Bai X, Chen W, Ma T, Hu Q, Liang
C, Xie S, Chen C, Hu L, Xu S, et al: Wnt/β-catenin signaling
enhances hypoxia-induced epithelial-mesenchymal transition in
hepatocellular carcinoma via crosstalk with hif-1α signaling.
Carcinogenesis. 34:962–973. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jiang BH and Liu LZ: PI3K/PTEN signaling
in angiogenesis and tumorigenesis. Adv Cancer Res. 102:19–65. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Agani F and Jiang BH: Oxygen-independent
regulation of HIF-1: Novel involvement of PI3K/AKT/mTOR pathway in
cancer. Curr Cancer Drug Targets. 13:245–251. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jung YJ, Isaacs JS, Lee S, Trepel J and
Neckers L: IL-1beta-mediated up-regulation of HIF-1alpha via an
NFkappaB/COX-2 pathway identifies HIF-1 as a critical link between
inflammation and oncogenesis. FASEB J. 17:2115–2117.
2003.PubMed/NCBI
|
|
38
|
Kasuno K, Takabuchi S, Fukuda K,
Kizaka-Kondoh S, Yodoi J, Adachi T, Semenza GL and Hirota K: Nitric
oxide induces hypoxia-inducible factor 1 activation that is
dependent on MAPK and phosphatidylinositol 3-kinase signaling. J
Biol Chem. 279:2550–2558. 2004. View Article : Google Scholar
|
|
39
|
Laughner E, Taghavi P, Chiles K, Mahon PC
and Semenza GL: HER2 (neu) signaling increases the rate of
hypoxia-inducible factor 1alpha (HIF-1alpha) synthesis: Novel
mechanism for HIF-1-mediated vascular endothelial growth factor
expression. Mol Cell Biol. 21:3995–4004. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wu Y, Deng J, Rychahou PG, Qiu S, Evers BM
and Zhou BP: Stabilization of snail by NF-kappaB is required for
inflammation-induced cell migration and invasion. Cancer Cell.
15:416–428. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhou BP, Deng J, Xia W, Xu J, Li YM,
Gunduz M and Hung MC: Dual regulation of Snail by
GSK-3beta-mediated phosphorylation in control of
epithelial-mesenchymal transition. Nat Cell Biol. 6:931–940. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Larue L and Bellacosa A:
Epithelial-mesenchymal transition in development and cancer: Role
of phosphatidylinositol 3′ kinase/AKT pathways. Oncogene.
24:7443–7454. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wang Y, Shi J, Chai K, Ying X and Zhou BP:
The role of Snail in EMT and tumorigenesis. Curr Cancer Drug
Targets. 13:963–972. 2013. View Article : Google Scholar : PubMed/NCBI
|