Introduction
Estrogen receptor (ER) is an important prognostic
marker and therapeutic target of breast cancer. There are two
classes of ER: ERα and ERβ (1). ERα
is predominantly expressed in breast ductal epithelial cells. It
plays a crucial role in both mammary carcinogenesis and breast
cancer progression (2,3). ERα is a member of the nuclear receptor
superfamily of transcription factors whose activity is primarily
regulated by estrogen (E2) binding. It regulates the transcription
of target genes (4,5). ERα-positive breast cancer accounts for
70% of all breast cancer cases. Patients with these tumors are
candidates for anti-estrogen therapy after surgical treatment. Such
therapy is through blockage of binding of E2/ERα with selective ER
modulators (SERMs) such as tamoxifen or by inhibiting E2
biosynthesis using aromatase inhibitors (AIs) (6). Only 60% of all ERα-positive breast
cancers are responsive to tamoxifen. Unfortunately, the majority of
these patients who do respond well initially often develop
resistance to tamoxifen therapy and have relapse during their
clinical courses (7). Although
acquisition of tamoxifen resistance (TR) is due to a variety of
factors, the mechanisms underlying this phenomenon remain poorly
understood.
As a biguanide derivative, metformin
(1,1-dimethylbiguanide hydrochloride) can suppress insulin levels
(8), but increase insulin
sensitivity of peripheral tissues (9). Accordingly, it has been approved to
treat type II diabetes mellitus. Notably, several meta-analyses
recently confirmed that metformin therapy could reduce the
incidence of cancers, including breast and colorectal cancer,
hepatocarcinoma and cancer-related mortality (10–14).
Moreover, metformin has been reported to be able to inhibit
proliferation and induce apoptosis in triple-negative breast cancer
cell lines (15,16). The antitumor properties of metformin
have been ascribed to its ability to activate adenosine
monophosphate kinase (AMPK), thus, inhibiting the mammalian target
of rapamycin (mTOR), a promoter of cell growth and proliferation
(8,17,18).
Based on these properties, metformin has gained increasing
attention as a potential anticancer agent (19). Metformin has been shown to be able
to reduce ER expression in endometrial tumors of women with type II
diabetes mellitus (20). However,
its effect on the expression and function of ERα in ERα-positive
breast cancers regardless of diabetes is currently unclear.
Therefore, the objective of the present study was to investigate
the anticancer activity of metformin in relation to ERα expression
and its signaling pathway in ERα-positive MCF-7 and MDA-MB-361
breast cancer cells, and TR MCF-7 breast cancer cells. Notably, it
was found that metformin may be more effective at inhibiting ERα
signaling by estrogenic stimulation compared to tamoxifen for
ERα-positive breast cancers and that metformin may be an effective
therapeutic agent for treating tamoxifen-resistant breast
cancer.
Materials and methods
Cell lines and reagents
Human breast cancer cell lines MCF-7 and MDA-MB-361
were purchased from the American Type Culture Collection (ATCC;
Manassas, VA, USA). MCF-7 cells were cultured in RPMI-1640 medium
supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin
and 100 µg/ml streptomycin (Gibco, Grand Island, NY, USA).
MDA-MB-361 cells were cultured in Leibovitz's L-15 medium (Gibco)
supplemented with 20% FBS at 37°C in a 5% CO2 humidified
incubator. Metformin, 17β-estradiol (E2) and 4-hydroxytamoxifen
(4-OHT) were purchased from Sigma (St. Louis, MO, USA). Metformin
was dissolved in sterile water. E2 and 4-OHT were dissolved in
ethanol. They were immediately stored at −80°C.
Establishment of the tamoxifen-resistant
(TR) MCF-7 cell line
MCF-7 cells were cultured in phenol red-free
RPMI-1640 medium supplemented with 10% charcoal stripped FBS (PAA
Laboratories, Morningside, Australia) to deplete estrogen. The
tamoxifen-resistant (TR) MCF-7 cell line was established from MCF-7
cells following continuous exposure to 10−6 M 4-OHT, an
active metabolite of tamoxifen (21,22).
Under these conditions, the growth rates of MCF-7 cells were
reduced. However, after ~2 months, cell growth was gradually
increased, indicating an acquisition of resistance to growth
inhibition of 4-OHT. Designated TR MCF-7 cells were cultured for an
additional 4 months in medium containing 4-OHT before
characterization studies.
Isolation of RNA and quantitative
real-time RT-PCR (RT-qPCR)
Total RNA was isolated using the RNeasy Mini kit
(Qiagen, Hilden, Germany). For cDNA synthesis, 1 µg of total
RNA was subjected to reverse transcription-polymerase chain
reaction (RT-PCR) assay using CycleScript RT PreMix kit (Bioneer
Corporation, Daejeon, Korea). RT-qPCR was performed with Power
SYBR-Green PCR Master Mix on an ABI 7300 real-time PCR system (both
from Applied Biosystems, Warrington, UK) using the following
cycling conditions: 50°C for 2 min, 95°C for 5 min, followed by 40
cycles of 95°C for 30 sec, 55°C for ERα and pS2 or 60°C for cyclin
D1 for 30 sec, 72°C for 30 sec. The following primers were used:
5′-AGCACCCAGTGAAGCTACT-3′ (ERα-forward) and
5′-TAGGGCACACAAACTCCT-3′ (ERα-reverse);
5′-TATGAATCACTTCTGCAGTGAG-3′ (pS2-forward) and
5′-GAGCGTTAGATAACATTTGCC-3′ (pS2-reverse); 5′-CGCCCCACCCCTCCAG-3′
(cyclin D1-forward) and 5′-CCGCCCAGACCCTCAGACT-3′ (cyclin
D1-reverse); and 5′-ATCATCCCTGCCTCTACTGG-3′ (forward) and
5′-CCCTCCGACGCCTGCTT-CAC-3′ (reverse) for GAPDH as internal
standard. Cycle threshold values were normalized to those of GAPDH.
The relative fold-change was calculated using the 2−ΔΔCt
method.
Western blot analysis
Protein lysates were prepared with RIPA buffer (20
mM Tris-HCl pH 7.5, 2 mM EDTA, 150 mM NaCl, 1 mM sodium vanadate,
10 mM NaF, 2.5 mM sodium pyrophosphate, 1% sodium deoxycholate,
0.1% SDS, 1% NP-40) supplemented with protease inhibitor cocktail
(Roche, Mannheim, Germany). Protein concentrations were determined
using a BCA protein assay kit (Thermo Scientific, Rockford, IL,
USA). Protein samples (30 µg) were resolved by SDS-PAGE and
transferred onto polyvinylidene fluoride (PVDF) membranes (Bio-Rad
Laboratories). Membranes were blocked with skim milk and incubated
with the primary antibodies. Following washing, the membranes were
incubated with horseradish peroxidase-conjugated anti-mouse or
anti-rabbit secondary antibody and developed with ECL Plus Western
Blot Detection System reagent (GE Healthcare Biosciences,
Piscataway, NJ, USA). Rabbit anti-ERα (1:2,000) antibody was
purchased from Millipore (Billerica, MA, USA). Mouse anti-cyclin D1
(1:1,000), rabbit anti-TFF1/pS2 (1:1,000), rabbit
anti-phospho-AMPKα (Thr172, 1:1,000) and rabbit anti-AMPKα
(1:1,000) antibodies were purchased from Cell Signaling Technology
Inc. (Beverly, MA, USA). Mouse anti-c-Myc (1:1,000) and rabbit
anti-progesterone receptor (PR) (1:1,000) antibodies were purchased
from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). A monoclonal
anti-β-actin (1:5,000) antibody obtained from Sigma was used to
determine protein loading. Protein levels were quantified using
ImageJ software (NIH, Bethesda, MD, USA).
Cell proliferation assay
Cells were seeded into 12-well plates at a density
of 5×104 cells/well (500 µl/well). After
incubation for 24 h, the cells were treated with metformin, 4-OHT
and E2 in estrogen-depleted RPMI-1640 medium and incubated for an
additional 72 h. After that, the cells were trypsinized and counted
after staining with trypan blue dye solution using the TC10™
Automated Cell Counter (Bio-Rad Laboratories). The number of viable
cells in each well was calculated. Results are presented as
relative percentage to the control of each group from three
independent experiments in triplicates.
Luciferase assay
The transcriptional activity of ERα was analyzed by
luciferase assay using pGL2-3X ERE TATA luc plasmid (Addgene,
Cambridge MA, USA). Cells were seeded into 12-well plates and grown
to confluency. After co-transfection with pGL2-3X ERE TATA luc and
pRL-TK-luc control plasmid (Promega, Madison, WI, USA) using
Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA) for 24
h, the cells were treated with metformin, 4-OHT and E2 in
estrogen-depleted RPMI-1640 medium. After an additional incubation
for 24 h, luciferase activity was measured using the
Dual-Luciferase reporter assay system (Promega) and normalized to
pRL-TK-luc activity to correct for differences in transfection
efficiency. Results are presented as fold-change relative to the
control of each group from three independent experiments in
triplicates.
Statistical analysis
Each experiment was performed independently at least
three times. Data are presented as mean ± standard deviation for
each experiment. Comparisons between two groups were performed
using the Student's t-test. P<0.05 and P<0.01 were considered
to indicate a statistically significant result.
Results
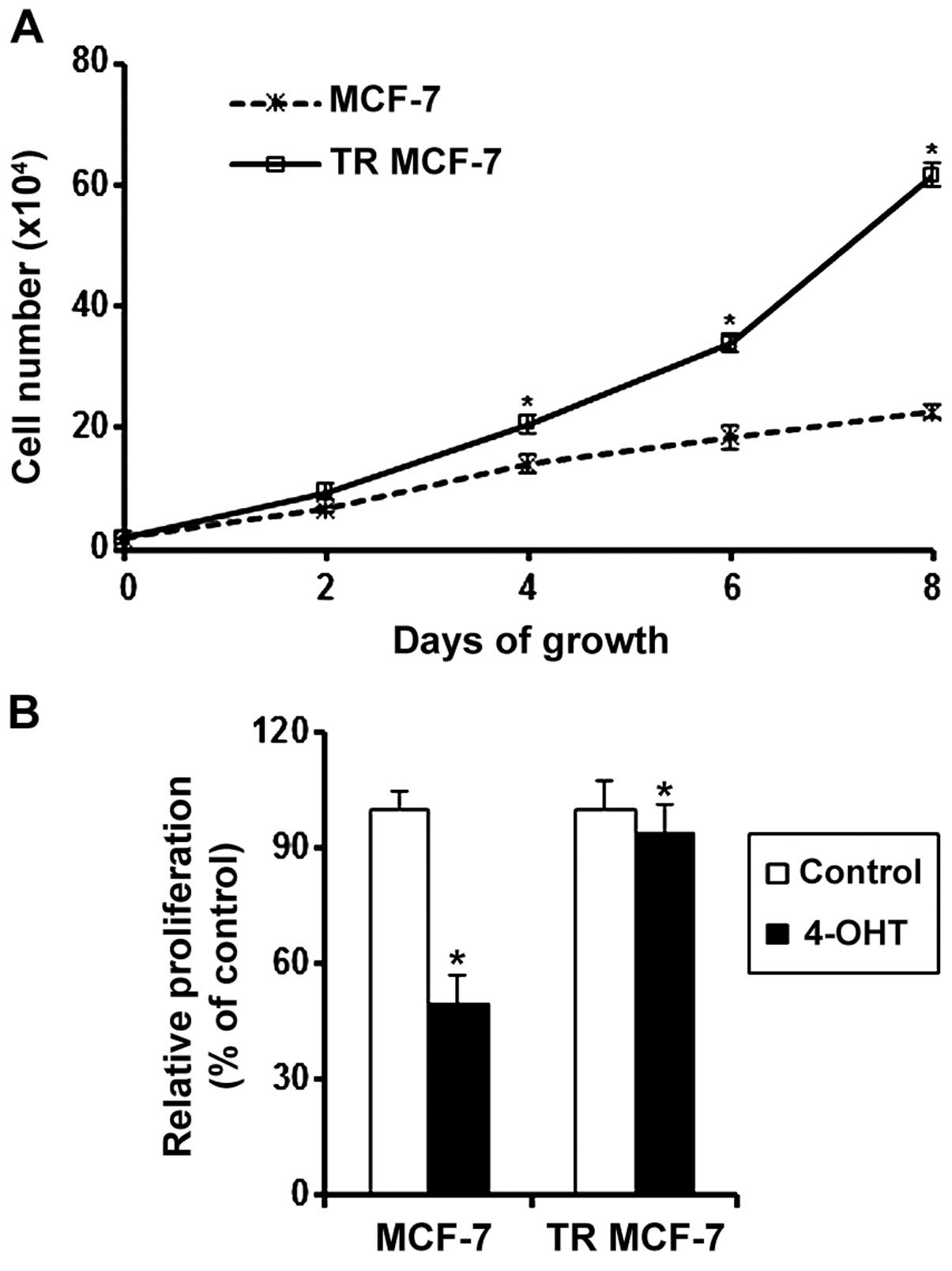
Effect of 4-OHT on the proliferation of
MCF-7 and TR MCF-7 cells
To evaluate the acquisition of resistance to the
anti-estrogenic action of 4-OHT, we measured the cell proliferation
rate of proliferation MCF-7 and the TR MCF-7 cells every 2 days by
counting cell numbers. The proliferation rate of proliferation TR
MCF-7 cells was significantly higher than that of the MCF-7 cells
in the presence of 4-OHT (Fig. 1A).
We also determined the proliferation of MCF-7 and TR MCF-7 cells
treated with 4-OHT or the control vehicle (Ctrl) for 5 days. The
proliferation of the 4-OHT-treated MCF-7 cells was inhibited by 50%
compared to that of the Ctrl cells. A slight decrease in the
proliferation was observed in the 4-OHT-treated TR MCF-7 cells
(Fig. 1B).
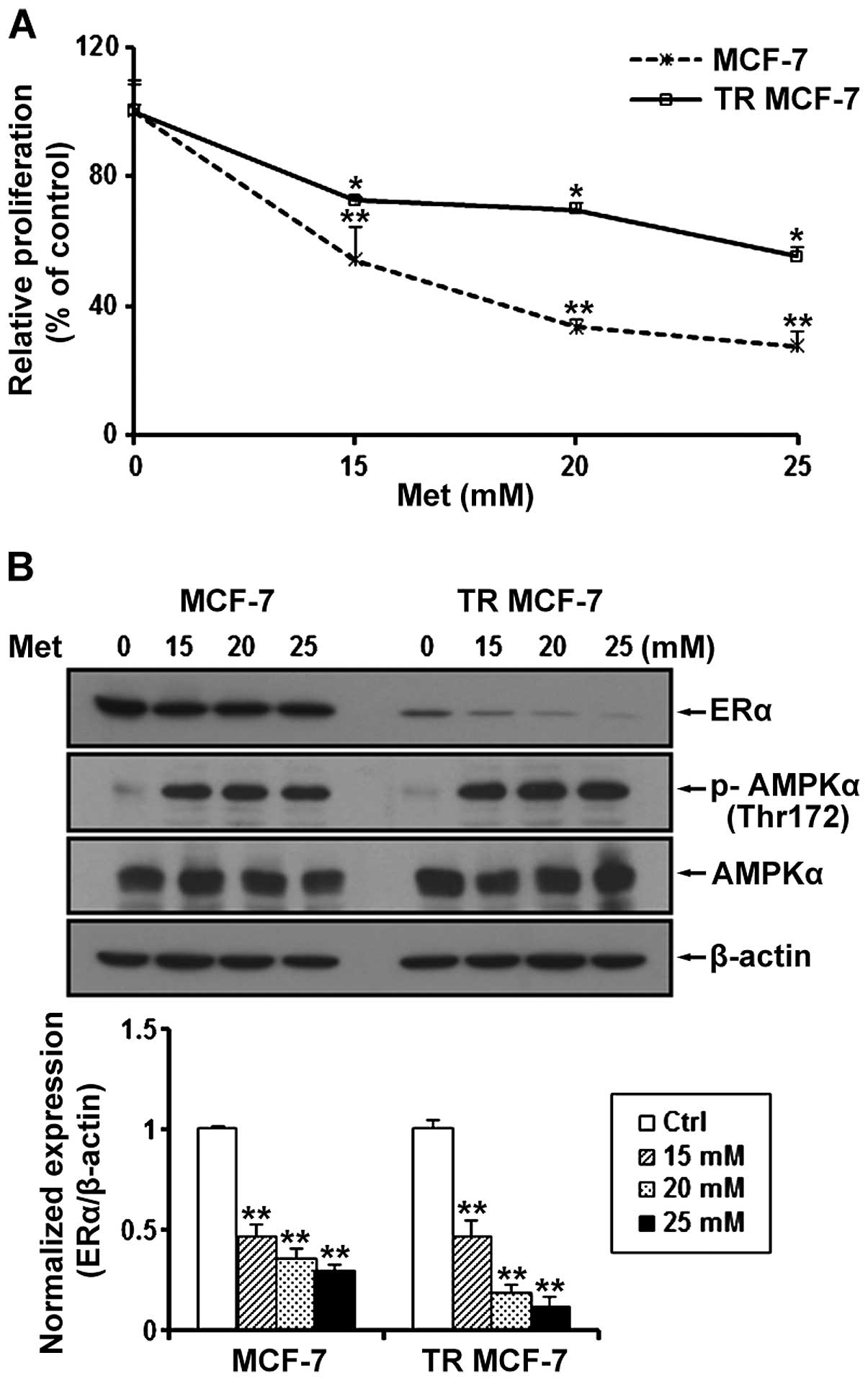
Metformin inhibits cell proliferation and
ERα protein levels in MCF-7 and TR MCF-7 cells
We determined the proliferation of MCF-7 and TR
MCF-7 cells treated with metformin at a concentration of 15, 20 and
25 mM for 72 h. Compared to the untreated control cells, metformin
inhibited the cell proliferation of the MCF-7 and TR MCF-7 cells in
a dose-dependent manner (Fig. 2A).
Compared to the untreated cells, the cell proliferation of the
MCF-7 and TR MCF-7 cells was inhibited 70 and 50%, respectively, by
25 mM metformin, indicating that the TR MCF-7 cells were less
sensitive at the same concentration of metformin compared to the
MCF-7 parental cells for the antiproliferative effect of metformin.
Next, we performed western blot analysis to evaluate the effect of
metformin on ERα protein levels. We found that the protein levels
of ERα were reduced in a dose-dependent manner by treatment with
15, 20 and 25 mM metformin in both cell lines (Fig. 2B). The concentrations of metformin
used in the present study are high, but to focus on the anticancer
effects of metformin on TR MCF-7 cells, based on these results,
subsequent experiments were carried out using 25 mM metformin. We
also determined AMPKα phosphorylation (Thr172) and total AMPKα
levels to demonstrate that metformin was present and active during
our experiments.
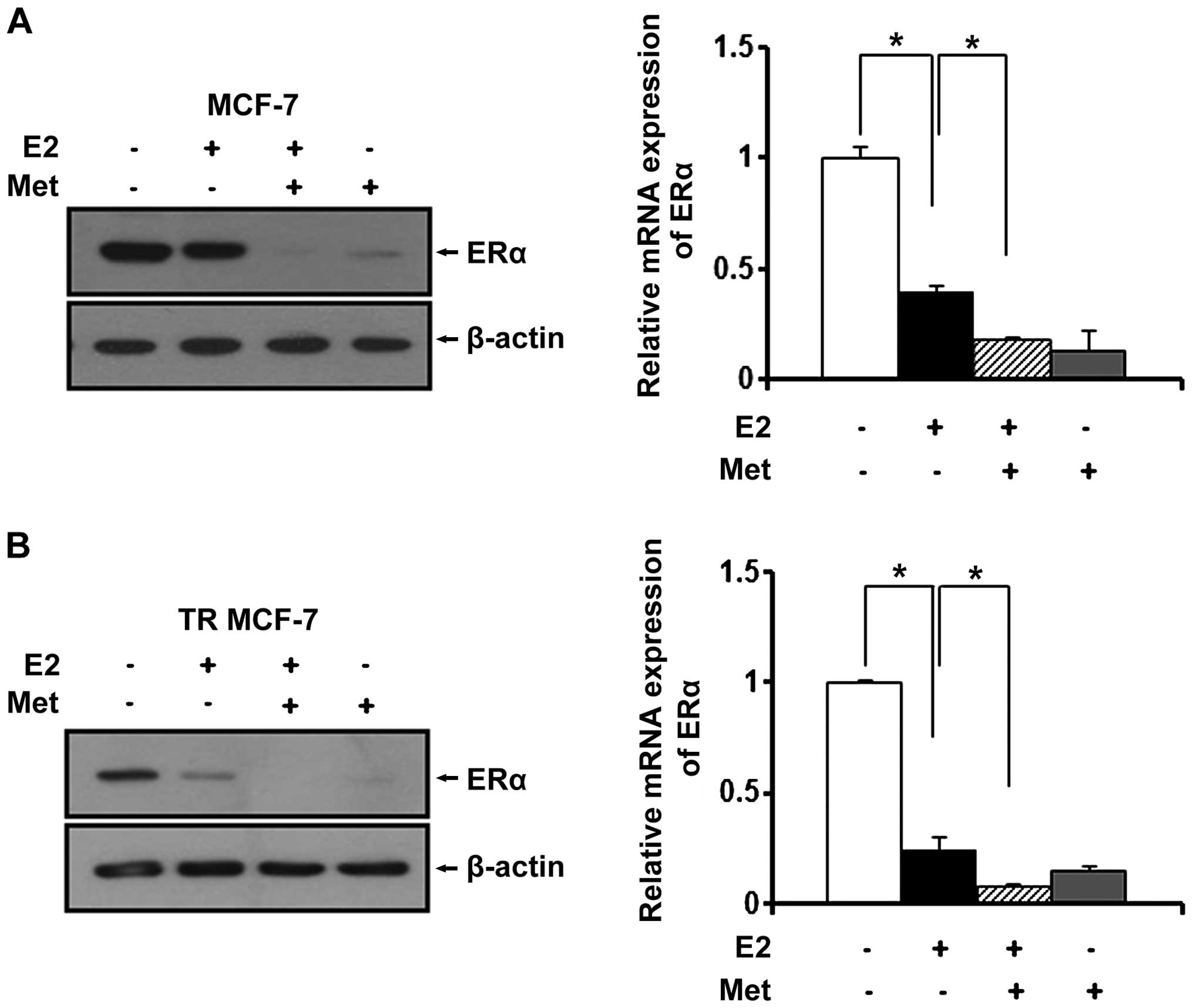
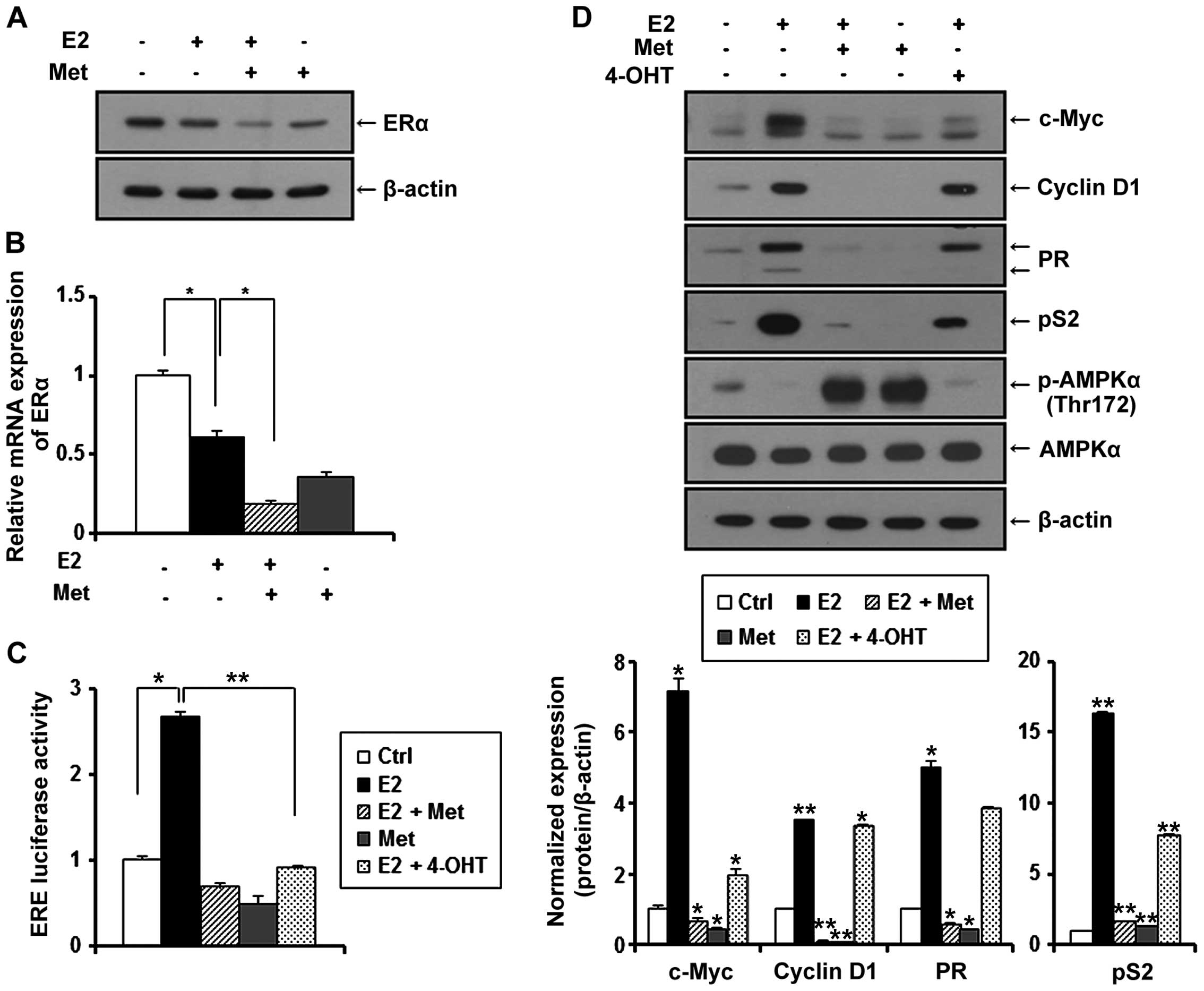
Metformin inhibits the expression of ERα
in MCF-7 and TR MCF-7 cells under E2
Since estrogen (E2) is a stimulatory signal for
breast cancer development and progression, we treated the MCF-7 and
TR MCF-7 cells with metformin in an E2-exposed physiological
condition. After treatment with E2, changes in the ERα protein
levels were evaluated by western blot analysis. E2 has been
reported to be able to rapidly reduce the levels of ERα protein
transiently expressed in cells (23). As shown in Fig. 3A and B (left panels), the E2-treated
cells had decreased ERα protein levels compared to these levels in
the untreated control cells. The protein levels of ERα were
decreased more significantly after treatment with metformin. Based
on RT-qPCR, we found that metformin also repressed the E2-induced
mRNA level of ERα in the MCF-7 cells (0.6-fold decrease compared to
E2-treated only cells) and the TR MCF-7 cells (0.7-fold decrease
compared to E2-treated only cells) (Fig. 3A and B, right panels).
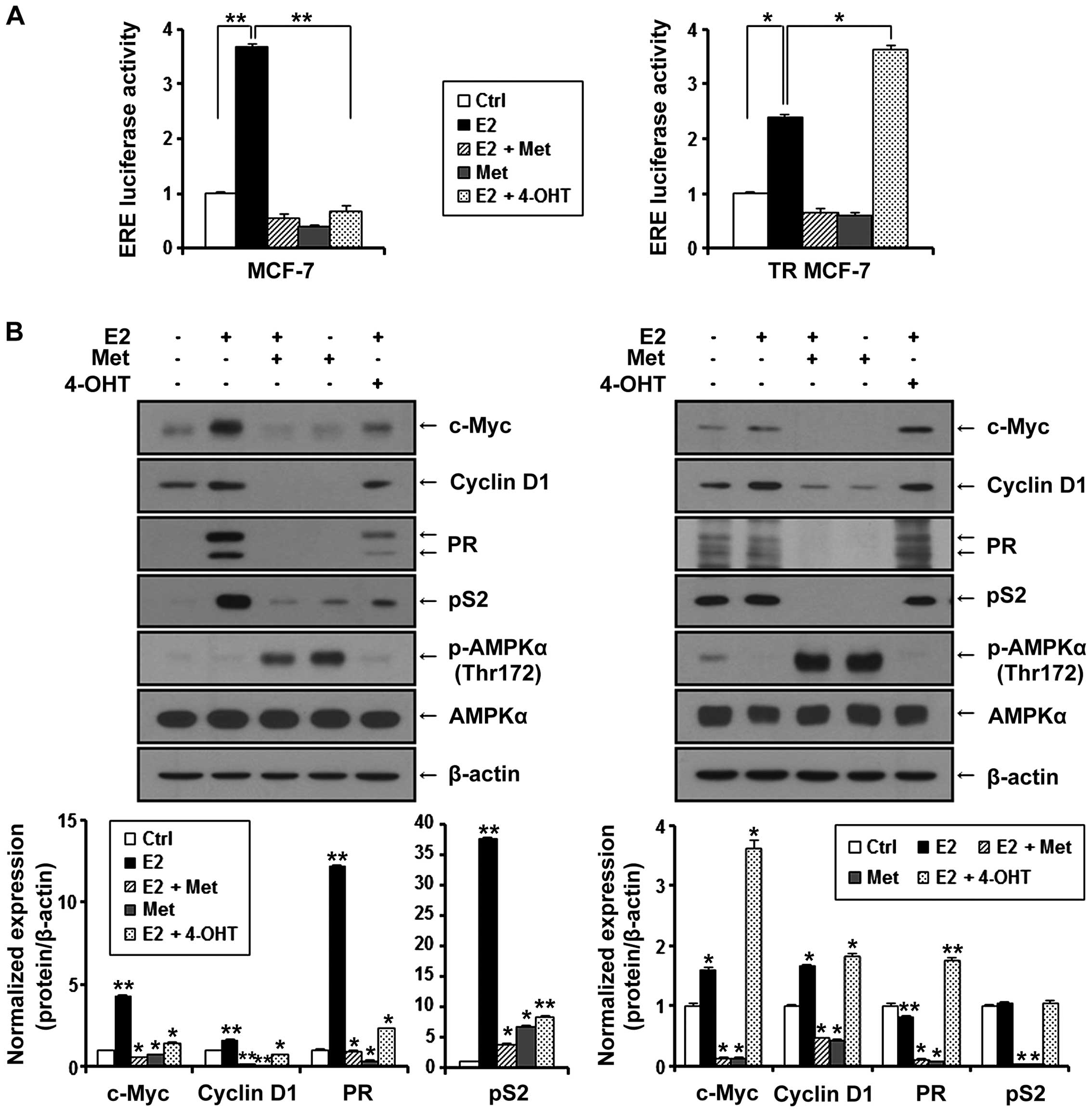
Metformin inhibits E2-inducible ERE
luciferase activity and the expression of ERα target genes in MCF-7
and TR MCF-7 cells
ERα is known to interact with the estrogen response
element (ERE) site of target genes and activate the transcription
of regulated genes in response to E2. Therefore, we evaluated the
effect of metformin on the transcriptional activity of ERα compared
to anti-estrogenic agent 4-OHT. MCF-7 and TR MCF-7 cells were
transiently co-transfected with E2-inducible luciferase reporter
gene (pGL2-3X ERE TATA luc) and pRL-TK-luc control plasmid.
Transfected cells were treated with metformin and 4-OHT under
stimulation of E2 or with only metformin for 24 h. As shown in
Fig. 4A, E2-induced ERE luciferase
activity was inhibited by the treatment of metformin in both the
MCF-7 and TR MCF-7 cells. Compared to the E2 only-treated cells,
metformin (85% decrease) was as effective as 4-OHT (81% decrease)
in regulating the transcriptional activity of ERα in MCF-7 cells.
However, 4-OHT stimulated the ERE luciferase activity in the TR
MCF-7 cells. Consequently, we compared the inhibitory effect of
metformin and 4-OHT on cellular levels of proteins encoded by
E2/ERα-regulated genes, including c-Myc, cyclin D1, PR and pS2 in
both the MCF-7 (Fig. 4B, left) and
the TR MCF-7 (Fig. 4B, right)
cells. Metformin inhibited the protein levels of E2-induced c-Myc,
cyclin D1, PR and pS2 to a greater extent than 4-OHT. However,
4-OHT exhibited no inhibitory effect on the expression of these
target genes in the TR MCF-7 cells. AMPKα phosphorylation (Thr172)
and total AMPKα levels were also examined by western blotting. In
addition, then we proceeded for RT-qPCR. E2-induced mRNA levels of
ERα target genes, cyclin D1 and pS2 were inhibited by the treatment
of metformin (Fig. 4C). These
results suggest that metformin inhibited ERα-mediated transcription
levels of its target genes through inhibiting the transcriptional
activity of ERα activated by E2 in both the MCF-7 and TR MCF-7
cells.
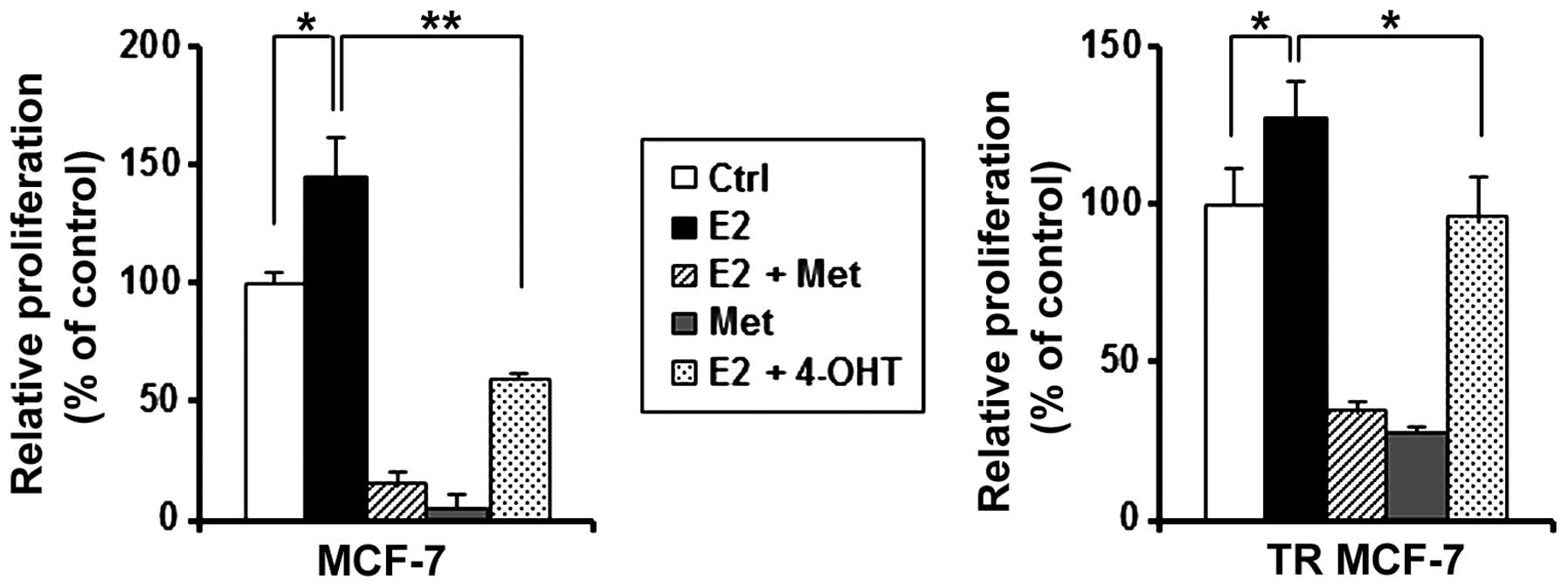
Metformin inhibits E2-stimulated cell
proliferation of the MCF-7 and TR MCF-7 cells
Cell counting using trypan blue staining was
performed to compare the antiproliferative effect of metformin and
4-OHT on E2-treated MCF-7 and TR MCF-7 cells. As shown in Fig. 5, metformin inhibited the cell
proliferation stimulated by estrogen in both the MCF-7 and TR MCF-7
cells compared to the E2 only-treated cells. Metformin inhibited
cell proliferation of MCF-7 cells (90 vs. 60% decrease compared to
E2 only-treated cells) and TR MCF-7 cells (74 vs. 25% decrease
compared to E2 only-treated cells) to a greater extend than 4-OHT.
These results suggest that metformin is likely to have an
inhibitory effect on the proliferation of MCF-7 and TR MCF-7 cells
through the inhibitory function of ERα.
Metformin inhibits E2-induced expression,
function of ERα and cell proliferation of MDA-MB-361 breast cancer
cells
In general, ERα and HER2 co-expression in breast
cancer may result in the treatment failure of tamoxifen therapy. To
properly evaluate the clinical potential of metformin in ERα- and
HER2-positive breast cancer (luminal B subtype), we investigated
the anticancer effect of metformin using the MDA-MB-361
(ERα+/HER2+) cell line. Similar to our
results in the MCF-7 and TR MCF-7 cells, metformin reduced the ERα
protein (Fig. 6A) and mRNA levels
(Fig. 6B). As shown in Fig. 6C, metformin and 4-OHT resulted in a
75 and 67% decrease in E2-induced ERE luciferase activity,
respectively. In addition, E2-induced expression of c-Myc, cyclin
D1, PR and pS2 were inhibited by the treatment of metformin.
Moreover, we showed AMPKα phosphorylation (Thr172) and total AMPKα
levels to demonstrate that metformin was present and active during
our experiments (Fig. 6D). Next, we
performed RT-qPCR. It was found that E2-induced mRNA levels of
cyclin D1 and pS2 were inhibited by the treatment of metformin
(Fig. 6E). Metformin resulted in
70% inhibition of the proliferation of MDA-MB-361 cells compared to
cells treated by E2 only (Fig.
6F).
Discussion
Estrogen (E2) plays a vital role in the pathogenesis
of breast cancer through estrogen receptor α (ERα) (24). Blocking the E2/ERα signaling pathway
is the first-line therapeutic strategy for patients with
ERα-positive breast cancer. Although anti-estrogenic therapy using
tamoxifen is still an important and major modality to manage
ERα-positive breast cancer (25),
its usefulness is greatly limited by de novo and acquired
resistance (26). Therefore, new
therapeutic strategies are required to overcome tamoxifen
resistance (TR). In the present study, we showed the effectiveness
of metformin by targeting ERα using ERα-positive as well as
tamoxifen-resistant breast cancer cells, thus providing a possible
mechanism underlying the anticancer effect of metformin.
Numerous in vitro and in vivo studies
have demonstrated that metformin treatment can result in the
inhibition of cancer cell growth (27–30). A
variety of mechanisms have been invoked to explain the antitumor
effect of metformin, including activation of AMPK and inhibition of
mTOR (31,32). We focused on research related to the
expression and signaling pathway of ERα. Our results revealed that
metformin inhibited E2-induced expression, ERE luciferase activity,
expression of ERα target genes, and cell proliferation of MCF-7 and
TR MCF-7 cells. Collectively, our data indicated that the
anticancer effect of metformin could be due to the repression of
expression and transcriptional function of ERα.
In addition to MCF-7 and TR MCF-7 breast cancer
cells, we also assessed the antiproliferative effect of metformin
on MDA-MB-361 (ERα+/HER2+) breast cancer
cells. HER2 is a transmembrane tyrosine kinase and a member of the
human epidermal growth factor receptor (EGFR) family. It leads to
the activation of the signaling pathway that promotes cell
proliferation, migration, and survival. HER2 amplification and/or
overexpression in breast cancer are correlated to poor patient
survival or resistance to tamoxifen therapy (33–37).
Consistent with our results in the MCF-7 and TR MCF-7 breast cancer
cells, metformin also inhibited E2-induced expression and function
of ERα as well as the cell proliferation of MDA-MB-361 cells.
E2-induced ERE luciferase activity, expression of ERα target genes,
and cell proliferation were also inhibited by tamoxifen in MCF-7
cells, although the effect of tamoxifen was less than that of
metformin. Overall, metformin inhibited the ERE luciferase
activity, the expression of ERα target genes, and the cell
proliferation to a greater extend than 4-OHT in the MCF-7, TR MCF-7
and MDA-MB-361 cells. These effects could be due to the fact that
4-OHT blocked the binding of E2/ERα without suppressing the
expression of ERα itself, suggesting that treatment with metformin
may be useful for patients with ERα-positive breast cancer.
In conclusion, these results suggest that metformin
exhibited a superior antiproliferative effect by inhibiting ERα
signaling than tamoxifen in ERα-positive MCF-7, TR MCF-7 and
MDA-MB-361 cells. Currently, there is no alternative standard
treatment for tamoxifen-resistant breast tumors except aromatase
inhibitors. Therefore, we suggest that metformin may be one of the
effective therapeutic agents for treating tamoxifen-resistant
breast cancer. Moreover, combination strategies with metformin may
be useful for enhancing the treatment efficacy of other cytotoxic
chemotherapies or targeted therapies (38). Further experiments including animal
studies and clinical trials are warranted.
Acknowledgments
The present study was supported by a grant
(HI14C3405) from the Korea Health Technology R&D Project
through the Korea Health Industry Development Institute (KHIDI),
funded by the Ministry of Health and Welfare (MOHW), Republic of
Korea.
References
|
1
|
Ali S and Coombes RC: Estrogen receptor
alpha in human breast cancer: Occurrence and significance. J
Mammary Gland Biol Neoplasia. 5:271–281. 2000. View Article : Google Scholar
|
|
2
|
Hall JM, Couse JF and Korach KS: The
multifaceted mechanisms of estradiol and estrogen receptor
signaling. J Biol Chem. 276:36869–36872. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Couse JF and Korach KS: Estrogen receptor
null mice: What have we learned and where will they lead us? Endocr
Rev. 20:358–417. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Colditz GA: Relationship between estrogen
levels, use of hormone replacement therapy, and breast cancer. J
Natl Cancer Inst. 90:814–823. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hankinson SE, Colditz GA and Willett WC:
Towards an integrated model for breast cancer etiology: The
lifelong interplay of genes, lifestyle, and hormones. Breast Cancer
Res. 6:213–218. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Uray IP and Brown PH: Chemoprevention of
hormone receptor-negative breast cancer: New approaches needed.
Clinical Cancer Prevention. Springer; pp. 147–162. 2011
|
|
7
|
Osborne CK: Tamoxifen in the treatment of
breast cancer. N Engl J Med. 339:1609–1618. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shaw RJ, Lamia KA, Vasquez D, Koo SH,
Bardeesy N, Depinho RA, Montminy M and Cantley LC: The kinase LKB1
mediates glucose homeostasis in liver and therapeutic effects of
metformin. Science. 310:1642–1646. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bailey CJ and Turner RC: Metformin. N Engl
J Med. 334:574–579. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bowker SL, Majumdar SR, Veugelers P and
Johnson JA: Increased cancer-related mortality for patients with
type 2 diabetes who use sulfonylureas or insulin. Diabetes Care.
29:254–258. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Evans JM, Donnelly LA, Emslie-Smith AM,
Alessi DR and Morris AD: Metformin and reduced risk of cancer in
diabetic patients. BMJ. 330:1304–1305. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Decensi A, Puntoni M, Goodwin P, Cazzaniga
M, Gennari A, Bonanni B and Gandini S: Metformin and cancer risk in
diabetic patients: A systematic review and meta-analysis. Cancer
Prev Res. 3:1451–1461. 2010. View Article : Google Scholar
|
|
13
|
Bosco JLF, Antonsen S, Sørensen HT,
Pedersen L and Lash TL: Metformin and incident breast cancer among
diabetic women: A population-based case-control study in Denmark.
Cancer Epidemiol Biomarkers Prev. 20:101–111. 2011. View Article : Google Scholar
|
|
14
|
Beck E and Scheen AJ: Metformin, an
antidiabetic molecule with anti-cancer properties. Rev Med Liege.
68:444–449. 2013.In French. PubMed/NCBI
|
|
15
|
Liu B, Fan Z, Edgerton SM, Deng XS,
Alimova IN, Lind SE and Thor AD: Metformin induces unique
biological and molecular responses in triple negative breast cancer
cells. Cell Cycle. 8:2031–2040. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jiralerspong S, Gonzalez-Angulo AM and
Hung M-C: Expanding the arsenal: Metformin for the treatment of
triple-negative breast cancer? Cell Cycle. 8:2681–2684. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Marx J: Medicine. Cancer-suppressing
enzyme adds a link to type 2 diabetes. Science. 310:1259. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Goodwin PJ, Pritchard KI, Ennis M, Clemons
M, Graham M and Fantus IG: Insulin-lowering effects of metformin in
women with early breast cancer. Clin Breast Cancer. 8:501–505.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dowling RJ, Goodwin PJ and Stambolic V:
Understanding the benefit of metformin use in cancer treatment. BMC
Med. 9:332011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Markowska A, Pawałowska M, Filas V, Korski
K, Gryboś M, Sajdak S, Olejek A, Bednarek W, Spiewankiewicz B,
Lubin J, et al: Does Metformin affect ER, PR, IGF-1R, β-catenin and
PAX-2 expression in women with diabetes mellitus and endometrial
cancer? Diabetol Metab Syndr. 5:762013. View Article : Google Scholar
|
|
21
|
Yoo YA, Kim YH, Kim JS and Seo JH: The
functional implications of Akt activity and TGF-beta signaling in
tamoxifen-resistant breast cancer. Biochim Biophys Acta.
1783:438–447. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Knowlden JM, Hutcheson IR, Jones HE,
Madden T, Gee JM, Harper ME, Barrow D, Wakeling AE and Nicholson
RI: Elevated levels of epidermal growth factor receptor/c-erbB2
heterodimers mediate an autocrine growth regulatory pathway in
tamoxifen-resistant MCF-7 cells. Endocrinology. 144:1032–1044.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dauvois S, Danielian PS, White R and
Parker MG: Antiestrogen ICI 164,384 reduces cellular estrogen
receptor content by increasing its turnover. Proc Natl Acad Sci
USA. 89:4037–4041. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Clark GM, Osborne CK and McGuire WL:
Correlations between estrogen receptor, progesterone receptor, and
patient characteristics in human breast cancer. J Clin Oncol.
2:1102–1109. 1984.PubMed/NCBI
|
|
25
|
Honig SF: Tamoxifen for the reduction in
the incidence of breast cancer in women at high risk for breast
cancer. Ann NY Acad Sci. 949:345–348. 2001. View Article : Google Scholar
|
|
26
|
Osborne CK and Schiff R: Mechanisms of
endocrine resistance in breast cancer. Annu Rev Med. 62:233–247.
2011. View Article : Google Scholar
|
|
27
|
Zakikhani M, Dowling R, Fantus IG,
Sonenberg N and Pollak M: Metformin is an AMP kinase-dependent
growth inhibitor for breast cancer cells. Cancer Res.
66:10269–10273. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dowling RJ, Zakikhani M, Fantus IG, Pollak
M and Sonenberg N: Metformin inhibits mammalian target of
rapamycin-dependent translation initiation in breast cancer cells.
Cancer Res. 67:10804–10812. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ben Sahra I, Laurent K, Loubat A,
Giorgetti-Peraldi S, Colosetti P, Auberger P, Tanti JF, Le
Marchand-Brustel Y and Bost F: The antidiabetic drug metformin
exerts an antitumoral effect in vitro and in vivo through a
decrease of cyclin D1 level. Oncogene. 27:3576–3586. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Buzzai M, Jones RG, Amaravadi RK, Lum JJ,
DeBerardinis RJ, Zhao F, Viollet B and Thompson CB: Systemic
treatment with the antidiabetic drug metformin selectively impairs
p53-deficient tumor cell growth. Cancer Res. 67:6745–6752. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gonzalez-Angulo AM and Meric-Bernstam F:
Metformin: A therapeutic opportunity in breast cancer. Clin Cancer
Res. 16:1695–1700. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhou G, Myers R, Li Y, Chen Y, Shen X,
Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, et al: Role of
AMP-activated protein kinase in mechanism of metformin action. J
Clin Invest. 108:1167–1174. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gullick WJ, Berger MS, Bennett PL,
Rothbard JB and Waterfield MD: Expression of the c-erbB-2 protein
in normal and transformed cells. Int J Cancer. 40:246–254. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
King CR, Kraus MH and Aaronson SA:
Amplification of a novel v-erbB-related gene in a human mammary
carcinoma. Science. 229:974–976. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Riese DJ II, van Raaij TM, Plowman GD,
Andrews GC and Stern DF: The cellular response to neuregulins is
governed by complex interactions of the erbB receptor family. Mol
Cell Biol. 15:5770–5776. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Slamon DJ, Clark GM, Wong SG, Levin WJ,
Ullrich A and McGuire WL: Human breast cancer: Correlation of
relapse and survival with amplification of the HER-2/neu oncogene.
Science. 235:177–182. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ross JS and Fletcher JA: The HER-2/neu
oncogene in breast cancer: Prognostic factor, predictive factor,
and target for therapy. Stem Cells. 16:413–428. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kim J, Lee J, Kim C, Choi J and Kim A:
Anti-cancer effect of metformin by suppressing signaling pathway of
HER2 and HER3 in tamoxifen-resistant breast cancer cells. Tumour
Biol. Nov 18–2015.Epub ahead of print. View Article : Google Scholar
|