Introduction
Esophageal cancer is one of the most common fatal
types of cancer worldwide. In China, esophageal cancer ranks as the
fourth leading cause of cancer-related mortality. Esophageal
squamous cell carcinoma (ESCC) and esophageal adenocarcinoma are
the two major histological types of esophageal carcinoma (1), and ESCC is the main histological
subtype in the so-called Asian belt, which includes Turkey,
northeastern Iran, Kazakhstan and northern and central China
(2). Although several strategies
have been developed for the treatment of ESCC, including surgery,
chemotherapy, radiation and comprehensive treatment, the 5-year
overall survival rate for ESCC remains as low as 20–30% (3). Therefore, more effective and targeted
therapies for ESCC are urgently needed.
Loss of the phosphatase and tensin homolog (PTEN)
protein, a tumor suppressor, has been reported as a prognostic
factor in numerous types of cancer such as endometrial carcinomas,
gliomas, prostate carcinomas, gastric cancers, lung and breast
cancers (4–9). Recent studies have revealed that the
expression of PTEN is correlated with prognosis in patients with
ESCC (10–12). Increased activity of the
PTEN/PI3K/Akt signaling pathway was found to inhibit ESCC cell
proliferation both in vitro and in vivo (13,14).
Conversely, ESCC cell proliferation is enhanced by transfection
with a PTEN anti-sense oligonucleotide (15). Therefore, delivery of the tumor
suppressor gene PTEN represents a powerful strategy for ESCC
therapy. However, to date, there have been few reports of
PTEN transfer-based gene therapies for the treatment of
ESCC. A previous study demonstrated that the adenovirus-mediated
transfer of PTEN inhibits the growth of esophageal cancer
cells in vitro and in vivo (16). Since virus-mediated gene therapy is
associated with safety problems, the use of non-viral vectors for
cancer gene therapy is a potential alternative. Of these,
cell-penetrating peptides (CPPs) have attracted considerable
attention for their potential utility in the delivery of exogenous
molecules into living cells. Tegument protein VP22 of herpes
simplex virus type 1 (HSV-1) is a CPP that is capable of
transporting heterologous proteins, such as p53, p27, cytosine
deaminase, and Hsp70, across the cell membrane, thereby enhancing
the biological functions of these proteins (17–20).
However, the mechanisms involved in the delivery of these proteins
by VP22 have not been fully characterized.
We previously reported that VP22-mediated
intercellular delivery of PTEN enhances the antitumor effects of
PTEN in the breast tumor cell line BT549 (21). In the present study, the
intercellular delivery and the antitumor activity of the fusion
gene PTEN-VP22 were examined in an ESCC cell line both in
vitro and in vivo.
Materials and methods
Cell culture
Human esophageal carcinoma cells (Eca109) were
cultivated in Dulbecco's modified Eagle's medium (DMEM; Gibco-Life
Technologies, Carlsbad, CA, USA) supplemented with 10% bovine serum
(FBS; Gibco), 100 U/ml penicillin and 100 mg/ml streptomycin at
37°C with 5% CO2.
Eukaryotic expression vector
construction
The pcDNA3-VP22, pcDNA3-PTEN and pcDNA3-PTEN-VP22
vectors were utilized in the present study for the expression of
wild-type human PTEN protein, HSV-1 VP22 protein, and the
N-terminal VP22-fused PTEN protein (PTEN-VP22), respectively, as
previously described (21).
Cell transfection
Eca109 cells were grown to 70% confluency and washed
twice with phosphate-buffered saline (PBS). Washed cells were
transfected with the plasmids diluted in serum-free DMEM containing
Lipofectamine 2000 reagent (Invitrogen-Life Technologies, Waltham,
MA, USA), according to the manufacturer's instructions. Prior to
the start of the experiment, transfection efficiency was determined
using pSV-β-galactosidase (Promega, Madison, WI, USA).
Western blot analysis
Eca109 cells were transfected with 5 µg/ml of
pcDNA3, pcDNA3-PTEN, pcDNA3-PTEN-VP22, or pcDNA3-VP22. After 48 h,
total protein was extracted from the cells. Protein concentrations
were determined using the Bradford assay (Bio-Rad Laboratories,
Inc., Hercules, CA, USA). Proteins were separated by 10% SDS-PAGE
and immunoblotting was performed using primary antibodies specific
to PTEN (Santa Cruz Biotechnology, Inc., Dallas, TX, USA),
phospho-Akt (Ser473; Cell Signaling Technology, Inc., Danvers, MA,
USA), or phospho-FAK (Tyr576/577; Cell Signaling Technology).
Horseradish peroxidase-conjugated anti-IgG (Santa Cruz
Biotechnology) was used as the secondary antibody for the DAB
detection system (Wuhan Boster Biological Technology, Ltd., Wuhan,
China). Antibodies specific to total Akt (Cell Signaling
Technology), total FAK (Cell Signaling Technology), or β-actin
(Wuhan Boster Biological Technology) were used as loading controls.
Each test was repeated in triplicate.
Immunofluorescence quantitation
Eca109 cells were transfected with 5 µg/ml of
each plasmid. After 48 h, cells were fixed in cold methanol for 10
min at 25°C and then permeabilized by incubation with 0.2% Triton
X-100 for 90 min at 25°C. After blocking for 30 min in 5% non-fat
milk at 25°C, cells were incubated with rabbit anti-PTEN antibody
(Santa Cruz Biotechnology) at 4°C overnight. After three washes,
cells were incubated with fluorescein isothiocyanate-conjugated
sheep anti-rabbit IgG (Santa Cruz Biotechnology) for 1 h at 25°C
and analyzed using a Fluoroskan Ascent FL microplate reader (Thermo
Fisher Scientific, Waltham, MA, USA). Each test was repeated in
triplicate.
Analysis of cell proliferation by Cell
Counting kit-8 assay
Eca109 cells were transfected with 10 µg/ml
of pcDNA3 and varying concentrations (2, 4, 6, 8 or 10
µg/ml) of pcDNA3-VP22, pcDNA3-PTEN or pcDNA3-PTEN-VP22.
Different quantities of pcDNA3 were added to each well to ensure
that the same overall amount of DNA was present in every well.
Transfected cells were harvested at 10 h and plated in 96-well
plates (Corning Life Sciences, Tewksbury, MA, USA) at a density of
3,000 cells/well for each treatment condition. At 24, 48 and 72 h
after transfection, 10 µl of WST-8 dye (Beyotime Institute
of Biotechnology, Shanghai, China) was added to each well. Plates
were incubated at 37°C for 1 h and the absorbance (A) at 450 nm was
measured using an iMark biomicroplate reader (Bio-Rad
Laboratories). Cell survival was determined as
Atreated/Acontrol. Each test was repeated in
triplicate.
Cell cycle analysis
Eca109 cells were transfected with 6 µg/ml of
each vector. Forty-eight hours later, cells were harvested by
trypsinization, fixed with 70% ethanol and stored at −20°C
overnight. Cell nuclei were stained by incubation for 30 min with
0.2 mg/ml RNase solution (Beyotime Institute of Biotechnology) and
0.05 mg/ml propidium iodide (PI). Analysis was performed using a
FACSVantage SE flow cytometer (BD Biosciences, Franklin Lakes, NJ,
USA). Each test was repeated in triplicate.
Analysis of apoptosis by Annexin V/PI
double staining
Eca109 cells were transfected with 6 µg/ml of
each vector. Forty-eight hours later, cells were double stained
using an Annexin V/PI apoptosis detection kit (Beyotime Institute
of Biotechnology), according to the manufacturer's instructions.
Cell apoptosis was then evaluated using a FACSVantage SE flow
cytometer (BD Biosciences). Each test was repeated in
triplicate.
Animal experiments
The present study was carried out in accordance with
the Guide for the Care and Use of Laboratory Animals (2011). All
animal experiments were conducted in the Experimental Animal Center
of Chongqing Medical University. The use of animals and the
experimental protocols were approved by the Animal Care and Use
Committee of Chongqing Medical University. Female 4- to 6-week-old
BALB/c nude mice (Animal Laboratory Center of Chongqing Medical
University) were maintained in specific pathogen-free,
environmentally controlled conditions on standard laboratory chow.
Eca109 cells were suspended in serum-free medium at a density of
1×107/ml, and 0.1 ml of the suspension was injected
subcutaneously into the dorsal area of each nude mouse. Thirty-two
tumor-bearing mice were divided randomly into four groups
(n=8/group), and treatments were carried out when the tumors had
grown to 6 mm in diameter. A mixture of 100 µg of the vector
and 100 µl of Lipofectamine 2000 reagent (Invitrogen Life
Technologies) were injected intratumorally into each mouse from
each group, followed by another injection the next day. One group
was injected with pcDNA3 and was designated the control group. The
other groups were treated with pcDNA3-PTEN, pcDNA3-VP22, or
pcDNA3-PTEN-VP22. Tumor growth was measured using calipers every
other day after the second injection, and tumor volume was
calculated using the following formula: Tumor volume
(mm3) = length × width2 × 0.5. After 15 days,
the mice were sacrificed using diethyl ether and tumor specimens
were harvested for western blot or immunohistochemical
analysis.
Microvessel counting by
immunohistochemistry
Tumor tissue specimens were formalin-fixed and
paraffin-embedded (FFPE). Sections from FFPE tissues were subjected
to immunohistochemical staining according to a standard method.
Briefly, 4-µm thick sections were obtained using a
microtome, transferred onto adhesive slides and dried at 59°C for
60 min. After deparaffinization and rehydration, the sections were
treated with a 3% hydrogen peroxide solution for 10 min to block
endogenous peroxidase and then pretreated for antigen retrieval in
10 mM citrate buffer (pH 6.0) in a microwave oven for 20 min.
Tissue sections were incubated at room temperature for 1 h with a
CD31-specific antibody (Santa Cruz Biotechnology) and then a
horseradish peroxidase-labeled anti-immunoglobulin for 30 min.
Sections were then developed with 3,3′-diaminobenzidine. To
quantify the microvessel density of the tumor sections, the
microvessels in three randomly selected fields of one randomly
selected section of each mouse were counted at a magnification of
×20 (Eclipse 50i microscope; Nikon, Tokyo, Japan). Every single
brown-stained cell and cell cluster was calculated as a blood
vessel.
Statistical analysis
Data are expressed as means ± standard error of the
mean (SEM). Statistical analysis was performed across multiple
groups using analysis of variance (ANOVA) and confirmed between
individual groups using the Student-Newman-Keuls method. P<0.05
was considered statistically significant.
Results
VP22 mediates PTEN intercellular delivery
in Eca109 cells
We previously reported that VP22 mediates the
intercellular delivery of PTEN in the breast tumor cell line BT549
(21). In the present study,
trafficking of the PTEN-VP22 fusion protein was examined in Eca109.
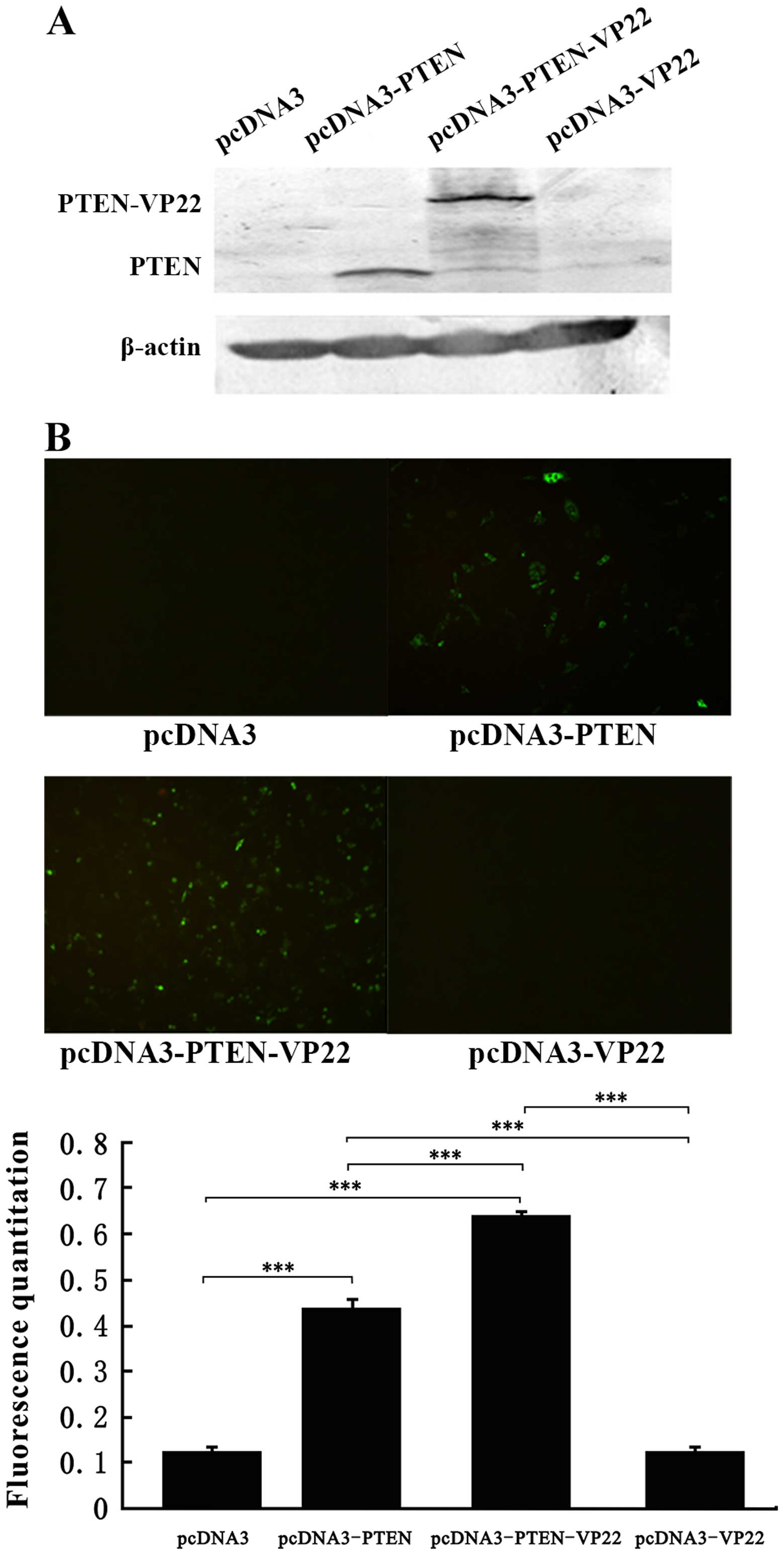
Western blot analysis showed that the pcDNA3-PTEN-transfected cells
exhibited high levels of PTEN expression (~60 kDa), and that
pcDNA3-PTEN-VP22-transfected cells exhibited higher expression of
PTEN-VP22 (~90 kDa). Except for those transfected with pcDNA3-PTEN,
each of the cell populations expressed very low endogenous levels
of PTEN at 48 h after treatment (Fig.
1A). Plasmid-transfected Eca109 cells were observed by
immunofluorescence microscopy and the levels of PTEN expression
were determined by fluorescence quantitation (Fig. 1B). At 48 h after transfection, there
was no difference in PTEN-specific fluorescence between the pcDNA3
(negative control)- and the pcDNA3-VP22-transfected cells. High
levels of fluorescence were detected in the pcDNA3-PTEN (P<0.001
vs. pcDNA3) and pcDNA3-PTEN-VP22 (P<0.001 vs. pcDNA3) groups.
Additionally, the pcDNA3-PTEN-VP22 group exhibited a higher level
of fluorescence than the pcDNA3-PTEN group (P<0.001). These
results suggest that VP22 may mediate the intercellular delivery of
PTEN, resulting in increased distribution of the latter within
Eca109 cells.
VP22 enhances PTEN-mediated
antiproliferative activity in Eca109 cells
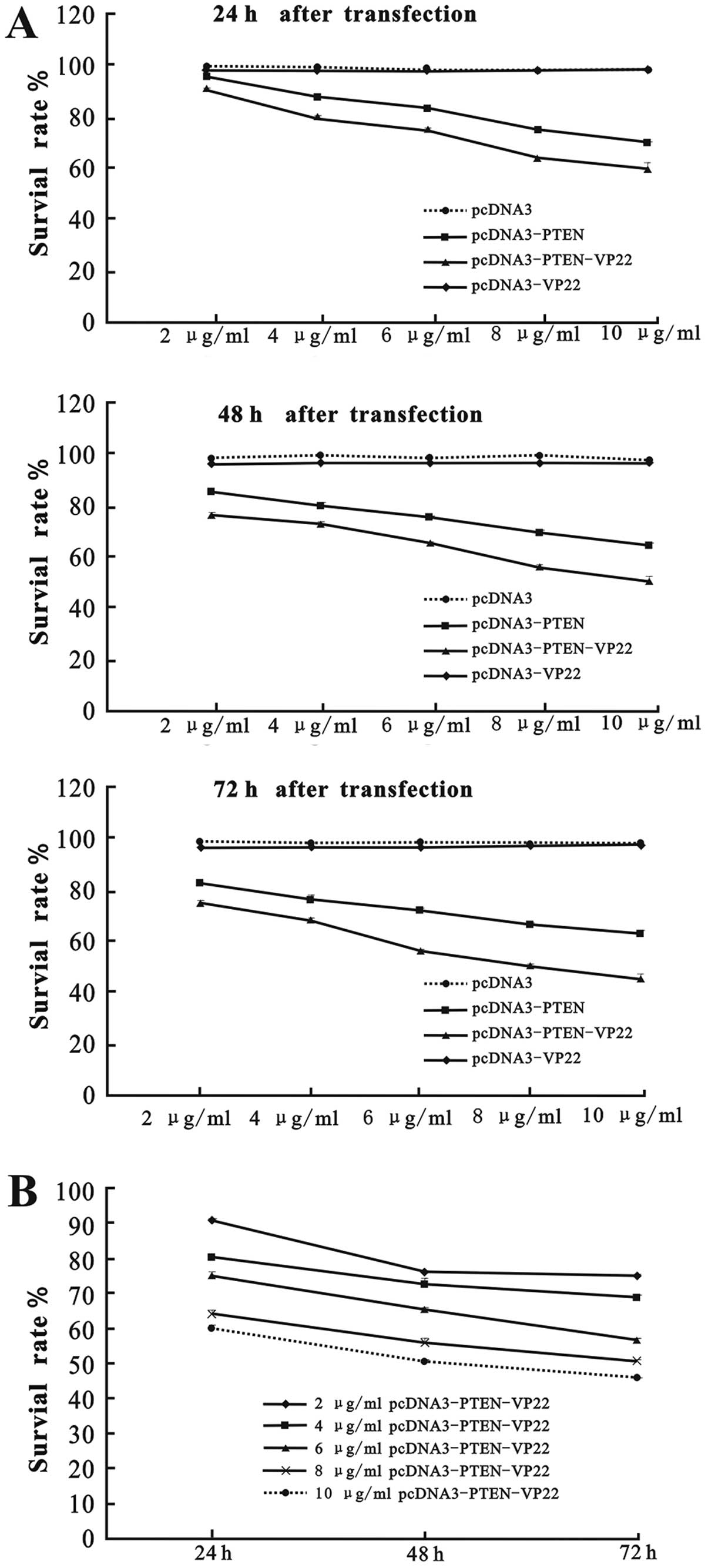
Eca109 cells were transfected with various doses (2,
4, 6, 8 and 10 µg/ml) of pcDNA3-PTEN, pcDNA3-PTEN-VP22 or
pcDNA3-VP22, and cell survival rates were evaluated at 24, 48 and
72 h after transfection. As shown in Fig. 2, there was no difference in the cell
survival rates of the pcDNA3 and the pcDNA3-VP22 group (P>0.05),
indicating that VP22 alone did not exert antiproliferative effects
on Eca109 cells. In contrast, cell proliferation was inhibited in
the pcDNA3-PTEN and pcDNA3-PTEN-VP22 groups in a dose-and
time-dependent manner. Indeed, both pcDNA3-PTEN and
pcDNA3-PTEN-VP22 exhibited a significantly greater
antiproliferative activity compared with pcDNA3 at identical
concentrations (2–10 µg/ml) at 24, 48 and 72 h
post-transfection (P<0.001; Fig.
2A). Furthermore, the efficacy of inhibition of proliferation
by pcDNA3-PTEN-VP22 was greater than that of pcDNA3-PTEN
(P<0.001 at the same concentrations at 24, 48 and 72 h; Fig. 2A). Higher concentrations of
pcDNA3-PTEN-VP22 were found to yield significantly lower levels of
Eca109 proliferation than lower concentrations (2–10 µg/ml)
at each time-point (P<0.001; Fig.
2B). Therefore, these results suggest that VP22 enhances the
antiproliferative activity of PTEN in Eca109 cells.
VP22 enhances PTEN-mediated cell cycle
arrest at G1 phase in Eca109 cells
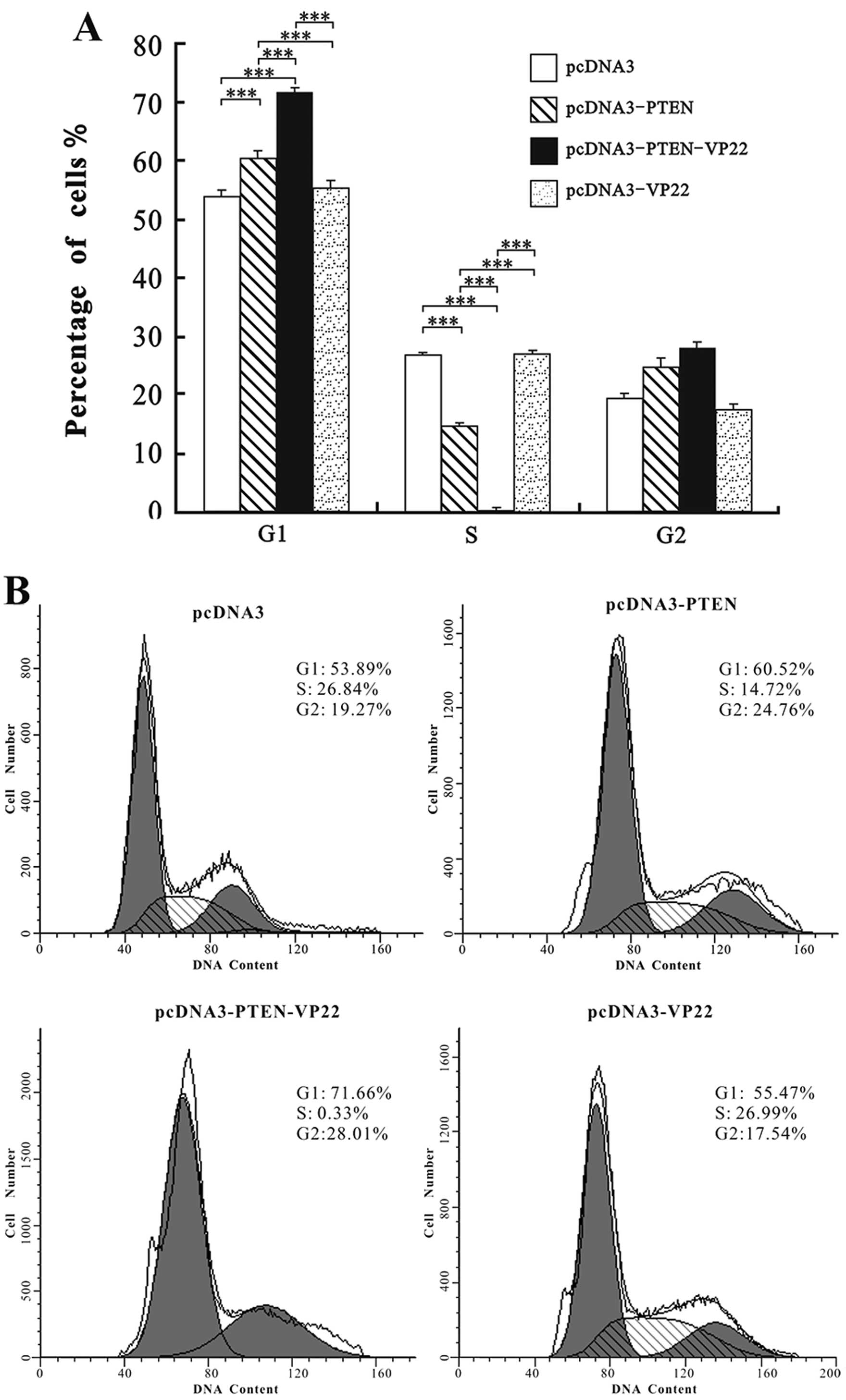
The cell cycle distribution of Eca109 cells was
examined at 48 h after transfection with 6 µg/ml of each
plasmid. As shown in Fig. 3, the
cell cycle distribution of the pcDNA3 group did not differ from
that of the pcDNA3-VP22 group (P>0.05), indicating that VP22
alone did not affect cell cycle progression in Eca109 cells.
Compared with pcDNA3, treatment with pcDNA3-PTEN and
pcDNA3-PTEN-VP22 resulted in significant increases in the
percentage of cells in G1 phase (P<0.001) and a significant
concomitant decrease in the number of cells in the S phase
(P<0.001; Fig. 3). However,
pcDNA3-PTEN-VP22 induced higher rates of G1 arrest than pcDNA3-PTEN
(P<0.001). These findings suggest that VP22 enhances the rate of
PTEN-mediated G1 cell cycle arrest in Eca109 cells.
VP22 enhances PTEN-mediated apoptotic
induction in Eca109 cells
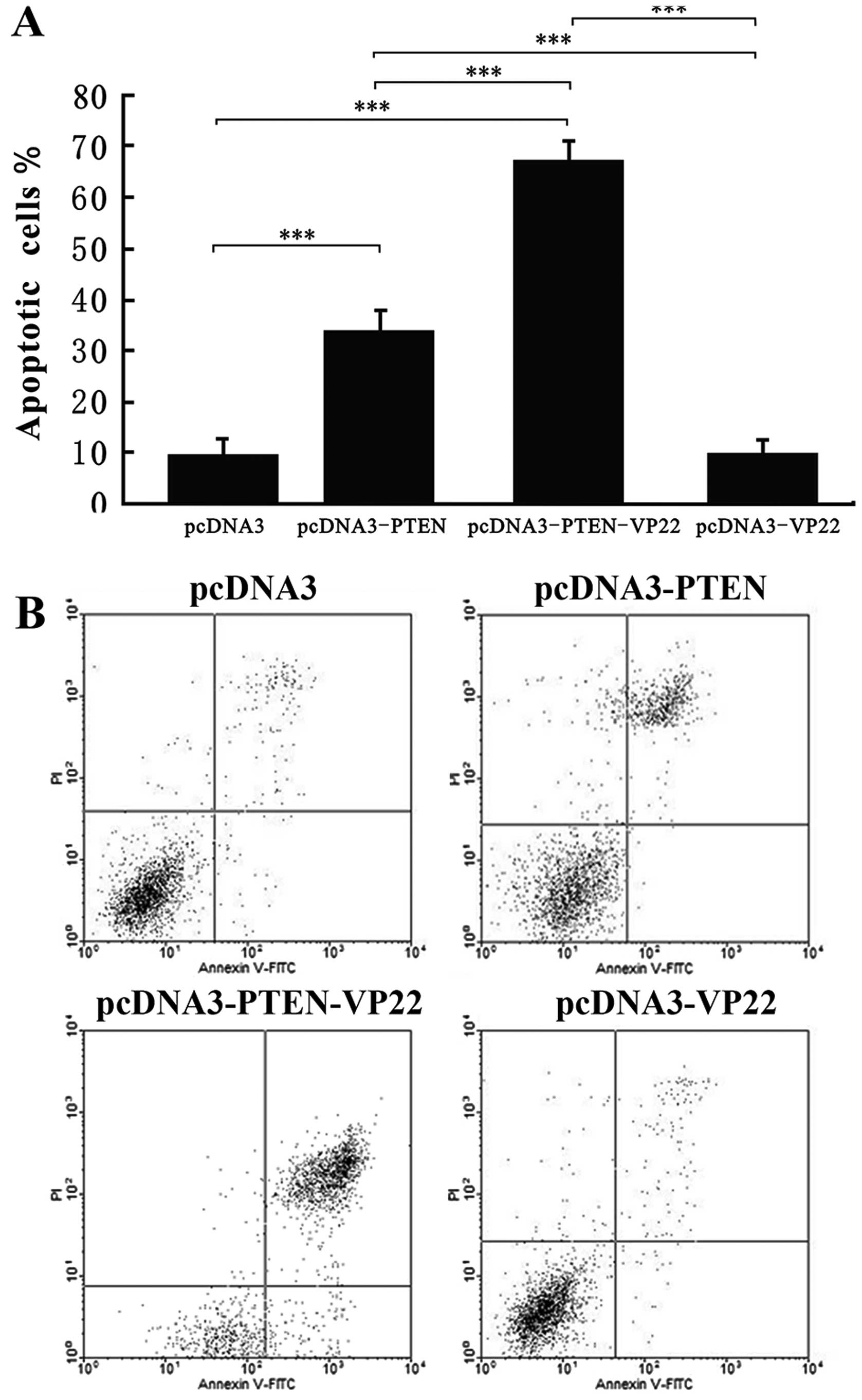
To determine whether the antitumor activity of
PTEN-VP22 in Eca109 cells occurs via the induction of apoptosis,
cells were stained with Annexin V and PI and visualized by
fluorescence photomicrography. Cells were examined at 48 h after
transfection with 6 µg/ml of various plasmids. Compared with
pcDNA3 (the negative control), pcDNA3-VP22 did not induce higher
levels of apoptosis; however, the rate of apoptosis of cells
transfected with pcDNA3-PTEN differed significantly from those
transfected with pcDNA3 (P<0.001; Fig. 4). In addition, a significant
increase in apoptosis was detected in cells transfected with
pcDNA3-PTEN-VP22 vs. pcDNA3-PTEN (P<0.001), indicating that VP22
enhances PTEN-mediated induction of apoptosis in Eca109 cells.
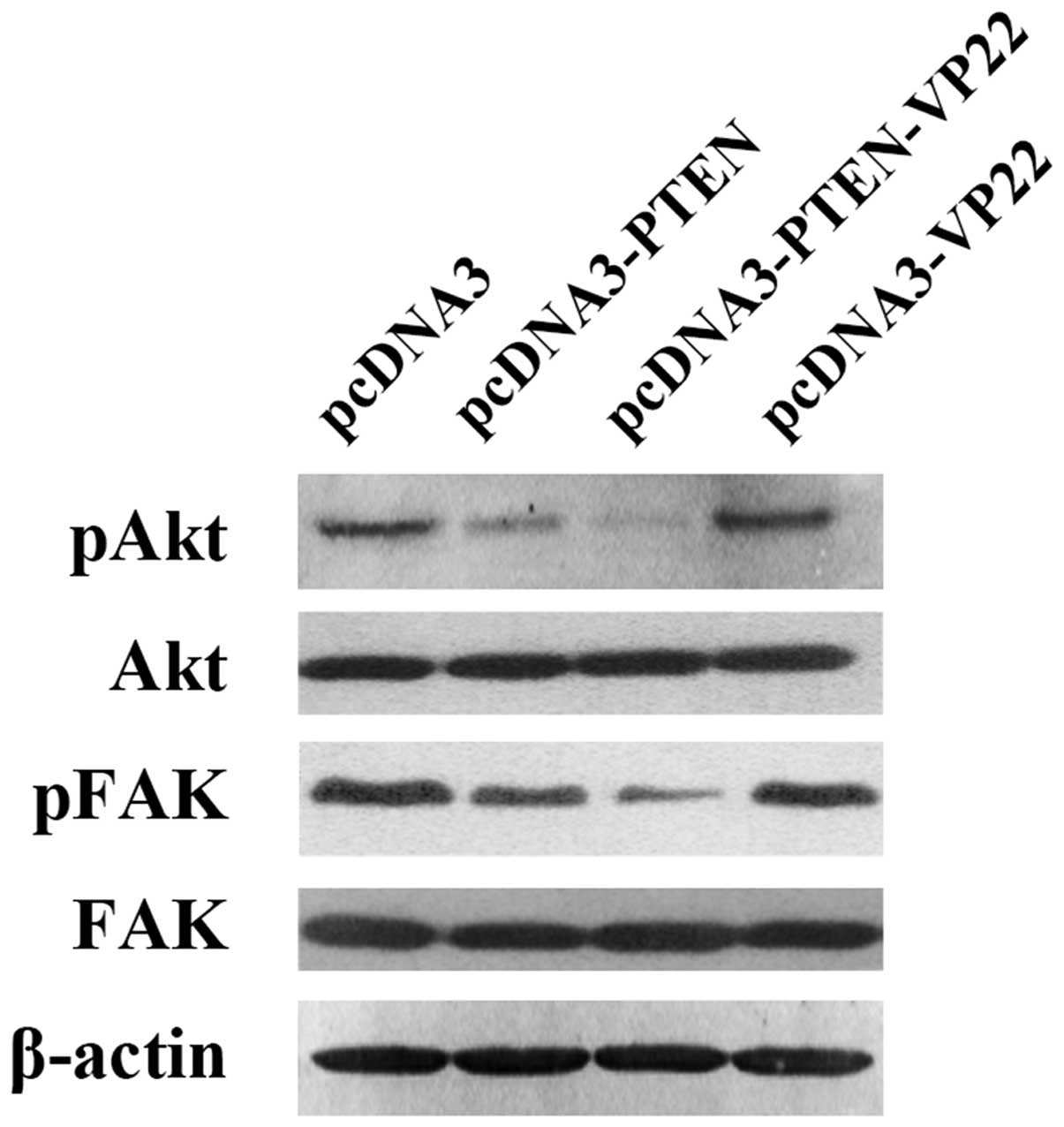
VP22 enhances PTEN-mediated decrease in
the level of phosphorylated Akt and FAK in Eca109 cells
Depending on the specific plasmids transfected, we
observed different levels of Akt phosphorylation at Ser473 and FAK
phosphorylation at Tyr576/577 (Fig.
5). Phospho-Akt and phospho-Fak were highly expressed in the
pcDNA3 pcDNA3-VP22 groups. In contrast, these proteins were
expressed at low levels in the pcDNA3-PTEN group and at very low
levels in the pcDNA3-PTEN-VP22 group. These results indicate that
VP22 alone does not affect the levels of phosphorylated Akt and
FAK, but enhances the PTEN-mediated decrease in the levels of
phospho-Akt and phospho-FAK.
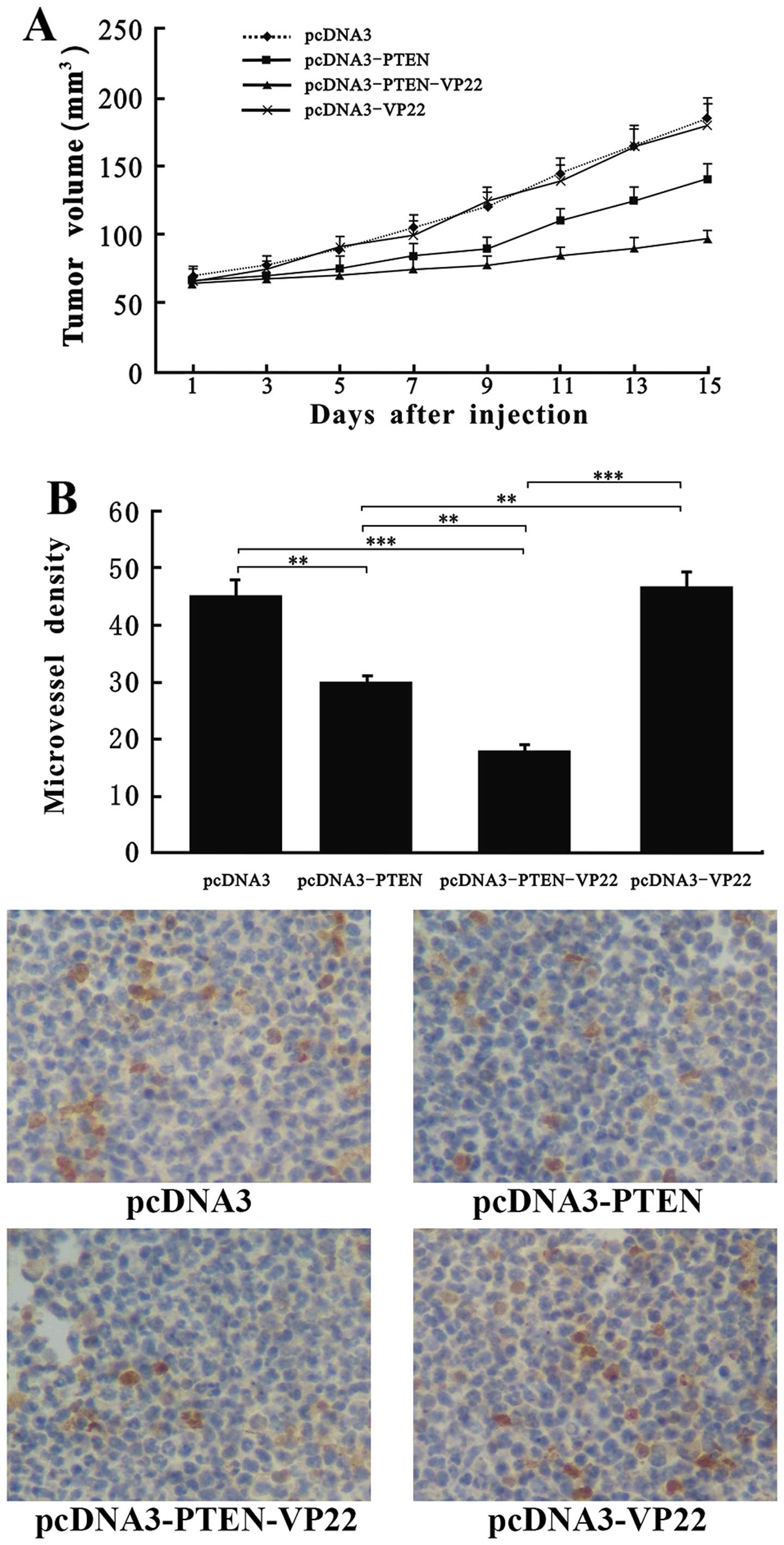
VP22 enhances PTEN-mediated antitumor
efficacy in vivo
Results from our present in vitro experiments
indicate that PTEN-VP22 and PTEN exert antitumor effects on Eca109
cells in vitro. To further these studies, we examined the
antitumor effects of PTEN-VP22 in vivo using a transplanted
tumor nude mouse model. Exposure of mice to pcDNA3-PTEN-VP22 and
pcDNA3-PTEN resulted in significant suppression of tumor growth
over a 15-day observation period (P<0.01), compared with the
control group (Fig. 6A).
Additionally, significant suppression of tumor growth was detected
in the pcDNA3-PTEN-VP22-treated mice vs. the pcDNA3-PTEN-treated
mice (P<0.01). However, no obvious difference in tumor growth
was observed between the pcDNA3-VP22-treated mice and the control
mice (P>0.05). Additionally, we examined the antiangiogenic
effect of PTEN-VP22 on Eca109 tumors. CD31 was used to analyze
microvessel density. CD31-positive vessels were abundant in pcDNA3-
and pcDNA3-VP22-treated Eca109 tumors (P>0.05). In contrast, the
microvessel density was significantly decreased in the pcDNA3-PTEN-
and pcDNA3-PTEN-VP22-treated Eca109 tumors (P<0.01; Fig. 6B), and was much lower in the
pcDNA3-PTEN-VP22-treated tumors than in those treated with
pcDNA3-PTEN (P<0.01). These findings demonstrate that VP22 alone
does not exert antitumor effects, but that VP22 may enhance
PTEN-mediated antitumor efficacy in vivo.
Discussion
Research during the past decade has demonstrated
that PTEN comprises a potential biological marker and therapeutic
target for ESCC (10–15). However, since the efficiency and
targeting of gene transfer approaches remain low, there have been
only a few reports on PTEN gene therapy for the treatment of ESCC.
Regardless, CPPs that are capable of carrying large macromolecules
across cellular membranes with high efficiency and minimal toxicity
have recently been found to overcome the cell membrane
impermeability issues associated with gene transfer (22).
VP22 interacts with the cell membrane more easily in
a partially unfolded state (23).
The motif responsible for the ability of the HSV-1 VP22 protein to
penetrate the cell membrane lies in the C-terminal 34-amino acids
'DAA TATRGRSAASRPTERPRAPARSASRPRRPVD' (24). Additionally, our previous findings
(21) demonstrated that the
C-terminus of VP22 is essential for intercellular delivery. In this
study, expression vectors for the wild-type PTEN gene and
the PTEN-VP22 fusion gene were constructed and their
antitumor activities were compared in the Eca109 ESCC cell line
with low endogenous expression of PTEN (Fig. 1).
High expression levels of PTEN and higher expression
levels of the PTEN-VP22 fusion protein were observed in the
pcDNA3-PTEN group and the pcDNA3-PTEN-VP22 group, respectively
(Fig. 1A). Furthermore, the
pcDNA3-PTEN-VP22 group exhibited a higher level of PTEN
fluorescence than the pcDNA3-PTEN group. The results of our
previous study demonstrated (21)
that VP22 transports PTEN across the cell membrane in BT549 cells
with a typical VP22 pattern (primary transfected cells with
cytoplasmic and nuclear staining surrounded by recipient cells with
nuclear staining), resulting in a wider distribution, without
affecting the subcellular localization of this protein. These
results suggest that VP22 may increase PTEN distribution via
intercellular delivery in Eca109 cells as well. Several reports
have indicated that proliferation of ESCC cells is inhibited via
increased PTEN activity both in vitro and in vivo
(13,14). In this study, to demonstrate whether
the VP22-mediated increase in PTEN distribution enhances the
antitumor activity of PTEN in Eca109 cells, we investigated the
effects of PTEN-VP22 on the behavior of Eca109 cells both in
vitro and in vivo. The PTEN-VP22 fusion protein induced
a stronger, time- and dose-dependent antiproliferative effect than
PTEN alone in vitro (Fig.
2). Conversely, VP22 did not display antiproliferative effects
in Eca109 cells (Fig. 2A),
suggesting that the observed increase in antiproliferative activity
is due to the delivery of PTEN by VP22.
To elucidate the mechanisms by which PTEN-VP22
induces these antitumor effects, we investigated whether higher
levels of protein delivery were correlated with cell cycle arrest
or increased apop totic activity in Eca109 cells. Cell cycle
analysis demonstrated that treatment with PTEN-VP22 resulted in
enhanced cell cycle arrest at G1 phase compared with PTEN.
Conversely, VP22 did not alter the cell cycle distribution in
Eca109 cells (Fig. 3). Previous
studies have shown that transduction of the wild-type PTEN
gene into cancer cells induces apoptosis (25,26).
Here, we observed that PTEN-VP22 enhanced apoptosis relative to
PTEN alone, whereas VP22 had no effect on apoptosis levels
(Fig. 4). These data suggest that
VP22 enhances PTEN-mediated G1 cell cycle arrest and apoptosis in
Eca109 cells, which may be induced by the VP22-mediated
intercellular distribution of PTEN.
PTEN is involved in the regulation of a variety of
signal transduction pathways, e.g. PTEN suppresses the PI3K/Akt
pathway via dephosphorylation of PIP3 (phosphatidylinositol
3,4,5-triphosphate) (27), thereby
inhibiting tumor proliferation and migration and inducing apoptosis
(28,29). PTEN is also involved in the
dephosphorylation and inactivation of FAK, thereby additionally
regulating tumor angiogenesis, migration, invasion and metastasis
(30,31). In the present study, we investigated
whether higher PTEN delivery levels were correlated with decreased
levels of phosphorylated Akt or FAK in Eca109 cells. The
phospho-Akt and phospho-FAK levels were significantly lower in the
presence of PTEN-VP22 than of PTEN in Eca109 cells, but were not
altered by VP22 alone (Fig. 5).
Together, these data demonstrate that VP22 enhances PTEN-mediated
suppression of the PI3K/Akt and FAK pathways, consequently
enhancing the antitumor activity of PTEN.
Finally, to empirically investigate the inhibitory
effect of PTEN-VP22 on tumor growth directly, we developed an in
vivo tumor model involving the intratumoral administration of
pcDNA3, pcDNA3-PTEN, pcDNA3-PTEN-VP22 or pcDNA3-VP22. While
treatment with PTEN alone resulted in moderate inhibition of tumor
growth and angiogenic effects in Eca109-bearing mice, PTEN-VP22
markedly suppressed tumor growth and exerted strong angiogenic
effects. In contrast, tumor growth and angiopoiesis were not
altered by VP22 alone.
In summary, VP22 alone does not directly exert
antitumor activity, but mediates the intercellular delivery of
PTEN, thereby causing increased number of cells containing PTEN,
which results in cells achieving a therapeutic steady state. This
leads to an overall increase in the antitumor activity of PTEN,
which is correlated with increased antiproliferative effects, cell
cycle arrest at G1, induction of apoptosis and antiangiogenic
effects. Therefore, our findings show that VP22-mediated
intercellular delivery of PTEN enhances the antitumor effects of
the latter, providing further experimental data for enhancing the
efficacy of PTEN-based gene therapy against cancer. In
future studies, we plan to focus on the development of targeting
delivery systems based on PTEN-VP22 gene therapy and assess
the safety of these gene therapies.
Acknowledgments
The present study was supported by grants from the
National Natural Science Foundation of China (81102288) and
Chongqing Science and Technology Commission
(cstc2014j-cyjA10009).
References
|
1
|
Enzinger PC and Mayer RJ: Esophageal
cancer. N Engl J Med. 349:2241–2252. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pennathur A, Gibson MK, Jobe BA and
Luketich JD: Oesophageal carcinoma. Lancet. 381:400–412. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kamangar F, Dores GM and Anderson WF:
Patterns of cancer incidence, mortality, and prevalence across five
continents: Defining priorities to reduce cancer disparities in
different geographic regions of the world. J Clin Oncol.
24:2137–2150. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Terakawa N, Kanamori Y and Yoshida S: Loss
of PTEN expression followed by Akt phosphorylation is a poor
prognostic factor for patients with endometrial cancer. Endocr
Relat Cancer. 10:203–208. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kondo Y, Hollingsworth EF and Kondo S:
Molecular targeting for malignant gliomas (Review). Int J Oncol.
24:1101–1109. 2004.PubMed/NCBI
|
|
6
|
Koksal IT, Dirice E, Yasar D, Sanlioglu
AD, Ciftcioglu A, Gulkesen KH, Ozes NO, Baykara M, Luleci G and
Sanlioglu S: The assessment of PTEN tumor suppressor gene in
combination with Gleason scoring and serum PSA to evaluate
progression of prostate carcinoma. Urol Oncol. 22:307–312. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Im SA, Lee KE, Nam E, Kim DY, Lee JH, Han
HS, Seoh JY, Park HY, Cho MS, Han WS, et al: Potential prognostic
significance of p185(HER2) overexpression with loss of PTEN
expression in gastric carcinomas. Tumori. 91:513–521. 2005.
|
|
8
|
Tang JM, He QY, Guo RX and Chang XJ:
Phosphorylated Akt overexpression and loss of PTEN expression in
non-small cell lung cancer confers poor prognosis. Lung Cancer.
51:181–191. 2006. View Article : Google Scholar
|
|
9
|
Toft DJ and Cryns VL: Minireview:
Basal-like breast cancer: from molecular profiles to targeted
therapies. Mol Endocrinol. 25:199–211. 2011. View Article : Google Scholar :
|
|
10
|
Tachibana M, Shibakita M, Ohno S, Kinugasa
S, Yoshimura H, Ueda S, Fujii T, Rahman MA, Dhar DK and Nagasue N:
Expression and prognostic significance of PTEN product protein in
patients with esophageal squamous cell carcinoma. Cancer.
94:1955–1960. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li P, Mao WM, Zheng ZG, Dong ZM and Ling
ZQ: Down-regulation of PTEN expression modulated by dysregulated
miR-21 contributes to the progression of esophageal cancer. Dig Dis
Sci. 58:3483–3493. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sun Z, Ji N, Bi M, Zhang Z, Liu X and Wang
Z: Negative expression of PTEN identifies high risk for
lymphatic-related metastasis in human esophageal squamous cell
carcinoma. Oncol Rep. 33:3024–3032. 2015.PubMed/NCBI
|
|
13
|
Zhao H, Yang J, Fan T, Li S and Ren X:
RhoE functions as a tumor suppressor in esophageal squamous cell
carcinoma and modulates the PTEN/PI3K/Akt signaling pathway. Tumour
Biol. 33:1363–1374. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ou Y, Ma L, Ma L, Huang Z, Zhou W, Zhao C,
Zhang B, Song Y, Yu C and Zhan Q: Overexpression of cyclin B1
antagonizes chemotherapeutic-induced apoptosis through PTEN/Akt
pathway in human esophageal squamous cell carcinoma cells. Cancer
Biol Ther. 14:45–55. 2013. View Article : Google Scholar :
|
|
15
|
Sun MM, Zhang MZ, Chen Y, Li SL, Zhang W,
Ya GW and Chen KS: Effect of PTEN antisense oligonucleotide on
oesophageal squamous cell carcinoma cell lines. J Int Med Res.
40:2098–2108. 2012. View Article : Google Scholar
|
|
16
|
Zhou YA, Zhang T, Zhao JB, Wang XP, Jiang
T, Gu ZP, Wang XN and Li XF: The adenovirus-mediated transfer of
PTEN inhibits the growth of esophageal cancer cells in vitro and in
vivo. Biosci Biotechnol Biochem. 74:736–740. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wills KN, Atencio IA, Avanzini JB,
Neuteboom S, Phelan A, Philopena J, Sutjipto S, Vaillancourt MT,
Wen SF, Ralston RO, et al: Intratumoral spread and increased
efficacy of a p53-VP22 fusion protein expressed by a recombinant
adenovirus. J Virol. 75:8733–8741. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zavaglia D, Favrot MC, Eymin B, Tenaud C
and Coll JL: Intercellular trafficking and enhanced in vivo
antitumour activity of a non-virally delivered P27-VP22 fusion
protein. Gene Ther. 10:314–325. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jin G, Zhou Y, Chai Q, Zhu G, Xu F and Liu
F: VP22 and cytosine deaminase fusion gene modified
tissue-engineered neural stem cells for glioma therapy. J Cancer
Res Clin Oncol. 139:475–483. 2013. View Article : Google Scholar
|
|
20
|
Nishikawa M, Otsuki T, Ota A, Guan X,
Takemoto S, Takahashi Y and Takakura Y: Induction of tumor-specific
immune response by gene transfer of Hsp70-cell-penetrating peptide
fusion protein to tumors in mice. Mol Ther. 18:421–428. 2010.
View Article : Google Scholar :
|
|
21
|
Yu X, Xu Z, Lei J, Li T and Wang Y: VP22
mediates intercellular trafficking and enhances the in vitro
antitumor activity of PTEN. Mol Med Rep. 12:1286–1290.
2015.PubMed/NCBI
|
|
22
|
Regberg J, Srimanee A and Langel U:
Applications of cell-penetrating peptides for tumor targeting and
future cancer therapies. Pharmaceuticals (Basel). 5:991–1007. 2012.
View Article : Google Scholar
|
|
23
|
Kueltzo LA, Normand N, O'Hare P and
Middaugh CR: Conformational lability of herpesvirus protein VP22. J
Biol Chem. 275:33213–33221. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Elliott G and O'Hare P: Intercellular
trafficking and protein delivery by a herpesvirus structural
protein. Cell. 88:223–233. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Persad S, Attwell S, Gray V, Delcommenne
M, Troussard A, Sanghera J and Dedhar S: Inhibition of
integrin-linked kinase (ILK) suppresses activation of protein
kinase B/Akt and induces cell cycle arrest and apoptosis of
PTEN-mutant prostate cancer cells. Proc Natl Acad Sci USA.
97:3207–3212. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li Z, Liu GX, Liu YL, Chen X, Huang XL and
Gan HT: Effect of adenovirus-mediated PTEN gene on ulcerative
colitis-associated colorectal cancer. Int J Colorectal Dis.
28:1107–1115. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Maehama T and Dixon JE: The tumor
suppressor, PTEN/MMAC1, dephosphorylates the lipid second
messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem.
273:13375–13378. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cully M, You H, Levine AJ and Mak TW:
Beyond PTEN mutations: The PI3K pathway as an integrator of
multiple inputs during tumorigenesis. Nat Rev Cancer. 6:184–192.
2006. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Leslie NR, Yang X, Downes CP and Weijer
CJ: PtdIns(3,4,5) P(3)-dependent and -independent roles for PTEN in
the control of cell migration. Curr Biol. 17:115–125. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Schwock J, Dhani N and Hedley DW:
Targeting focal adhesion kinase signaling in tumor growth and
metastasis. Expert Opin Ther Targets. 14:77–94. 2010. View Article : Google Scholar
|
|
31
|
Sieg DJ, Hauck CR, Ilic D, Klingbeil CK,
Schaefer E, Damsky CH and Schlaepfer DD: FAK integrates
growth-factor and integrin signals to promote cell migration. Nat
Cell Biol. 2:249–256. 2000. View
Article : Google Scholar : PubMed/NCBI
|